
Abstract
Dogs provide a more clinically relevant model of human disease than rodents, particularly with respect to hereditary diseases. Thus, the availability of canine stem cells will greatly facilitate the use of the dog in the development of stem cell-based gene therapies and regenerative medicine. In this study we describe the production of canine induced pluripotent stem cells (ciPSCs) from adult dermal fibroblasts. These cells have a morphology resembling previously described canine embryonic stem cells, a normal karyotype, and express pluripotency markers including alkaline phosphatase, Nanog, Oct4, Telomerase, SSEA1, SSEA4, TRA1-60, TRA1-81, and Rex1. Furthermore, the inactive X chromosome is reactivated indicating a ground-state pluripotency. In culture they readily form embryoid bodies, which in turn give rise to cell types from all 3 embryonic germ layers, as indicated by expression of the definitive endoderm markers Cxcr4 and α-fetoprotein, mesoderm markers Collagen IIA and Gata2, and ectoderm markers βIII-tubulin, Enolase, and Nestin. Of particular significance is the observation that these ciPSCs are dependent only on leukemia inhibitory factor (LIF), making them similar to mouse and canine embryonic stem cells, but strikingly unlike the ciPSCs recently described in two other studies, which were dependent on both basic fibroblast growth factor and LIF in order to maintain their pluripotency. Thus, our ciPSCs closely resemble mouse ESCs derived from the inner cell mass of preimplantation embryos, while the previously described ciPSCs appear to be more representative of cells from the epiblast of mouse postimplantation embryos.
Introduction
E
Dogs are a more applicable model for human disease than are rodents for a number of reasons, but perhaps most practically, because the lifespan of dogs is considerably longer than that of rodents, allowing for more long-term studies than what is currently afforded by a dependency on rodent models of disease and therapeutics—this is particularly important when examining the survival and long-term safety and efficacy of transplanted stem cells. Dogs also more closely approximate the size of humans than do rodents and so the demands on cell proliferation are more similar. A further consideration is that the genetic backgrounds of both dogs and humans are heterogeneous, whereas those of mouse models are more homogeneous; the effects of this can be seen in gene-knockout studies in the mouse where the resultant phenotype can vary depending on the strain of mouse used and does not always recapitulate the equivalent human condition [2]. Approximately 481 hereditary diseases have been identified in the dog and, significantly, around half of these are orthologs of human diseases, including Alzheimer's disease, various cardiomyopathies, retinal atrophy, muscular dystrophy, prostate cancer and breast cancer [3,4]. And finally, the construction of a high-quality draft of the canine genome is now complete [5], which further expands the potential of the dog as a model for genetic and acquired disease, and makes possible the genetic manipulation of canine stem cells such as ESCs and iPSCs.
The benefit of canine iPSCs (ciPSCs) over ESCs is that, being patient-specific, they are more suitable for transplantation back into the study animal from which they were derived without having to address issues of immune rejection. Furthermore, the production of ciPSCs from individuals with genetically based diseases allows for the in vitro dissection of the events at the genomic, proteomic, and epigenetic levels that are involved in the disease process and, ultimately, also allows for the modeling of gene therapies and the testing of drug efficacies.
Here we report the generation of ciPSCs from adult dermal fibroblasts, which express key pluripotency markers and maintain their pluripotency and ability for self-renewal over many passages. Significantly, the ciPSCs in this study are leukemia inhibitory factor (LIF)-only dependent, which distinguishes them from the ciPSCs that have been described in 2 recent studies that require both LIF and basic fibroblast growth factor (bFGF) in order to remain pluripotent [6,7]. Thus, our ciPSCs closely resemble mouse ESCs derived from the inner cell mass of preimplantation embryos, while the previously described ciPSCs appear to be more representative of cells from the epiblast of mouse postimplantation embryos.
Materials and Methods
All use of animals, and tissues obtained from animals, was approved by the University of Queensland Animal Ethics Committee.
Derivation of dermal fibroblasts
Canine dermal fibroblasts were derived from a 6-mm diameter skin punch biopsy, taken from the ventral abdomen of an adult female mixed-breed dog, according to standard protocols. Briefly, tissue was minced into approximately 1.0-mm3 pieces and cultured under coverslips in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Invitrogen Australia) with 10% (v/v) fetal calf serum (FCS) (JRH Biosciences), 0.1 mM nonessential amino acids (NEAA) (Gibco), 6 mM
Lentivirus production and transduction of canine fibroblasts
Commercial pseudotyped lentiviruses expressing human OCT4, SOX2, KLF4, c-MYC, LIN28, and NANOG were obtained from Addgene. 293FT cells were transfected in 293FT medium consisting of DMEM high glucose (Gibco) with 10% (v/v) FCS, 0.1 mM NEAA, 6 mM
Cell culture of canine iPSCs
Putative ciPSC colonies were maintained in mouse ESC medium, with the supplements described above, at 37°C, 5% CO2, until they were large enough to be mechanically isolated, at which point they were subcultured onto feeder layers of irradiated mouse embryonic fibroblasts (MEFs) at a density of 20,000 MEFs per 1 mL volume organ culture plate (Costar, Corning Life Sciences). Once colonies were transferred to MEFs, they were maintained in mouse ESC medium with one of three types of supplementation: (1) with all supplements as described above (ie, 1 mM valproic acid, 1,000 U/mL murine LIF, 6 ng/mL human bFGF, 3 μM GSK3β inhibitor, 0.5 μM MEK inhibitor and 0.25 μM TGF-β antagonist); (2) with 1,000 U/mL murine LIF and 6ng/mL human bFGF only; or (3) with 1,000 U/mL murine LIF alone. Colonies were mechanically isolated and subcultured onto fresh MEFs approximately every 7 days. Medium was changed daily.
Alkaline phosphatase staining and fluorescence immunocytochemistry
Alkaline phosphatase staining was performed using the Quantitative Alkaline Phosphatase Detection Kit (Millipore) as per the manufacturer's instructions.
For fluorescence immunocytochemistry, colonies were washed in phosphate buffered saline (PBS) (Gibco) three times for 1 min each, followed by fixation in 4% paraformaldehyde for 10 min at room temperature (RT). Colonies were then washed three times for 1min each before permeabilization with 0.5% Triton X-100 (v/v) (Ajax Finechem, Thermo Fisher Scientific) in PBS for 15 min. After three further 1 min washes in PBS, colonies were blocked with 10% (v/v) goat serum (Gibco) in 0.1% Triton X-100-PBS for 1 h at RT. Blocking solution was removed and colonies were then incubated overnight at 4°C with primary antibody diluted in 10% goat serum/0.01% Tween-PBS. The primary antibodies used, and their respective dilutions, are as follows: anti-stage-specific embryonic antigen (SSEA)1 (MAB4301, Millipore), 1:20; anti-SSEA4 (MAB4304, Millipore), 1:50; anti-TRA1-60 (MAB4360, Millipore), 1:50; anti-TRA1-81 (MAB4381, Millipore), 1:50; anti-OCT4 (MAB4401, Millipore), 1:50; anti-REX1 (ab50828-50, Abcam, Sapphire Bioscience), 1:50; and anti-H3K27me3 (ab6002, Abcam), 1:50. Negative control colonies were incubated with 10% goat serum/0.01% Tween-PBS in place of primary antibody. The following day, colonies were given three 1-min washes with 0.1% Tween-PBS before applying the secondary antibody diluted 1:1,000 into 10% goat serum/0.01% Tween-PBS, and incubated at RT for 1 h. The secondary antibodies used were Alexa Fluor goat anti-mouse immunoglobin G, H & L chain (IgGH & L) (Invitrogen), Alexa Fluor goat anti-mouse IgM, and Alexa Fluor donkey anti-rabbit IgGH & L. Colonies were visualized and photomicrographs taken on an Olympus 1X51 inverted fluorescence microscope with Q Imaging camera and software.
Karyotyping
Canine iPSCs were commercially karyotyped by Sullivan Nicolaides Pathology (Taringa, Qld). Fifteen G-banded metaphase spreads were examined for each canine iPSC line.
RNA isolation, cDNA synthesis, and polymerase chain reaction
Cultures of canine dermal fibroblasts, ciPSC colonies, and embryoid bodies (EBs) were washed once with PBS and then lysed directly on the tissue culture plate using the lysis buffer from the Qiagen RNeasy Mini kit (Qiagen). Total RNA was then extracted as per the manufacturer's protocol. Complementary DNA was synthesized using the Bio-Rad iScript Reverse Transcriptase kit (Bio-Rad Laboratories) according to the manufacturer's instructions.
Polymerase chain reaction (PCR) was performed using Thermopol Taq DNA polymerase (New Zealand Biolabs, Thermo Fisher Scientific) and the following cycle parameters: denaturation at 94°C, 3 min; amplification for 35 cycles with melting at 95°C for 15 s, annealing at 58°C for 20 s, and extension at 72°C for 35 s; followed by a final extension at 72°C for 5 min. The primers used, and their product size, are listed in Supplementary Table S1; Supplementary Data are available online at
Teratoma formation
Colonies were mechanically passaged to remove any differentiated fibroblast-like cells from the population before trypsinization (TrypLE, Gibco) to produce small aggregates of ciPSCs for injection. Approximately 1×106 ciPSCs at passages 5 and 26, from 2 different clones, were injected into the thigh muscles of each of 8 female NOD.CB17-Prkdcscid /J mice. Mice were examined for a period of up to 16 weeks for the development of tumors, at which point any tumors present were resected, fixed in 4% paraformaldehyde, and processed for routine histology. Paraffin sections were cut at 7 μm, mounted onto glass slides, and stained with haematoxylin and eosin as per standard protocols. Slides were visualized on an Olympus 1X51 inverted microscope with Q Imaging camera and software.
In vitro differentiation
Canine iPSC colonies were mechanically passaged to remove any differentiated fibroblast-like cells from the population and then passaged with trypsin (TrypLE, Gibco) to produce small aggregates of ciPSCs. Cells were cultured in Costar Ultra-low Attachment plates (Corning Life Sciences) in mouse ESC medium supplemented with 5% FCS but without growth factors. After 7 days, EBs were transferred to 0.1% gelatin-coated tissue culture plates. Differentiating EBs were maintained in mouse ESC medium supplemented with 5% FCS for 4 weeks before being used for total RNA extraction.
Results
Generation of canine iPSCs
We have successfully reprogrammed adult canine dermal fibroblasts into ciPSCs using lentiviruses expressing human OCT4, SOX2, KLF4, c-MYC, NANOG, and LIN28. Canine iPSC colonies were first noted 12 days after transduction (Fig. 1A) and displayed a characteristic ESC-like morphology, being slightly domed, tightly-packed, and highly refractile, with cells having a high nuclear to cytoplasmic ratio (Fig. 1A, B). A total of 10 primary ciPSC clones were isolated from a total of 1.5×106 transduced fibroblasts, giving an efficiency of reprogramming of 0.0007%. Three clones were fully characterized and maintained in culture to beyond passage 40. All 10 clones retained their stem cell-like morphology after successive passages (Fig. 1B). After approximately passage 8, all 10 clones began to exhibit a corona of fibroblast-like cells at the periphery of the colony; typically, these cells appear at around 2 days after mechanical passage in which only undifferentiated cells are selected (Fig. 1C).
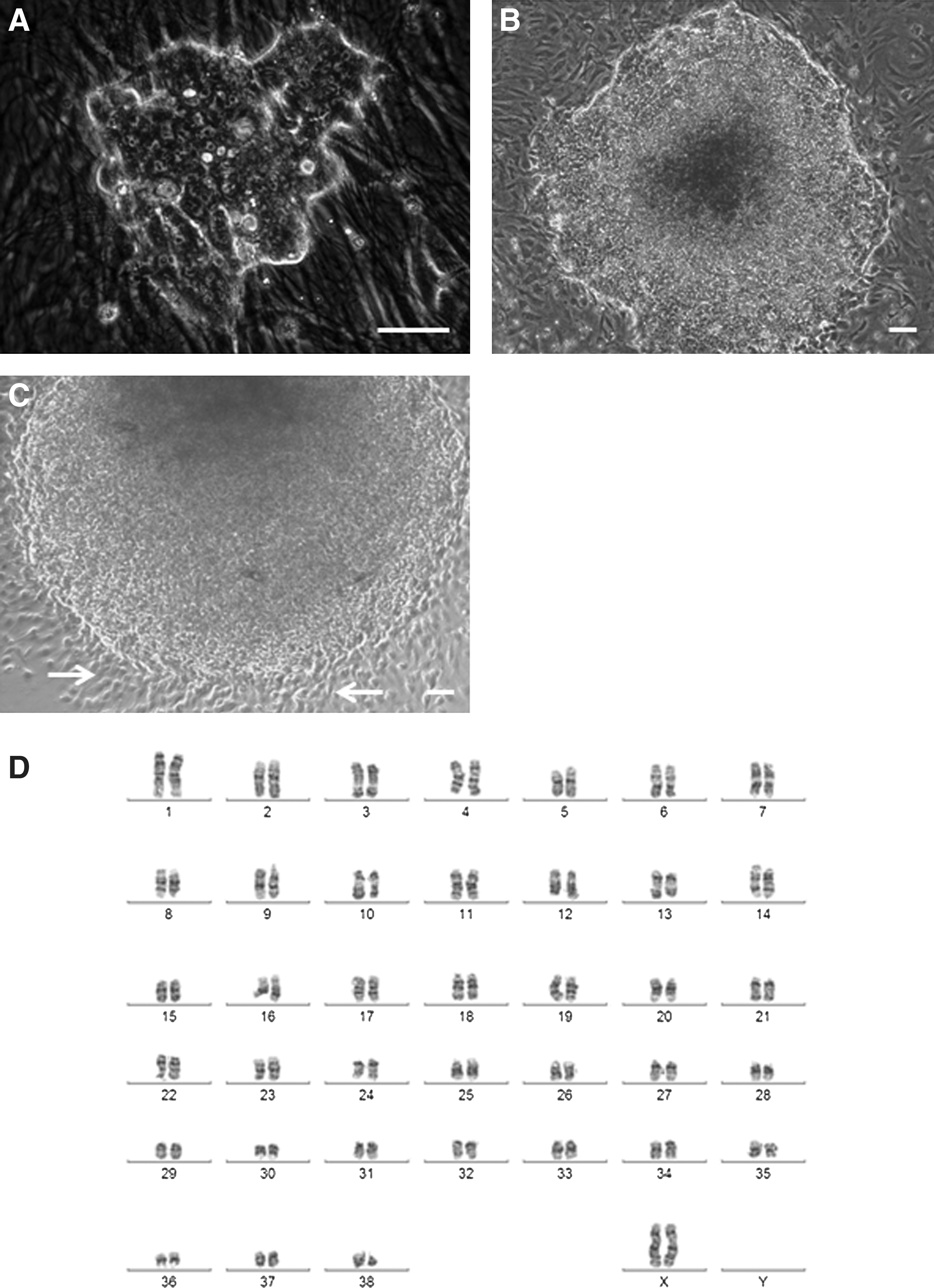
Morphology and karyotype of canine induced pluripotent stem cells (iPSCs).
Karyotype analysis of the 3 clones used for further characterization confirmed a normal 38, XX karyotype at passage 5 (Fig. 1D), with no gross structural rearrangements identifiable.
Canine iPSCs express pluripotency factors
Canine iPSCs show intense staining for alkaline phosphatase (Fig. 2A); significantly, fibroblast-like cells at the periphery of colonies and colonies that have spontaneously differentiated are negative for alkaline phosphatase activity (Fig. 2B).
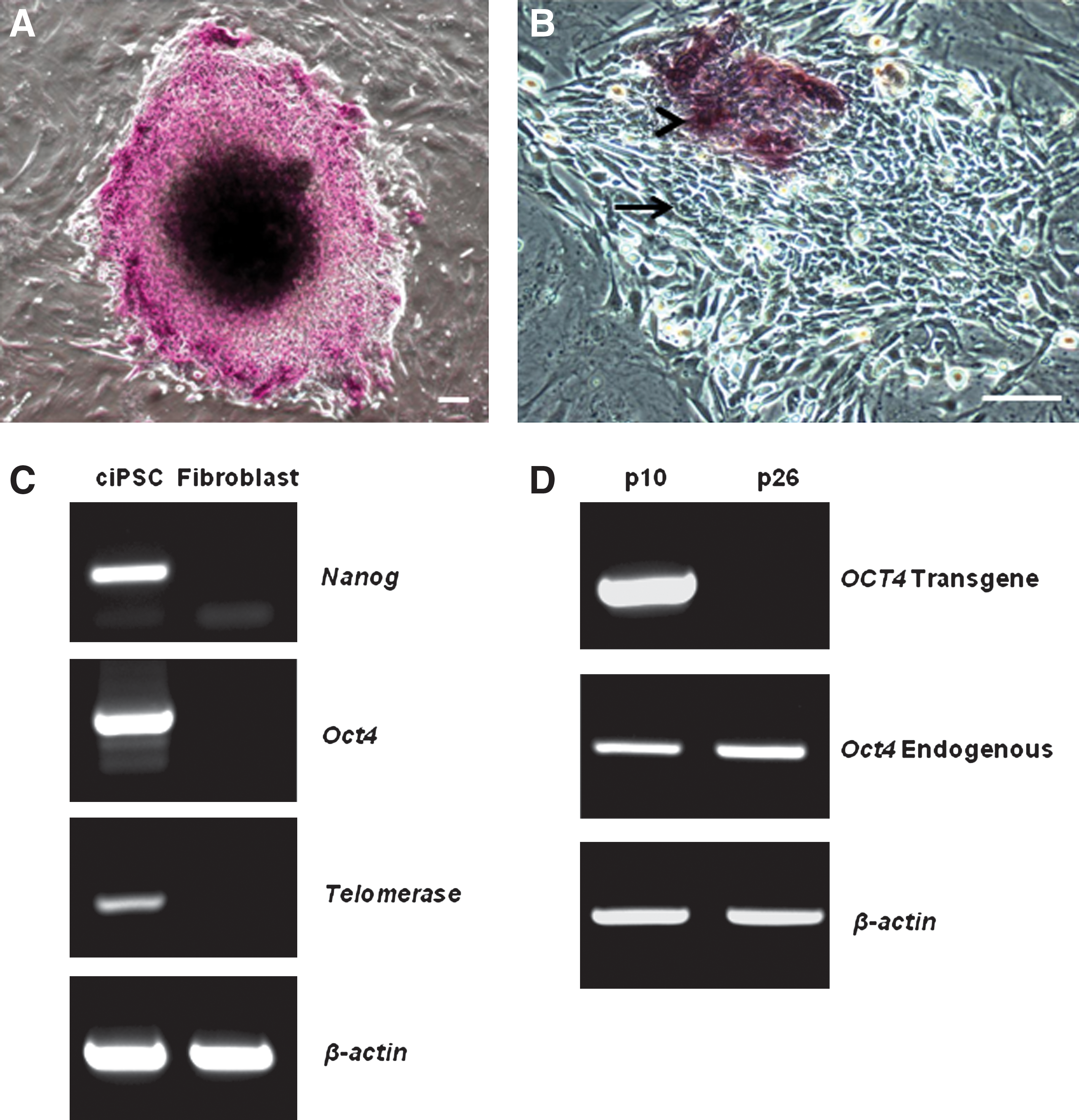
Expression of pluripotency factors by canine iPSCs.
Reverse transcriptase PCR (RT-PCR) was used to examine the expression of pluripotency factors by ciPSCs. Canine-specific primers for Nanog and Oct4 were designed to enable differentiation between transcripts of the human NANOG and OCT4 transgenes used for reprogramming and endogenous expression of canine Nanog and Oct4. Canine iPSCs express Nanog, Oct4, and Telomerase (Fig. 2C). In contrast, the canine dermal fibroblasts, from which the ciPSCs were generated, do not express Nanog, Oct4, or Telomerase. All water and reverse transcriptase-negative controls lacked amplicons (data not shown).
At passage 10, all 6 transgenes were still expressed at very high levels in all clones. However, by passage 26, expression of all of the transgenes was barely detectible, or absent, in the 3 clones analyzed. Expression of endogenous canine Nanog and Oct4 was present at passage 10 and at passage 26 when expression of the human NANOG (data not shown) and OCT4 transgenes had been extinguished (Fig. 2D).
Fluorescence immunocytochemistry for Oct4, SSEA1, SSEA4, TRA1-60, TRA1-81, and Rex1 was used to further confirm pluripotency in all 3 clones after passage 26, when expression of the human transgenes was known to be silent or barely detectable by RT-PCR. Canine iPSCs showed intense positive nuclear immunostaining for Oct4; positive staining was also observed in the fibroblast-like cells at the periphery of the colonies (Fig. 3A). Rex1 immunostaining was similarly very strong and localized to the nucleus of both ciPSCs within the colonies and also in the peripheral fibroblast-like cells (Fig. 3B). SSEA1 was detected at very low levels in colonies (Fig. 3C); it was not possible to be certain if peripheral fibroblast-like cells were definitively negative, because immunostaining may have been below a detectable threshold given that they were only a single layer of cells. Immunostaining for SSEA4 (Fig. 3D), TRA1-60 (Fig. 3E), and TRA1-81 (Fig. 3F) was present in ciPSC colonies, while peripheral fibroblast-like cells showed only background levels of immunostaining. Colonies incubated with buffer rather than primary antibody confirmed the specificity of the secondary antibodies (data not shown).
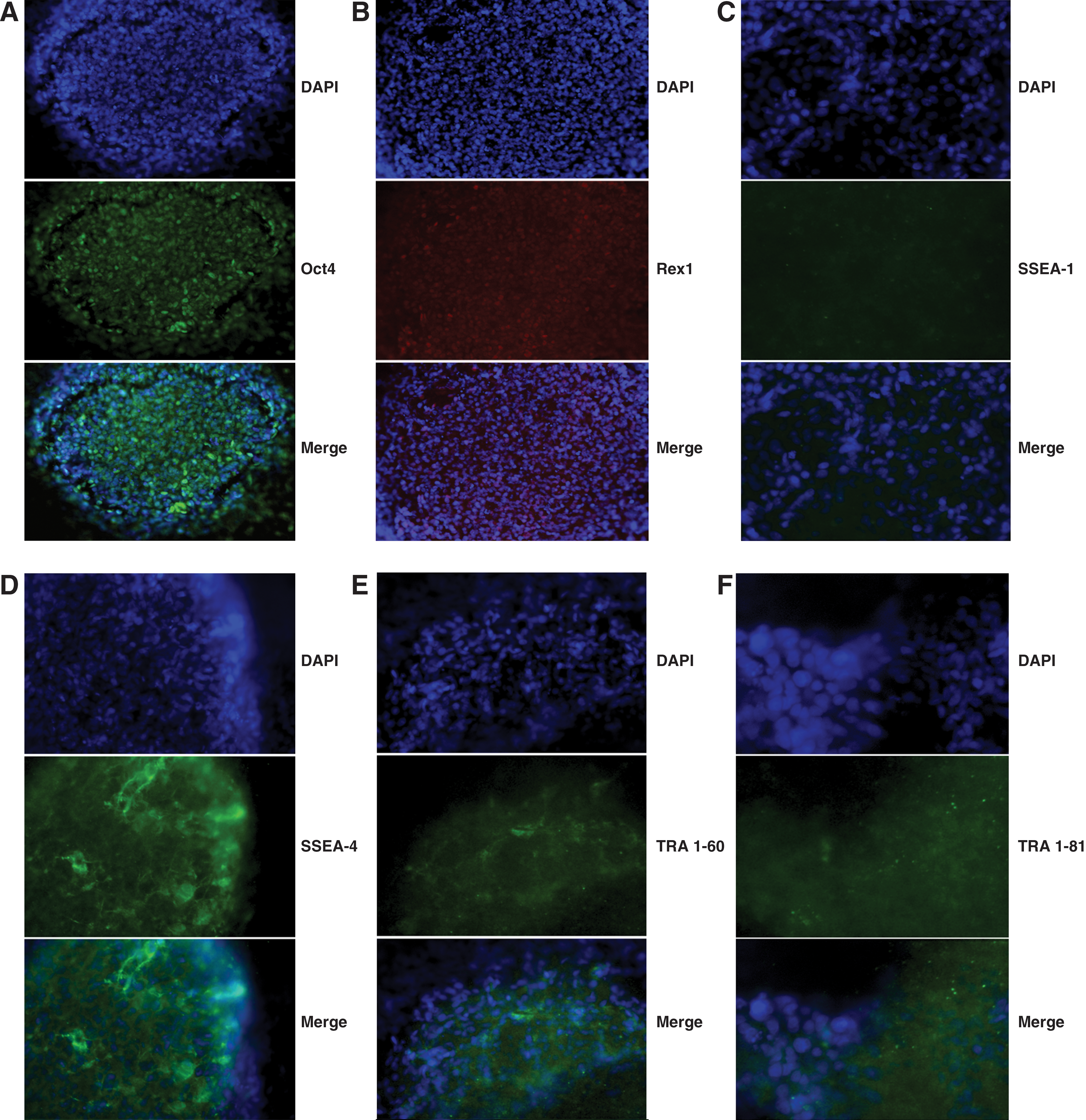
Immunofluorescence for expression of pluripotency factors by canine iPSCs at passage 26.
Canine iPSCs undergo X chromosome reactivation
Fluorescence immunocytochemistry for trimethylation of histone H3K27 was used to identify the inactive X chromosome in the female canine dermal fibroblasts that were used for reprogramming, in ciPSCs, and in the female fetus-derived MEFs upon which the ciPSCs were maintained. The canine dermal fibroblasts showed clear immunolabeling of the inactive X chromosome in its characteristic position at the periphery of the nucleus (Fig. 4A). Similarly, female MEFs also showed strong immunostaining of the inactive X chromosome at the periphery of the nucleus (Fig. 4B). In contrast, ciPSCs from all 3 clones, including the autologous fibroblast-like cells at the periphery of the colonies, consistently failed to show any immunostaining within the nucleus apart from the diffuse labeling (Fig. 4C) also seen in the fibroblasts and MEFs.
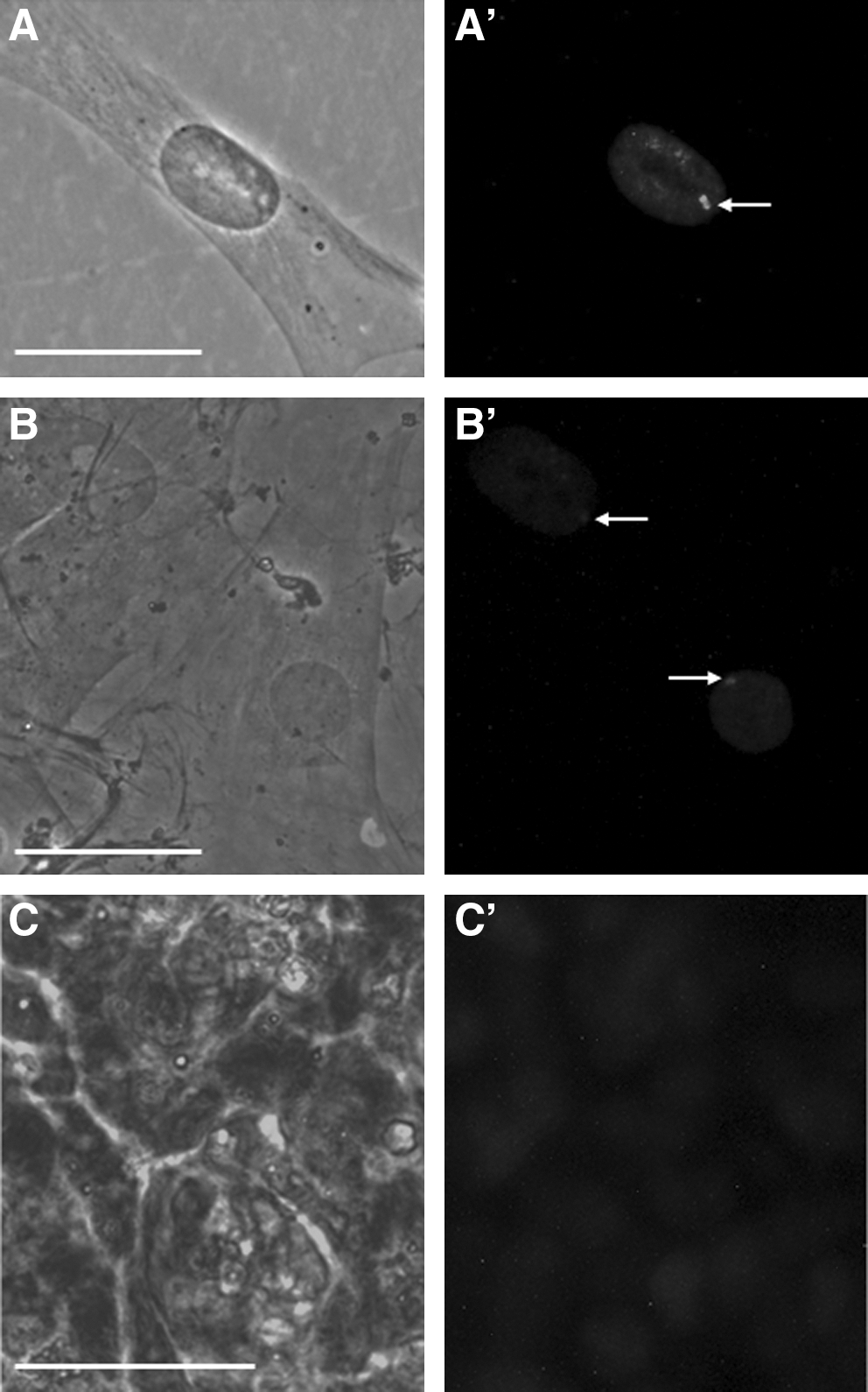
Immunofluorescence for trimethylation of histone H3K27, a marker of X inactivation.
Canine iPSCs form germ cell-like tumors in vivo
Of the 8 mice injected, only 2 yielded tumors, both from cells at passage 5 when the transgenes were still being transcribed, and from 2 different clones. Both of these contained discrete areas of cord-like structures which enclosed large cells that have the histological appearance of primordial germ cells (PGCs) (Fig. 5). Histological sections were submitted for examination by a specialist veterinary pathologist at the School of Veterinary Science, University of Queensland, and were determined to be “germ cell tumors consisting of organized cord-like structures delineated by a fibrovascular stroma with occasional collagenous connective tissue trabeculae, that are populated by large, undifferentiated cells that resemble primordial germ cells” (R. Allavena, written communication, March, 2011).

Canine iPSCs can form germ cell-like tumors in vivo when the transgenes are still being transcribed. Tumors contain cord-like structures (white arrows) which enclose large cells that have the appearance of primordial germ cells (black arrows), haematoxylin and eosin; 100×. Scale bar, 1.0 μm.
Canine iPSCs differentiate into cells from all 3 germ layers in vitro
Embryoid bodies cultured on 0.1% gelatin-coated plates in mouse ESC medium supplemented with 5% FCS, but without growth factors, differentiated into cell types from all 3 embryonic germ layers as determined by the expression of genes known to be indicative of ectoderm, mesoderm, and endoderm differentiation. After 4 weeks of culture, differentiated ciPSCs expressed ectoderm markers βIII-tubulin, Nestin, and Enolase (Fig. 6A), mesoderm markers Collagen IIA and Gata2 (Fig. 6A), and the endoderm markers AFP and Cxcr4 (Fig 6A). Colonies of undifferentiated ciPSCs and canine fibroblasts did not express any of the differentiation markers examined above (Fig. 6A). Following differentiation, expression of canine Oct4 was extinguished and that of canine Nanog was present at levels that were barely detectable (Fig 6B).

Expression of differentiation markers by canine iPSCs that have undergone spontaneous differentiation as embryoid bodies (EBs).
Canine iPSCs are dependent only on LIF
The requirement for each of LIF and bFGF for ciPSC maintenance was examined. When ciPSCs were grown in the absence of both LIF and bFGF, or with bFGF alone, they spontaneously differentiated into fibroblast-like cells. When colonies were cultured with both LIF and bFGF, they similarly differentiated into fibroblast-like cells or underwent cell death (Fig. 7). Colonies cultured with LIF alone maintained their pluripotency and self-renewal ability to beyond passage 40.
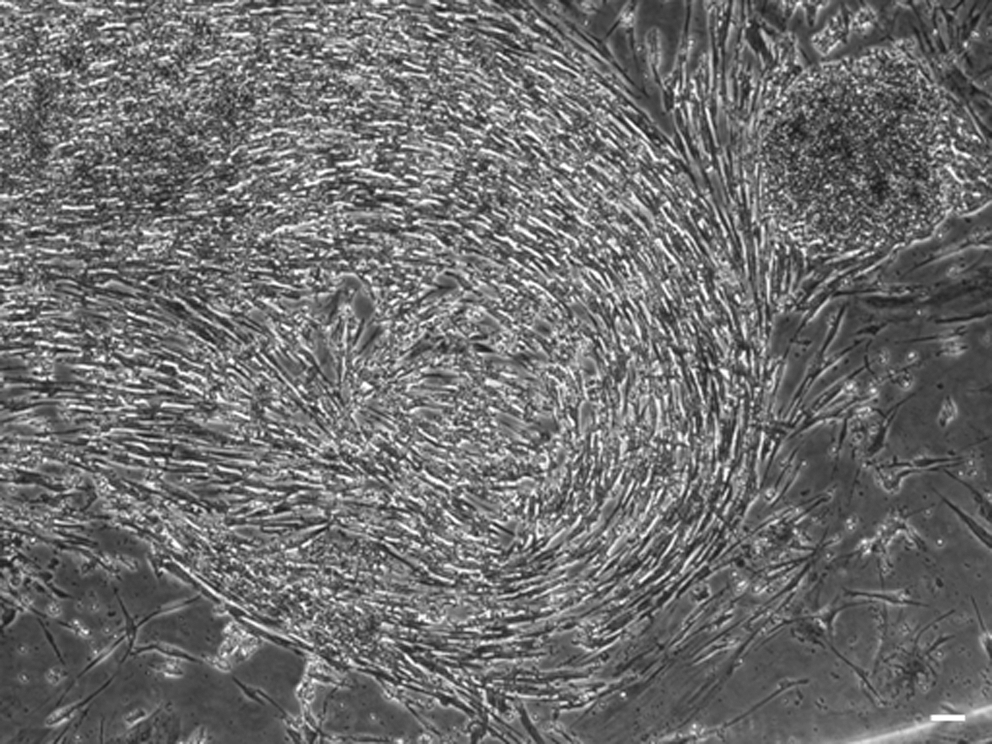
Canine iPSCs are dependent only on leukemia inhibitory factor (LIF) and become fibroblastic when cultured with LIF and basic fibroblast growth factor (bFGF); ×40. Scale bar, 1.0 μm.
Discussion
Dogs provide a clinically relevant model for the development of stem cell-based gene therapies and regenerative medicine, and hence, to date, 5 studies have described the isolation of canine ESCs [9 –13]. Wilcox and colleagues [13] clearly demonstrate 2 morphologically distinct types of canine ESC colonies dependent on the method by which the cells of the embryo were isolated. The first type, termed OVC.ID, was derived by mechanical dissociation of the inner cell mass of the blastocysts from the trophoblast, while the second type of colony, identified as OVC.EX, was derived from explant outgrowths of intact blastocysts [13]. Our canine iPSC colonies strongly resemble the OVC.EX colonies in having a slightly domed appearance that is more reminiscent of human ESCs than mouse ESCs [13]. This human ESC-like morphology has been used to describe the canine ESCs produced by other researchers [9,10,12]. In contrast, Hayes et al. [11] described their colonies as being particularly flat and resembling those of the mink. Remarkably, our ciPSC colonies further resemble the OVC.EX colonies described by Wilcox et al. [13] in that they also developed a layer of autologous fibroblast-like cells at the periphery of the colony.
A further differential characteristic that Wilcox and colleagues [13] describe between their two morphologically distinct cESC lines is the response of the OVC.EX lines to the presence of bFGF. When colonies of OVC.EX cESCs were cultured in medium supplemented with bFGF in addition to LIF, they differentiated into fibroblast-like cells [13]. In contrast, the OVC.ID cells remained unaffected. Our ciPSC colonies similarly differentiated into fibroblastic cells when cultured with bFGF and LIF. The cESCs produced by Hatoya et al. [9], Schneider at al. [10], and Hayes et al [11] were maintained in medium supplemented only with LIF. Vaags and colleagues [12], like Wilcox et al. [13], also obtained 2 morphologically distinct types of cESC colonies; however, both types were cultured in the presence of both LIF and bFGF with no indication of any adverse effects due to the presence of the bFGF. However, what is not clear from these studies where cESCs are cultured in medium containing both bFGF and LIF is whether the bFGF is required for the pluripotency and self-renewal of the cESCs, which is an altogether different situation to it simply not having any observable detrimental effects. Clearly, while the current lines of cESCs are similar in their LIF-dependency, they show a significant degree of heterogeneity morphologically and in their response to bFGF. Our ciPSCs resemble the majority of the cESC lines described in that they can be maintained in medium supplemented only with LIF, and are particularly similar to the OVC.EX cESC lines of Wilcox et al. [13] in their tendency to become fibroblastic when cultured in the presence of bFGF.
While mouse ESCs, derived from the inner cell mass of the blastocyst, are dependent only on LIF, a separate population of pluripotent stem cells isolated from the mouse postimplantation epiblast, termed EpiSCs, appear to require only bFGF [14 –16]. Based on their dependence on LIF alone, our ciPSCs are more similar to the mouse inner cell mass-derived ESCs, than the epiblast-derived EpiSCs. Further support for our ciPSCs being at a stage equivalent to the preimplantation mouse embryo is found in their expression of the ESC-specific transcription factor Rex1, which is not expressed in EpiSCs [17].
To date, 2 other studies have described the generation and characterization of canine iPSCs, and remarkably both report a codependency on both LIF and bFGF in order to maintain pluripotency and self-renewal [6,7]. Mouse FAB-SCs are another type of stem cell that can be generated from the mouse blastocyst, and which show a response to both LIF and bFGF [18]. While FAB-SCs express pluripotency markers, they are incapable of differentiation unless converted to ESCs via culture with LIF and bone morphogenetic protein 4 (BMP4) [17,18]. It has been argued that human ESCs, which have a requirement only for bFGF, are more similar to mouse EpiSCs than ESCs [17]. Thus, it is possible that canine ESCs and iPSCs that show a tolerance, or dependency, towards bFGF are similar to the mouse FAB-SCs and EpiSCs, in that they are stem cell types that are at a subtly different stage of development to those equivalent to mouse ESCs, possibly reflecting cells at the postimplantation stage of development rather those from the preimplantation embryo inner cell mass. Hence, our ciPSCs reflect a more inner cell mass-like population of stem cells, which in turn suggests that they have potentially been reprogrammed to a more naïve state than the previously published canine iPSCs [6,7] that have a strict requirement for bFGF. One likely explanation for this difference is that we used GSK3β and MEK inhibitors for reprogramming, in addition to LIF, a combination which has been shown to convert EpiSCs into ESCs in the mouse [17]. A further consideration is that we reprogrammed with 6, rather than 4, pluripotency factors.
When cultured in mouse ESC medium supplemented with 5% FCS, our ciPSCs were able to spontaneously give rise to cell types representative of all 3 embryonic germ layers, further indicating that our cells have been reprogrammed to pluripotency. While the majority of our ciPSCs did not survive to form teratomas when injected into immunodeficient mice, cells in which the transgenes were still being expressed at very high levels did differentiate into 2 neoplasms, both of which resembled germ cell tumors. Oct4, c-Kit, Sox2, Nanog, and Lin28 are all expressed in primordial germ cells where they are thought to play a critical role in the generation of the germ cell population [19]. Furthermore, they are also expressed in several types of germ cell tumor, and Lin28, in particular, appears to play a role in the differentiation, and metastatic behavior, of type 2 germ cell tumors [19]. Thus, it is quite possible that with high levels of Oct4, c-Kit, Sox2, Nanog, and especially Lin28 transgene expression, and in the absence of LIF and MEFs, our ciPSCs have been induced to differentiate into primordial germ cells, which, in a normal undifferentiated gonad, become organized into cord-like structures as seen in our ciPSC-derived tumors. Unfortunately, pluripotent stem cells, such as ESCs and iPSCs, and PGCs coexpress a significant number of pluripotency cell markers including Oct4, Sox2, Nanog, Lin28, and alkaline phosphatase [19], in addition to putative germ cell markers such as Stella, Fragilis, Piwil2, Dazl, Vasa, and Blimp1 [20], making it difficult to confirm the differentiation of PGCs from ciPSCs using RT-PCR or immunohistochemical methods. However, based on an histological analysis it appears that our ciPSCs are capable of differentiating into PGCs, and so it is tempting to suggest that in addition to the 3 germ lineages, our ciPSCs are potentially able to differentiate into the fourth embryonic lineage, that of the germ cell population.
A common issue of concern with most of the cESC and ciPSC studies is the poor survival of transplanted canine cells in immunodeficient mice. Among 6 studies—3 using cESCs [11 –13] and 3 using ciPSCs [this study, 6,7]—only 2 have succeeded in generating teratomas: one with cESCs [12] and one with ciPSCs [6]. Similar to other researchers, we have successfully—and routinely—produced teratomas from human and mouse iPSCs following injection into the thigh muscles of NOD/SCID immunodeficient mice, and yet we were unsuccessful in multiple attempts at generating teratomas from our ciPSCs. Perhaps significantly, as well as being unable to produce teratomas from their cESCs, Wilcox and colleagues [13] similarly failed to establish canine hematopoiesis in immunodeficient mice injected with canine CD34+ cells, whereas human CD34+ cells readily became engrafted. The factor(s) responsible for this apparent resistance of canine cells to become established in NOD/SCID immunodeficient mice is unclear. However, an important consideration is that while these NOD.CB17-Prkdcscid /J strains lack C5 complement, they do still have active C3 complement, in addition to increased numbers of neutrophils and monocytes/macrophages (The Jackson Laboratory); thus, it is possible that our ciPSCs are opsonized by the C3 complement and subsequently phagocytozed by neutrophils, macrophages, or monocytes. In support of this argument, Koch and co-workers [21] have very elegantly demonstrated that complement activity, via the alternative pathway, can lyse mouse ESCs and inhibit the formation of teratomas when transplanted onto immunodeficient Rag2-/-γc -/- mice.
Reactivation of the inactive X chromosome occurs in the female embryo at the time of the formation of the inner cell mass and is therefore a hallmark of pluripotency in stem cells. In the mouse, X chromosome reactivation is thought to be under the regulation of Oct4, Nanog, Sox2, KLf4, c-Myc, and Rex1 [22]. Trimethylation at H3K27 (H3K27me3) is a hallmark of X inactivation, and key events in the reactivation of the inactive X chromosome are the removal of Xist RNA and H3K27me3 from the entire chromosome [23]. In this study, canine XX fibroblasts and supporting MEFs showed clear immunolabeling of H3K27me3 in a characteristic location at the periphery of the nucleus, whereas ciPSCs consistently failed to show H3K27me3 immunolabeling, indicating that X reactivation had occurred. This is in keeping with the known expression of Oct4, Nanog, and Rex1 in our ciPSCs and further strengthens the argument that our ciPSCs have been reprogrammed to an inner cell mass-like ground state of pluripotency.
Conclusion
In this study we have generated ciPSCs that maintain a normal karyotype and express a range of key pluripotency markers including Rex1, a transcription factor restricted to the inner cell mass of the preimplantation mouse embryo. Furthermore, these cells show reactivation of the inactive X chromosome, a hallmark of pluripotency. In vivo, ciPSCs are able to spontaneously differentiate into cell types from all 3 embryonic germ layers, and in vivo appear capable of giving rise to the germ cell lineage. Of key importance is the observation that these ciPSCs are dependent solely on LIF for the maintenance of pluripotency and self-renewal, clearly distinguishing them from the ciPSCs recently described in 2 other studies, which display a codependency on both bFGF and LIF. Thus, we postulate that our iPSCs, unlike those that are dependent on both LIF and bFGF, have been reprogrammed to an inner cell mass-like ground state.
Footnotes
Acknowledgments
The authors are very grateful to Sam Nayler and Jane Sun for invaluable scientific discussions and also for technical assistance, especially in maintaining our cells during the Queensland floods. We also thank Rachel Horne for her technical assistance; the ASCC Stem Core for the provision of MEFs; Dr. Rachel Allavena, from the School of Veterinary Science, for the pathology report on the germ cell tumors; and Ross Brookwell, from Sullivan Nicolaides Pathology, for the canine karyotypes. This work was supported by grants from the Australian Companion Animal Health Foundation, the John and Mary Kibble Trust, the University of Queensland New Staff Research Start-up Fund, and the Centre for Companion Animal Health, The University of Queensland, to D.J.W.
Author Disclosure Statement
The authors each declare that there are no actual, or potential, conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.