
Abstract
High-quality embryos give rise to embryonic stem cells (ESCs) at greater efficiencies than poor-quality embryos. However, most embryos available for human ESC derivation are of a reduced quality as a result of culture in relatively simple media up to 10 years earlier, before cryopreservation, or before compaction. In the present study, we used a mouse model to determine whether a culture with insulin from the 8-cell stage could increase the number of ESC progenitor epiblast cells in blastocysts, as well as endeavor to determine the molecular mechanism of the insulin's effect. Culture in media containing 1.7 ρM insulin increased epiblast cell number (determined by Oct4 and Nanog co-expression), and proportion in day 6 blastocysts. The inhibition of phosphoinositide 3 kinase (PI3K) (via LY294002), an early second messenger of the insulin receptor, blocked this effect. The inhibition of glycogen synthase kinase 3 (GSK3) or p53, 2 s messengers inactivated by insulin signaling (via CT99021 or pifithrin-α, respectively), increased epiblast cell numbers. When active, GSK3 and p53 block the transcription of Nanog, which is important for maintaining pluripotency. A simultaneous inhibition of GSK3 and p53 had no synergistic effects on epiblast cell number. The induced activation of GSK3 and p53, via the inhibition of proteins responsible for their inactivation (PKA via H-89 and SIRT-1 via nicotinamide, respectively), blocked the insulin's effect on the epiblast.From our findings, we conclude that insulin increases epiblast cell number via the activation of PI3K, which ultimately inactivates GSK3 and p53. Furthermore, we suggest that the inclusion of insulin in culture media could be used as a strategy for increasing the efficiency with which the ESC lines can be derived from cultured embryos.
Introduction
W
The addition of insulin to embryo culture media for development from the zygote or 2-cell stage has been previously shown as having beneficial effects, with an increased blastocyst formation rate [16], accelerated development [16,17], increased blastocyst cell number [16], stimulated protein synthesis [18 –20], reduced protein degradation [19], and increased protein uptake [19]. While the capacity of insulin or any other growth factor—many of which have similar effects to those just described [21,22]—to increase the number of epiblast cells is unknown, insulin differs from most in that it increases total cell number (TCN) by increasing inner cell mass (ICM) cell number [16]. The aim of the present study, therefore, was to determine whether the addition of insulin from the 8-cell stage increases the number of epiblast cells in the expanded blastocyst. We then undertook a series of experiments to investigate the involvement of phosphoinositide 3 kinase (PI3K), glycogen synthase kinase 3 (GSK3), and p53 in insulin's effect on epiblast cell number, as these are known pathways for insulin stimulation in other tissues [23 –26].
Materials and Methods
Experimental design
To determine the effect of insulin on epiblast cell number, an embryo culture medium was supplemented with 0, 0.17, 1.7, and 1,700 ρM insulin from the 8-cell stage in Experiment 1. In Experiment, 2 the effect of LY294002 [27], an inhibitor of PI3K—a second messenger present in the mouse preimplantation embryo involved in the maintenance of pluripotency [25,28]—was examined in the presence or absence of insulin to determine whether insulin was acting via PI3K. Since the PI3K second messenger pathway involves the subsequent phosphorylation and inactivation of GSK3 [29], which has been shown to be beneficial for maintaining cells in a pluripotent state in outgrown ICMs [30], we hypothesized that the activation of GSK3 would inhibit the positive response of insulin, while inactivation would mimic the effects of insulin. This hypothesis was tested in Experiment 3 using H-89 [31] in the presence or absence of insulin and CT99021 [32] for GSK3 activation and inhibition, respectively. Stimulation of the PI3K second messenger pathway can also result in the ubiquitination, inactivation, and degradation of pro-apoptotic protein p53 [23,33,34]. In the nucleus, p53 is known to directly inhibit the transcription of Nanog [35] and, therefore, inhibit pluripotency. To establish whether the positive effects of insulin on the epiblast were mediated via p53, Experiment 4 was performed where p53 was indirectly activated via acetylation due to nicotinamide [36,37] in the presence or absence of insulin and inhibited by pifithrin-α [38]. Finally, p53 and GSK3 are known to have significant cross talk, with GSK3 inactivation leading to p53 build up [39] and resultant apoptosis [40], or loss of p53 activity in different circumstances [41 –43]. In Experiment 5, we inhibited both factors at once to determine whether there was synergy.
Insulin and inhibitors
Chemicals used were purchased from Sigma-Aldrich, (St. Louis, MO) except where otherwise noted. In this study, all the chemicals and consumables used for standard embryo culture were tested for compatibility with embryo development in a one-cell mouse embryo assay [44]. Bovine insulin was used at concentrations of 0, 0.17, 1.7, and 1,700 ρM. LY294002 was used at 50 μM to inhibit PI3K; this concentration has been shown to reduce the phosphorylation of Akt and P70S6k in mESCs, which are phosphorylated as a result of PI3K activity (in the same experiment, LY294002 was shown to have the same effect on blastocyst cells as ESCs (induce apoptosis)) [27]. LY294002 has also been directly shown to be effective in decreasing PI3K activity in blastocysts, as evidenced by the decreased Akt phosphorylation at 250 μM [45]. Additionally, the treatment of ESCs with LY294002 reduced the phosphorylation of Akt at 5–30 μM, as well as S6 and GSK3 at 10–60 μM, which are also phosphorylated as a result of PI3K activity [46]. In an in vitro assay, LY294002 was shown to completely abolish PI3K activity at 100 μM [47]. CT99021 (Axon Medchem, Groningen, The Netherlands) was used to inhibit GSK3 (the concentrations studied were 0.04, 0.3, 3.0, and 15 μM; initial experiments using CT99021 at 30 μM showed a significant negative effect on the development of embryos). In [48], CT99021 at 3 μM was shown to decrease the phosphorylation of β-catnin, a GSK3 target in ESCs. Furthermore, a culture of ESCs in which GSK3 α and β had been deleted with CT99021 did not produce the effects seen when wild-type ESCs were cultured with CT99021 [48]. This decrease in β-catnin phosphorylation was also observed in embryos (bovine 2-cell) cultured with CT99021 at 3 μM, an effect that was the same as LiCl, another recognized inhibitor of GSK3 [49]. CT99021 at 1 μM was shown to reduce GSK3β activity to 1% in an in vitro assay in which the inhibitor was shown to be highly specific [32]. H-89 at 10 μM was used to activate GSK3, as it is a robust activator of GSK3 whose inclusion in culture medium has been shown to prevent or inhibit the phosphorylation of GSK3 (which would inactivate GSK3) in embryonic kidney cells at 10 μM [31,50], muscle cells at 50 μM [51], spermatozoa at 100 μM [52], and glioma cells at 10 μM [53]. Pifithrin-α was used to inhibit p53 at 30 μM [38], as it has been established as an effective inhibitor of p53 that blocks the activation of p53 responsive LacZ in ConA cells, and inhibits p53-mediated apoptosis at 10–20 μM [54]. Pifithrin-α at 10–20 μM is also able to affect p53-dependent cell-cycle checkpoint control, as it prevents gamma irradiation-induced arrest, but not in cells with no functional p53 [54]. Embryos cultured in the presence of pifithrin-α at 10–30 μM reproduced the effect of p53 deletion, and reversed the effect of culture in conditions known to induce increased p53 activity [38]. Pifithrin-α lowers the level of nuclear p53 at 10–20 μM in vitro and 2.2 mg/mL when injected in vivo [54,55]. Nicotinamide at 10 μM was used to activate p53; our group has previously used this inhibitor in embryo culture [36], as it has been shown to increase the levels of acetylated p53 (active p53) in lung, breast, and bone cancer cells [37] and induce apoptosis in a p53-dependent manner in chronic lymphocytic leukemia cells as well as increase p53 acetylation [56]. Where dimethyl sulfoxide (DMSO) was used to dissolve inhibitors, a vehicle control of an equal quantity of DMSO was included.
Embryo collection, culture, and assessment
Approval for all procedures was obtained from The University of Adelaide Animal Ethics Committee, in compliance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Mice were fed ad libitum, and kept in a 14:10 light:dark cycle. C57BL/6 female mice aged 3–4 weeks were injected intraperitonealy with of 5 IU equine chorionic gonadotropin (Folligon, Intervet Australia Pty Ltd, Bendigo, Victoria), followed by 5 IU human chorionic gonadotropin (hCG; Pregnyl, Organon, Sydney, Australia) 48 h later, inducing ovulation. The ovulating females were placed with a male mouse of the same strain, and mating was assessed the next morning by the presence of a vaginal plug. Zygotes were collected 22 h post hCG in MOPS-G1 [57], and cumulus cells were removed with 50 IU/mL hyaluronidase. Zygotes were cultured in 20 μL of G1.2 medium [44], in groups of 10 at 37°C in 6% CO2, 5% O2, and 89% N2 for 48 h. Embryos that reached the 8-cell stage after 48 h of culture were transferred to 20 μL drops of G2.2 supplemented with the relevant treatment, and cultured individually at 37°C in 6% CO2, 5% O2, and 89% N2 from 48 to 115 h. Since embryos secrete exogenous factors, which affect development when they are cultured in groups [58,59], individual culture was used to minimize the potentially confounding paracrine growth-factor effect.
Embryo development was assessed at 94 and 115 h. Embryos were scored as being in the following stages of development: arrested (embryos that failed to develop from the 8-cell stage), early blastocysts (embryos where the blastocoel cavity was <2/3rds the volume), blastocysts (embryos with a blastocoel cavity >2/3rds the volume), hatching blastocysts (embryos with cells herniating from the zona pellucida), and hatched blastocysts (embryos that had hatched completely from the zona pellucida).
Immunohistochemistry
Oct4 and Nanog, whose co-expression marks epiblast cells, were assessed in blastocysts at 115 h of culture using immunohistochemistry and confocal microscopy. Blastocysts were fixed overnight in 4% paraformaldahyde at 4°C. After fixation, the embryos were incubated for 5 min in 0.1M glycine in phosphate-buffered saline (PBS) at room temperature (RT). Blastocysts were permeablized in PBS with 0.25% TritonX-100 (PBS-TX) for 30 min at RT, then blocked in 10% Normal Donkey Serum (Sapphire Bioscience, Redfern, New South Wales, Australia) for 1 h at RT. Embryos were incubated with Nanog rabbit anti-mouse polyclonal antibody (Sapphire, Cat#120-21603 or Cosmo Bio, Tokyo, Japan, Cat#REC-RCAB0002P-F) at 1:200 and Oct-3/4 goat anti mouse polyclonal antibody (Santa Cruz Biotechnology inc, Santa Cruz, CA; sc-8628) at 1:100 overnight in PBS-TX at 4°C. Blastocysts were then washed in PBS-TX, and incubated with donkey anti-rabbit secondary antibody (1:100) conjugated to FITC (Australian Laboratory Services, Homebush New South Wales, Australia) and donkey anti-goat secondary antibody (1:100) conjugated to Rhodamine (Jackson immunoResearch, West Grove, PA; 705-025-003) for 2 h at 37°C. Embryos were then incubated with 3 nM of 4′-6-diamidino-2-phenylindole (DAPI), a nuclear stain, for 2–3 min at RT, before being examined by confocal microscopy (Nikon, EZ-C1 software or Calcium Lecia SP5, Lecia SP5 software). The number of cell nuclei stained blue by DAPI gave TCN; ICM cell number was the number of cells whose nuclei were stained red by rhodamine, indicating the presence of Oct4, and epiblast cell number was the number of red ICM nuclei that were also stained green by FITC, indicating the presence of both Oct4 and Nanog. Trophectoderm cell number was TCN minus ICM cell number, and primitive endoderm cell number was ICM cell number minus epiblast cell number. A cell was counted as being positive for the flurophore in question when the intensity of the stain was sufficient for the nucleus to be clearly distinguished from the background. A negative control was assessed where the primary antibody was not applied.
Statistics
All data are expressed as mean±sem. The treatment group was fitted as a fixed factor, and the replicate was fitted as a cofactor in all analyses. Data were analyzed using Univariate General Linear Model using PASW Statistics 17 or chi-square tests. Between-treatment differences were assessed using the Least Significant Difference method. Values such as P<0.05 were considered significant.
Results
Experiment 1: effect of insulin on epiblast cell number
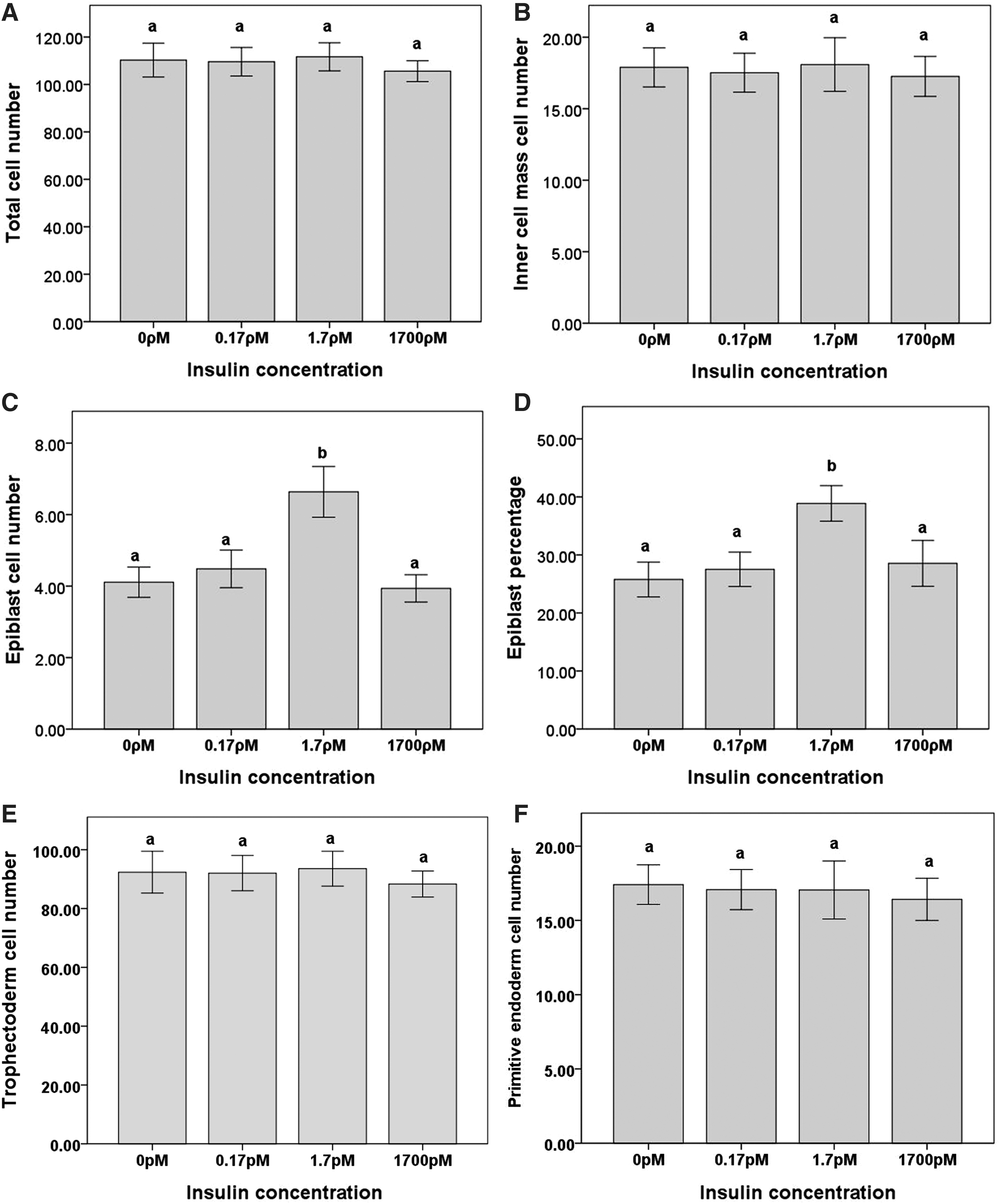
The supplementation of culture medium with insulin (0, 0.17, 1.7, and 1,700 ρM) from the 8-cell stage had no effect on the development at 94 h (blastocyst or hatching blastocysts for all treatments, range 81%−87%) or 115 h (hatching blastocyst or hatched blastocyst for all treatments, range 77%−86%). At 115 h of culture, there was no effect of the insulin on TCN, ICM, trophectoderm, or primitive endoderm cell numbers (Fig. 1A, B, E, and F). However, culture in the presence of 1.7 ρM insulin significantly increased epiblast cell number (P<0.05, Fig. 1C). Since the epiblast exists as a subpopulation of the ICM, this meant that there was an increase in the proportion of ICM cells which were ESC progenitor epiblast cells in the presence of 1.7 pM insulin (P<0.05, Fig. 1D). Representative images of blastocysts cultured in the control treatment or 1.7 ρM of insulin and stained for Oct4, Nanog, and TCN are supplied in Fig. 2.
Blastocyst culture in the presence of insulin.

Representative images of blastocysts stained for Oct4, Nanog, and TCN after culture in control medium and medium supplemented with 1.7 ρM insulin.
Experiment 2: effect of PI3K inactivation on epiblast cell number in embryos cultured in the presence or absence of insulin
To determine whether the effect of insulin was a result of PI3K stimulation, PI3K was inactivated with LY294002 at 50 μM [27] in the presence or absence of insulin. At 94 h of culture, LY294002 prevented hatching in either the presence or absence of insulin, and significantly increased the number of arrested or degenerate embryos in the presence of insulin (P<0.01, Table 1). At 115 h, the inhibition of PI3K in the presence of insulin significantly reduced the percentage of embryos that developed in the blastocyst stage, with hatching reduced in either the presence or absence of insulin (P<0.001, Table 2).
Mean percentage of embryos at stage of development after 94 h of culture in the specified treatment.
n≥55 per treatment.
Like pairs, a and b, are significantly different (P<0.01).
Mean percentage of embryos at stage of development after 115 h of culture in the specified treatment.
n≥55 per treatment.
Like pairs, a and b are significantly different (P<0.001).
PI3K inhibition significantly reduced TCN and ICM cell number, irrespective of the presence of insulin (P<0.01, Fig. 3A, B). However, while the inhibition of PI3K in the absence of insulin did not affect epiblast cell number compared with the control, PI3K inhibition prevented the stimulatory effect of insulin on epiblast cell number (P<0.01, Fig. 3C).

PI3K inhibition during blastocyst culture in the presence of insulin.
Interestingly, insulin in the presence of PI3K inhibition increased ICM size compared with embryos cultured in LY294002 alone (P<0.01, Fig. 3B), suggesting that insulin is able to affect ICM size through signaling pathways other than PI3K.
Experiment 3: effect of GSK3 activation/inhibition on epiblast cell number in embryos cultured in the presence or absence of insulin
GSK3 activation by the PKA inhibitor H-89 [31] had no effect on the development at 94 h of culture (blastocyst or hatching blastocysts for all treatments, range 84%−94%); however, it significantly reduced the number of blastocysts that had hatched in the presence or absence of insulin after 115 h of culture (P<0.01, Table 3). Furthermore, GSK3 activation did not alter TCN (Fig. 4A), but significantly decreased the ICM cell number with or without insulin (P<0.05, Fig. 4B). The activation of GSK3 in the absence of insulin did not affect epiblast cell numbers relative to the control, but insulin's stimulation of epiblast cell number was prevented by the activation of GSK3 (P<0.05, Fig. 4C). The percentage of the ICM that was epiblast was unchanged with GSK3 activation with or without insulin (Fig. 4D).

GSK3 activation during blastocyst culture in the presence of insulin.
Mean percentage of embryos at stage of development after 115 h of culture in the specified treatment.
n≥29 per treatment.
Like pairs, a and b are significantly different (P<0.01).
The inactivation of GSK3 with CT99021 had no effect on the development at 94 h (blastocyst or hatching blastocyst for all treatments, range 75%−85%) or 115 h (hatching blastocyst or hatched blastocyst for all treatments, range 54%−67%) of the culture. However, GSK3 inhibition significantly increased ICM cell number (20.1±1.2 compared with control 15.9±1.2 P<0.01; n≥50) and epiblast cell number (5.1±0.3 compared with control 3.2±0.3 P<0.001; n≥50), although the epiblast proportion (23.2±2.5 compared with control 23.8±2.5 NS; n≥50) and TCN (111.7±4.4 compared with control 107.2±4.4 NS; n≥50) were not affected.
Experiment 4: effect of p53 activation/inhibition on epiblast cell number in embryos cultured in the presence or absence of insulin
The activation of p53 by nicotinamide in the presence or absence of insulin had no effect on embryo development at 94 h (blastocyst or hatching blastocyst for all treatments, range 88%−98%) or 115 h (hatching blastocyst or hatched blastocyst for all treatments, range 69%−84%), nor TCN (Fig. 5A). However, the activation of p53 significantly reduced ICM cell number both with and without insulin (P<0.05, Fig. 5B). The activation of p53 did not affect epiblast cell numbers compared with control-treated embryos; however, insulin's increase of epiblast cell numbers was prevented (P<0.05, Fig. 5C). The inactivation of p53 had no significant effect on the percentage of the ICM that was epiblast (Fig. 5D).

p53 activation during blastocyst culture in the presence of insulin.
The inhibition of p53, with pifithrin-α (30 μM) [38], was able to mimic the effects of insulin with no effect on blastocyst development at 94 h (blastocyst or hatching blastocyst for all treatments, range 85%−86%) or 115 h (hatching blastocyst or hatched blastocyst for all treatments, range 75%−80%). However, TCN was increased (132±7.0 compared with control 111.7±6.6 P<0.05; n≥24) as was ICM (30.5±2.4 compared with control 18.1±2.2 P<0.001; n≥24) and epiblast cell number (7.1±0.6 compared with control 4.1±0.6 P<0.01; n≥24) that were increased by p53 inhibition, while the percentage of ICM that was epiblast was unchanged (14.8±3.7 compared with control 13.6±3.4 NS; n≥24).
Experiment 5: interaction of GSK3 and p53 signaling
Our results show that insulin acts on the epiblast via the inactivation of GSK3 and p53. The inactivation of one factor may reduce the other's activity [41 –43], but the inactivation of GSK3 can also lead to p53 accumulation [39] and apoptosis [40]. If the inactivation of GSK3 causes p53 accumulation, then inactivation of both GSK3 and p53 may have a synergistic effect.
The inactivation of GSK3 together with p53 had no effect on the development at 94 h (blastocyst hatching blastocyst for all treatments, range 89%−97%), but significantly increased the number of hatched blastocysts relative to the culture with either inhibitor alone (P<0.05, Table 4). However, there were no further synergistic effects on TCN, ICM, or proportion of epiblast compared with either inhibitor alone (Fig. 6).

Duel inhibition of GSK3 and p53.
Mean percentage of embryos at stage of development after 115 h of culture in the specified treatment.
n≥39 per treatment.
Like pairs, a and b are significantly different (P<0.05).
Discussion
The present study demonstrated that the culture of 8-cell embryos with 1.7 ρM insulin increases the number of epiblast cells in the ICM without affecting the size of the ICM itself and without increasing trophectoderm, primitive endoderm, or TCN. This suggests that rather than acting as a nonspecific mitogenic factor, insulin is specifically acting on the ICM to shift the ratio of epiblast and primitive endoderm toward a larger population of pluripotent cells. Since ESCs are derived from the pluripotent epiblast [13,60], the number of pluripotent cells in a blastocyst is a key determinant of the capacity of a blastocyst to give rise to an ESC line [7,48,61]. Therefore, it is desirable to have conditions for the development of the blastocyst that maximize the number of epiblast cells. Although treatments using inhibitors can produce larger epiblasts [7,62], inhibitors frequently have nonspecific activities [32,63], while insulin is present in vivo and a component of routine ESC culture, although not routine in embryo culture. As such, culture in insulin could be a useful strategy for improving the pluripotential of embryos cultured in vitro, such as human embryos that are often donated for hESC derivation at the cleavage stage after 5–10 years of cryopreservation and culture in media now known to be perturbing [12]—without the use of inhibitors.
Insulin binds to the insulin receptor (IR), which phosphorylates tyrosine residues on the insulin receptor substrates (IRS-1, IRS-2 and IRS-3), thus enabling the IRSs to activate PI3K via its SRC homology 2 domains [64,65]. PI3K is a second messenger known to be present in the mouse preimplantation embryo [25]. Previous studies have demonstrated that PI3K inhibition impairs embryo development and reduces TCN [27,45], a result that we reproduced in this study. Importantly, in ESCs, PI3K has been shown to play a role in maintaining pluripotency [28,46], and its inhibition in ESCs decreases Nanog protein levels [28]. Our results suggest that the activation of PI3K in the later-stage embryo is a part of a signaling pathway involved in insulin's stimulation of epiblast cell numbers.
An intermediate of the PI3K signaling pathway, GSK3, has been shown to decrease Nanog transcription and retention of pluripotency via inactivation of β-catenin [66 –68], Hedgehog [69,70], and c-Myc [71,72]. Additionally, active GSK3 protects the intracellular domain of Notch from degradation [73], increasing differentiation [74]. The indirect activation of GSK3 by H-89 [31]—which inhibits PKA whose activity would otherwise prevent GSK3 activation [75 –77]—was sufficient to block insulin's ability to increase epiblast cell number. We hypothesized that the inhibition of GSK3 would have the opposite outcome and replicate the effects of insulin on the epiblast. GSK3 inhibition increased epiblast cell number, similar to that seen with insulin supplementation. Taken together, these data suggest that insulin increases the proportion of epiblast cells in the ICM through mechanisms which involve the inactivation of GSK3.
This is in keeping with previous studies where GSK3 inactivation via BIO increased the percentage of epiblast in outgrown ICMs and increased ESC derivation efficiency [30]. Additionally, the GSK3 inhibitor CT99021 is one of the 3 inhibitors that make up the 3i culture system which improves ESC culture and derivation as well as increasing epiblast proportion of the ICM to close to 100% (the other 2 inhibitors inhibit FGFR and MEK signaling) [7]. However, embryos cultured with GSK3 inhibitor LiCl have reduced hatching and attachment rates [78] and often fail to develop past the 2-cell stage [79]. In our own results, we saw an increase in epiblast cell number at 0.3 μM CT99021, but this increase was reduced at 3 μM and lost at 15 μM (data not shown). Additionally, at 94 h, 15 μM reduced hatching, reproducing previous findings [78] but with a different inhibitor, thereby suggesting that negative effects are not due to nonspecific activity by the inhibitor at high concentrations, but that GSK3's broad influence [80,81] makes moderate inhibition the key to a successful embryo culture.
The activation of PI3K is also able to exert significant effects on p53 availability and activity. As a pro-apoptotic protein, active p53 causes cell death and differentiation [35,82]. Specifically, active p53 has been shown to cause differentiation in ESCs by binding to the Nanog promoter region and repressing expression [35]. Additionally, the in vitro culture of embryos has been shown to increase p53 activity, with p53-dependant negative effects on embryo development and viability [83], while culture with pifithrin-α, an inhibitor of p53, has been shown to improve the proportion of embryos that develop to the blastocyst stage [38]. When p53 was indirectly activated by inhibiting SIRT-1 [37] with nicotinamide, insulin's ability to increase epiblast cell number was, as with GSK3 activation and PI3K inhibition, ameliorated. Furthermore, the inhibition of p53 had the opposing effect of increasing TCN, ICM, and epiblast cell numbers. This shows that the inhibition of p53 can mimic insulin's effect on the epiblast, suggesting that p53 inactivation is involved in insulin-mediated increase of epiblast cell number.
The beta isoform of GSK3 (GSK3β) can phosphorylate and form a complex with p53, which increases the activity of both GSK3β and p53 [41 –43]; however, active GSK3 activates MDM2 [39], which targets p53 for degradation. Therefore, the inactivation of GSK3 can cause the accumulation of p53 [39] and apoptosis [40]. This makes it difficult to say whether the effect of GSK3's inhibition on epiblast cell number is helped or hindered by its role in p53 regulation, and suggested to us that GSK3 inactivation coupled with p53 inactivation may be necessary for optimal epiblast increases.
However, the inhibition of both GSK3 and p53 produced no additional synergistic effect on epiblast cell number, suggesting that the effect of GSK3 inhibition at 0.3 μM CT99021 on epiblast cell number is not being limited by an increase in p53 due to the inactive GSK3's inability to phosphorylate and activate MDM2.
Our results show that the culture of embryos from the 8-cell stage with insulin increases the number and percentage of epiblast cells in the ICM of blastocysts via the activation of PI3K, which, in turn, inactivates GSK3 and p53. While being involved in many signaling pathways with many different targets, in their active forms, both GSK3 and p53 inhibit Nanog transcription [28,35], making it likely that insulin's apparent inactivation of these factors increases Nanog transcription, resulting in more Nanog-positive epiblast cells. Since epiblast cells are the progenitor cells that give rise to ESCs, a culture with insulin offers a potential strategy, effective from the cleavage stage, for increasing the derivation efficiency of ESCs from in vitro cultured embryos, though this will require validation by the derivation of ESCs. This is of particular importance for hESC derivation, which often utilizes embryos cryopreserved at the cleavage stage after culture 5–10 years ago in relatively simple media.
Footnotes
Acknowledgments
The authors acknowledge the support of the NHMRC program grant for funding. Dr. Michelle Lane is a recipient of an NHMRC Senior Research Fellowship. The authors thank Alicia Filby, Sarah L. Wakefield, and Nicole Palmer for their technical assistance.
Author Disclosure Statement
No competing financial interests exist.