
Abstract
Human embryonic stem cells (hESCs) have the potential to revolutionize many biomedical fields ranging from basic research to disease modeling, regenerative medicine, drug discovery, and toxicity testing. A multitude of hESC lines have been derived worldwide since the first 5 lines by Thomson et al. 13 years ago, but many of these are poorly characterized, unavailable, or do not represent desired traits, thus making them unsuitable for application purposes. In order to provide the scientific community with better options, we have derived 12 new hESC lines at New York University from discarded genetically normal and abnormal embryos using the latest techniques. We examined the genetic status of the NYUES lines in detail as well as their molecular and cellular features and DNA fingerprinting profile. Furthermore, we differentiated our hESCs into the tissues most affected by a specific condition or into clinically desired cell types. To our knowledge, a number of characteristics of our hESCs have not been previously reported, for example, mutation for alpha thalassemia X-linked mental retardation syndrome, linkage to conditions with a genetic component such as asthma or poor sperm morphology, and novel combinations of ethnic backgrounds. Importantly, all of our undifferentiated euploid female lines tested to date did not show X chromosome inactivation, believed to result in superior potency. We continue to derive new hESC lines and add them to the NIH registry and other registries. This should facilitate the use of our hESCs and lead to advancements for patient-benefitting applications.
Introduction
H
Earlier studies created genetically differing hESC lines unintentionally, for example, by determining male and female gender after the lines were already established [5,6]. Later studies sought to deliberately derive lines with a particular genetic profile by, for example, using embryos that were found to harbor disease-specific genetic abnormalities by preimplantation genetic diagnosis (PGD) [7 –11] or by directed mutagenesis of hESCs [12,13]. However, in some cases, the mutation analysis results of the hESCs did not match the original PGD results [8]. In addition, many of the hESC lines that were derived worldwide since the first 5 lines by Thomson et al. 13 years ago [5] are poorly characterized in terms of their marker gene and protein expression profile, growth rate, differentiation potential, genetic status, and ethnicity and medical history of the embryo donors. In particular, genetic information such as inherited or cell culture-induced genetic abnormalities is often not available, and of the few lines for which ethnicity information is available, nearly all were of Northern and Western European and Middle Eastern provenience [14]. Similarly, little information is available on the genetic predispositions that those lines may carry for conditions other than certain cancers [15].
In order to improve upon this, we have derived 12 new hESC lines at New York University (NYUES1–12) from discarded genetically normal and abnormal embryos (Table 1). A majority of the lines were derived from embryos that underwent PGD and were found to carry chromosomal and single gene abnormalities. New hESC lines from PGD and non-PGD embryos will continue to be derived by us and added to the NIH Registry and other registries. To our knowledge, a number of genetic features of our hESCs have not been previously reported, for example, mutation for α-thalassemia X-linked mental retardation (ATRX) syndrome, unbalanced translocation of the long arms of chromosomes 8 and 15, and linkage to embryo donors with asthma and poor sperm morphology as conditions with a genetic component [16,17] as well as novel combinations of ethnic backgrounds such as Ashkenazi Jewish/English and Italian/Irish/German/Dutch. Furthermore, all our undifferentiated euploid female lines tested to date did not show signs of X chromosome inactivation (XCI), believed to result in superior potency [18].
For NYUES3–8 and NYUES12, disease diagnosis was achieved by preimplantation genetic diagnosis of embryos before their plating and for NYUES6 additionally by reverse transcription-polymerase chain reaction mutation analysis of human embryonic stem cells. For NYUES11, diagnosis of trisomy 14 was exclusively established by microarray analysis of hESCs at day 91 (∼102 cell doublings) and by fluorescence in situ hybridization at day 244 (∼275 cell doublings).
sister lines; 1donor used; 2inconclusive; 3carrier.
ND, not done; N/A, not applicable.
Materials and Methods
hESC line establishment, culture, and differentiation
All human embryos used to derive the NYUES lines were discarded by embryology laboratory staff independently from this project due to genetic or morphologic abnormalities or because they were no longer needed for pregnancy attempts. Some cleavage-stage embryos underwent PGD by testing biopsied blastomeres for genetic abnormalities [19 –22] independently from this project. Some embryos were stored frozen before thawing and derivation of hESCs. Collecting these discarded embryos and establishing and analyzing hESC lines were carried out with previous approval from the Institutional Review Board of New York University School of Medicine (NYUSoM IRB Nos. H-07-418, H-07-419) with proper informed consent from embryo donors. For hESC line establishment, embryos with a clearly visible cavity at the blastocyst stage (mostly 6 days after fertilization, ranging from 5 to 9 depending on speed of development) were mechanically hatched by a glass tool if they had not yet hatched by themselves. If embryos had a high-quality large and compact inner cell mass (ICM), it was microsurgically separated from most of the mural trophectoderm (TE) and plated onto irradiated mouse embryonic fibroblasts as feeder cells (ATCC, Manassas, VA). Otherwise, embryos were plated whole. Any TE and differentiating cells were regularly removed, and culture media were changed daily. Colonies were initially mechanically split and plated onto fresh feeder cells when reaching about 1 mm in diameter. After several weeks, colony splitting was performed by trypsin digestion once a week. hESCs were cultured according to a protocol after Klimanskaya et al. [23] in knockout Dulbecco's modified Eagle's medium (DMEM) containing glutamine, nonessential amino acids, β-mercaptoethanol, penicillin/streptomycin, knockout serum replacement (KSR) (all from Invitrogen, Carlsbad, CA), human plasma protein (Bayer, Wayne, NJ), leukemia inhibitory factor (LIF; Millipore, Temecula, CA) up to 20 ng/mL, and basic fibroblast growth factor (bFGF; Invitrogen) up to 40 ng/mL at 37°C with 5% CO2 and regular atmospheric oxygen pressure. Morphology of hESC colonies was documented by regularly taking bright field and phase-contrast microscopic images. Spontaneous in vitro differentiation was achieved at variable time points of hESC culture by embryoid body formation (using the hanging drop method [24 –26] and hESC culture media without bFGF, LIF, and human plasma protein, but with 15% KSR) or by overgrowth through omission of splitting [27]. Furthermore, differentiated cells were observed, as they sometimes spontaneously appear in culture without any changes to the culture protocol. hESCs were cryostored by vitrification as described [28,29], that is, transferring mechanically and enzymatically harvested hESCs into a solution with cryoprotectants propylene glycol, dimethyl sulfoxide (DMSO), and acetamide and immediately plunging them into liquid nitrogen. For thawing, hESCs were rapidly warmed in a 37°C water bath and plated onto fresh feeder cells. Growth rates of hESCs were determined by measuring specific colony areas on microscopic images by Adobe Photoshop tools (Adobe Systems, San Jose, CA).
Reverse transcription-polymerase chain reaction
For reverse transcription-polymerase chain reaction (RT-PCR), cells were washed with phosphate-buffered saline (PBS), and cDNA preparation and DNAse digestion was carried out as described [30,31]. PCRs for marker genes were performed with 1 μL cDNA template and primers human telomerase 5′ AGGAGCTGACGTGGAAGATG and 3′ TCAAGTGCTGTCTGATTCCAA (264 bp product), SRY 5′ TTGCTTCCCGCAGATCCCGC and 3′ AGTGGCTGTAGCGGTCCCGT (172 bp), RNF12 5′ AGAACCCGGCACCATGTGACA and 3′ ACCTGCCCGCTCAGAACGTG (333 bp), ATRX exon 35 (5′ part) 5′ AGCAGCAACAGCAACAAATG and 3′ CTTGGGAAGGTCCTGGATTT (207 bp), ATRX exon 35 (3′ part) 5′ CCTCATGATGCCAAAGCCCCCA and 3′ CATTGGGGGTGGTGCACGCT (133 bp), and FMR1 5′ TGTGCCAAAGAGGCGGCACA and 3′ TGCGCAGACTCCGAAAGTGCA (160 bp). Other primers were described: OCT4 [32,33] (218bp), REX1 [34] (306 bp), β-hCG 5′ primer [35] and 3′ primer [36] (144 bp), Nestin [37] (389 bp), α-fetoprotein (AFP) [38] (200 bp), CD34 [38] (200 bp), natriuretic peptide receptor type A (NPRA) [39] (491 bp), XIST [40] (151 bp), ATRX exon 34 [41] (79bp), KCNQ1 [42] (195bp) and β-actin [33] (243 bp; 0.5 μL template). Real-time PCR was carried out with SYBR Green PCR Buffer, 1U AmpliTaq Gold DNA polymerase, and 20 μL final volume in a StepOne Real-Time PCR System (all from Applied Biosystems, Foster City, CA). C T values were normalized against control β-actin values for each reaction, and results were analyzed by StepOne software (Applied Biosystems). All PCR experiments were controlled by β-actin mRNA expression and RT-negative and water samples. Select PCR products for OCT4, REX1, AFP, and KCNQ1 were sequenced to confirm amplification of the intended cDNAs.
Immunofluorescence
For immunofluorescence, cells were washed with PBS and fixed by transferring them onto Superfrost Plus positively charged slides (Thermo Scientific Shandon, Waltham, MA), air drying, immediate immersion in 90% ice cold acetone for 20 s, and air drying once more. Primary antibodies were diluted in dilution buffer [500 mL PBS, 5% KSR (Invitrogen) and 100 μL 2% sodium azide (Sigma Aldrich, St. Louis, MO)] and incubated with fixed cells for 45 min at room temperature. Cells were then washed once with dilution buffer at room temperature and incubated with secondary antibodies [anti-mouse IgG or IgM Cy3 (Millipore) diluted 1:100 in dilution buffer] for 2 h at room temperature under aluminum foil to block light. Cells were then washed once with dilution buffer and washed once in distilled water at room temperature, fixed with 100% ethanol for 2 min at room temperature, air dried, and mounted with Fluoromount G (Southern Biotech, Birmingham, AL). Signals were recorded by an Olympus AX70 fluorescent microscope and DP72 camera system (Center Valley, PA) and slides stored at −20°C. All immunofluorescence experiments were controlled by known positive tissue samples such as ICMs and negative samples such as feeder cells, by replacing primary antibodies with nonspecific immunoglobulins, or by omitting secondary antibodies.
Fluorescence in situ hybridization
For fluorescence in situ hybridization (FISH), cells were fixed as for immunofluorescence except using 3:1 methanol:acetic acid for 5 min at room temperature. FISH was performed according to the manufacturer's instructions (Abbott Molecular, Des Plaines, IL) except using room temperature for applying the probe onto the cells, counterstaining with Hoechst solution (bis-benzimide Hoechst 33342; Acros Organics, Geel, Belgium), and mounting with Fluoromount G. For FISH analysis of the Myosin heavy chain 7 (MYH7) locus, a specific probe was constructed from a BAC clone, tested, and applied onto fixed cells as described [43].
DNA fingerprinting
For DNA fingerprinting, hESCs were assessed for numbers of short tandem repeats (STRs) on each allele according to protocols by Cell Line Genetics (Madison, WI). STR markers examined consisted of the 13 and gender-determining amelogenin of the Combined DNA Index System by the United States Federal Bureau of Investigation plus Penta D and Penta E as 2 low-stutter, highly polymorphic pentanucleotide repeat loci. Samples were run in duplicate with positive and negative controls and blinded to the interpreter.
Microarray analysis
Microarray analysis for all 24 chromosomes was performed as described [44] by a 99% accurate informatics-based testing approach using an Illumina HapMap microarray (San Diego, CA) covering ∼300,000 SNPs.
Results
Derivation of hESC lines
We derived 12 independent, genetically diverse, and stable hESC lines (NYUES1–12) from discard embryos (Table 1) taking advantage of methods developed in our lab [30,33,45 –49]. The lines were derived from 59 embryos from 18 in vitro fertilization (IVF) cycles. Those 59 embryos were initially cultured into 21 ICM outgrowths (35.6% survival rate), which, in turn, were further cultured into the 12 lines (57.1% survival rate). Plating of whole embryos proved more successful for initial ICM outgrowth establishment than separating the ICM from TE (38.5% vs. 30.0% success rate). Additionally, frozen embryos performed better for this step than fresh ones (45.5% vs. 33.3%), which was likely due to the higher quality of the ICMs (27.3% vs. 13.3% top-graded). For hESC line establishment from initial ICM outgrowths, the success rate for frozen embryos was still superior to fresh embryos (66.7% vs. 53.3%). However, separating the ICM from TE now performed better than whole embryo plating (66.7% vs. 53.3%), possibly because there was an observed risk that left-over TE cells can overwhelm initial ICM outgrowth. None of these correlations reached statistical significance (Fisher's 2-tailed exact test [50]).
Figure 1A depicts an example of a colony of undifferentiated female hESCs (NYUES10) not carrying any genetic abnormalities as diagnosed by microarray analysis of hESCs. The colony is shown 3 months after derivation of the line, which corresponds to ∼92 cell doublings. Colony cells were found by RT-PCR to express mRNAs of hESC pluripotency markers OCT4 [51
–53], REX1 [54,55], and Telomerase (TERT) [56
–59] (Fig. 1B). Sequencing of RT-PCR products confirmed amplification of the intended cDNAs (data not shown). Furthermore, hESCs were found by immunofluorescence to express cell surface pluripotency markers SSEA3, SSEA4, Tra 1–60, and Tra 1–81, but not the differentiation marker SSEA1 [34,60
–63] (Supplementary Fig. S1; Supplementary Data are available online at

Characterization of hESC lines.
Gender and X chromosome activation status of hESCs
We next determined the gender of our hESCs independently from any PGD results if available by applying FISH for chromosomes X and Y and RT-PCR for the X chromosome marker ATRX and the Y chromosome marker SRY. Figure 1E and F show hESC line NYUES10 (for which no previous gender information was available; see Table 1) diagnosed to be female by these techniques (see also Supplementary Fig. S3). Later microarray analysis confirmed the female karyotype as well as the absence of any aneuploidies. These undifferentiated euploid hESCs did not show XCI as judged by the absence of upregulation of the noncoding mRNA XIST and the XCI-activator and E3 ubiquitin ligase RNF12 after 74 and 192 days of continuous culture, respectively, which is indicative of their highly undifferentiated state [18,64] (Fig. 1F). Similarly, other undifferentiated euploid female NYUES hESCs tested to date did not show these signs of XCI either, but some aneuploid or differentiated did (data not shown).
Differentiation of hESCs
To add further evidence to the pluripotent state of our hESCs in culture, we investigated whether they could spontaneously differentiate into morphologically distinct cell types representing all 3 embryonic germ layers. We found the occurrence of cells resembling erythrocytes (mesoderm), melanin-producing cells (prominently melanocytes and retinal pigment epithelium cells; ectoderm), and intestinal gland cells (endoderm) (Fig. 2A). Furthermore, we found mesodermal beating cardiomyocytes (Supplementary Video S1: continuously cultured NYUES10 cells 90 days after plating of the embryo; video plays at thrice the original recording speed). In addition, we could derive differentiated cells by omission of splitting; such overgrowth differentiation yielded early neural tissue (ectoderm), liver tissue (endoderm), and skeletal muscle tissue (mesoderm) (Fig. 2B–D). Finally, hESC differentiation via formation of serum-starved free-floating embryoid bodies showed that these cells had lost their pluripotent state as evident by upregulation of SSEA1 (Supplementary Fig. S4).

Differentiation of hESCs.
Single gene abnormalities of hESCs
Next, we examined the presence of single gene abnormalities in hESCs independently from any PGD results if available as well as the versatility of such mutation-bearing hESCs as disease models. In female NYUES6 hESCs, we could verify by RT-PCR the presence of a large exon 35 deletion in one of the alleles of the gene for the ATRX syndrome [65] as predicted by PGD linkage analysis (Fig. 3A). This X-linked recessive condition is characterized by severe psychomotor retardation and anemia; the ATRX gene codes for an ATPase/helicase of the SWI/SNF family of chromatin remodeling proteins [66] and ATRX mutations are known to cause diverse changes in DNA methylation pattern [67]. At least 2 cases with a large exon 35 deletion in the ATRX gene were previously described [68]. We further examined the skeletal muscle tissue derived from ICM outgrowth that was generated from an embryo inconclusively tested by PGD for myotonic muscular dystrophy type 1 (Fig. 2D; no hESC line was created). For this autosomal dominant triplet expansion disease, the dystrophia myotonica-protein kinase (DMPK) protein is significantly downregulated in skeletal muscle as compared with unaffected individuals [69,70]. Indeed, we saw a downregulation of DMPK protein expression in the hESC-derived skeletal muscle tissue (Fig. 3B), suggesting that these hESCs can be differentiated into disease-specific tissue as a disease model.

Analysis of single gene abnormalities in hESCs.
Chromosomal abnormalities of hESCs
Next, we sought to verify the microarray diagnosis of trisomy 14 in NYUES11 hESCs (all other chromosomes were found euploid by this analysis). Indeed, a FISH probe specific for the cardiac MYH7 gene locus on chromosome 14 (14q11.2) showed 3 signals, while a control chromosome 12 probe (12p12.3) showed 2 signals (Fig. 4A). Normal peripheral blood lymphocytes served as positive control cells (Supplementary Fig. S5). The MYH7 locus is known to be associated with a number of familial hypertrophic and dilated cardiomyopathies next to skeletal muscle conditions [71,72]. These heart conditions lead to a diminished pumping function and increase the risk for sudden heart failure [72]. On the chromosomal level, trisomy 14 itself is known to be associated with congenital heart defects, among them Fallot tetralogy, and is ranked in the top 25% of all single chromosome trisomies [73,74]. Those hESCs could, thus, serve as a model to study congenital heart defects; indeed, by RT-PCR, we could observe the expression of the potassium channel gene KCNQ1 in differentiating cells derived from them, mutations in which cause the cardiac condition long QT syndrome type 1 [75]. In the future, beating cardiomyocytes [25] similar to the ones shown in the Supplementary Video S1 could be generated from these trisomy 14 hESCs as well. In addition, we found that the trisomy 14 hESCs, which are male, exhibited a significantly decreased expression of the fragile X gene FMR1 compared with other hESCs (Supplementary Fig. S6). This may be a result of the intergenerational expansion of the CGG triplet repeats in the 5′ region of one of the FMR1 alleles on the X chromosomes of the female embryo donor (which was found at the borderline premutation level with 54 repeats) to the full-mutation level with more than 200 repeats. Such full mutations are known to cause epigenetic changes in the FMR1 gene promoter, leading to a decreased FMR1 expression as the pathogenic mechanism [76 –78]. Thus, these hESCs may also serve as a model for fragile X syndrome and for the disease progression from one generation to the next.
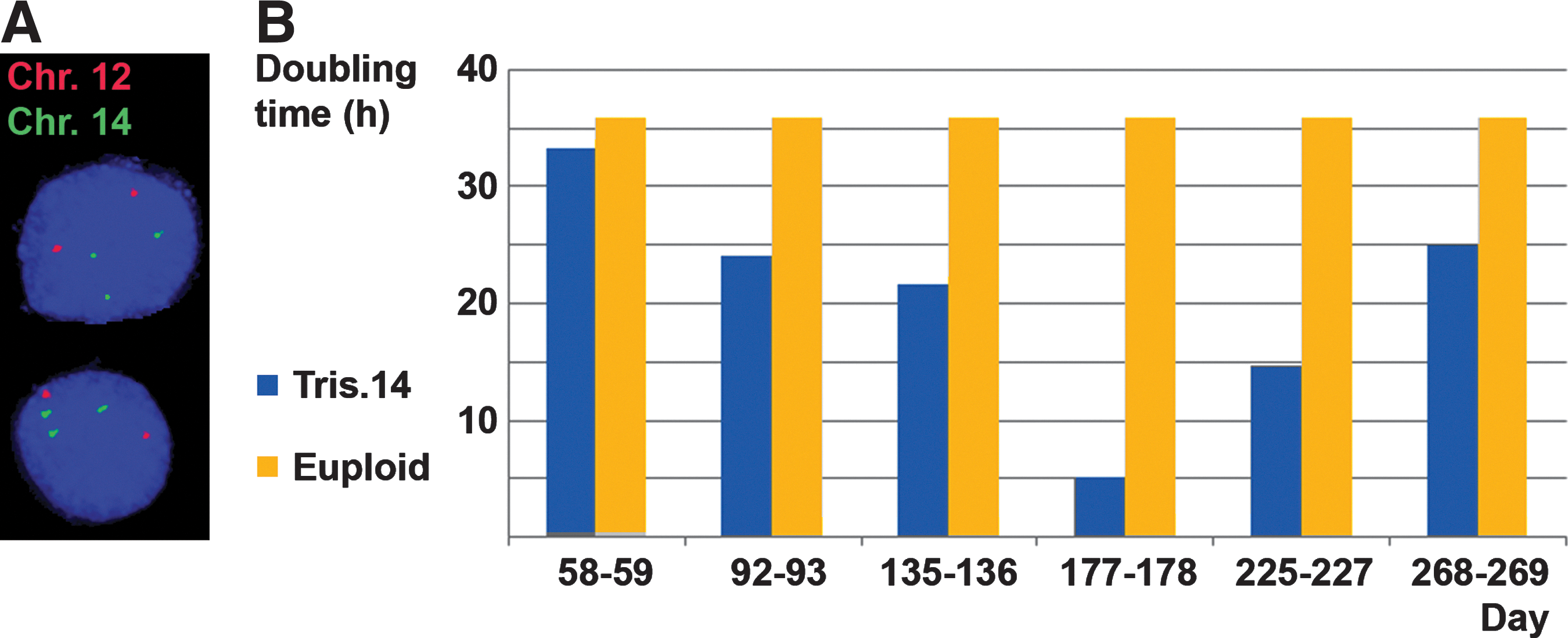
Analysis of chromosomal abnormalities in hESCs.
When analyzing the growth rates of these trisomy 14 cells, we noted that they showed a consistently accelerated proliferation pattern compared with the average 36 h doubling time for euploid hESCs that we and others have found [79], including a phase of very rapid growth around 6 months of culture (Fig. 4B). hESCs were found trisomic for chromosome 14 at days 91 and 244, rendering a relationship between the occurrence of this aneuploidy and the phase of rapid growth unlikely. DNA fingerprinting for these hESCs on day 244 revealed the duplication mentioned above of an amelogenin STR on the Y chromosome at Yp11.2 (Supplementary Table S1) that could have acted as a culture-induced trigger for the growth spurt. However, SRY expression levels were consistent from day 84 to 223 (data not shown), and no Y chromosome abnormalities were found on day 91 by microarray analysis (∼8 kb resolution), indicating that no widespread changes of the Y chromosome genome such as deletions, insertions, and unbalanced translocations occurred during culture. Nevertheless, it seems plausible that at least 1 of the 2 detected chromosomal abnormalities of these hESCs contributed to the overall accelerated growth pattern compared with unaffected hESCs. Another one of our aneuploid hESC lines that is trisomic for chromosomes 18, 21, 22, and X (NYUES3, Table 1) showed an altered growth pattern as well, a decreased proliferation rate of less than half the rate for unaffected hESCs (data not shown). Taken together, these findings may help explain why so many aneuploid concepti are lost at early stages of pregnancy, possibly due to a lack of sufficient coordination of cell proliferation to form vital organs at critical time points during development.
Discussion
Each pluripotent human cell line is unique in its genetic background as well as in its cellular and molecular features. This is due to the natural variability of many parameters such as the genetic makeup of the cells inherited, the method by which the lines were derived, and changes to the cells during culture. As a result, hESCs, for example, are known to differ in their marker gene and protein expression profiles, human leukocyte antigen (HLA) and global gene expression, epigenetic and karyotype stability, X chromosome activation status, propensities to preferably differentiate into specific target tissue, single-nucleotide polymorphisms, growth rate, and infectability by adenovirus [80 –93]. Indeed, cDNA microarray analysis of 3 different hESC lines showed no less than 30%–50% of all genes exhibiting differential expression patterns [94], and greater than 100-fold variations in lineage-specific gene expression were found in another study comparing 17 hESC lines [95]. Subtle molecular allocations of individual ICM cells to developmental fates more specific than the rather broad epiblast and primitive endoderm categories may be an important cause for these phenomena [45,46,48] as is the case for mice [96 –99].
One of the main aspects of genetic diversity of human pluripotent cells are disease-specific mutations, as they are relevant to disease modeling, drug discovery, and toxicity testing. While a variety of strategies is available to generate such cells, hESCs derived from disease-harboring embryos [7 –11] have some key advantages over other methods: First, the derivation efficiency is superior with up to 24.6% for whole embryo plating [100 –102] and about 17% for microsurgically or immunosurgically isolated ICM cells [5,6,52,103 –107]. This compares to very low efficiencies for the induced pluripotent stem (iPS) cell technology of 10–4 to 10–3 of the transfected cell population despite recent technological advances [108 –110] and of even lower 10−8 to 10−5 for genetic manipulation of hESCs [12,13]. Somatic cell nuclear transfer (SCNT) in mice using the preferred cumulus cells has a low success rate as well of less than 7% [111], and no cloned human embryos have been shown to produce unaffected or disease-specific hESC lines to date [112]. Second, many conditions such as nearly all aneuploidies, unbalanced chromosomal translocations, and diseases with an epigenetic component can only be properly reflected by hESCs derived from diseased embryos. These chromosomal conditions are frequently lethal in utero or shortly after birth, leading to a lack of target tissue for iPS and SCNT approaches, while epigenetic conditions such as fragile X syndrome are not correctly reprogrammed in iPS cells [113]. In humans, aneuploidy accounts for the majority of spontaneous miscarriages and is the main cause of hereditary birth defects such as Down syndrome [114,115], while, for example, fragile X syndrome is the main genetic cause of mental retardation and autism [116]. Third, the quality of hESCs is unsurpassed in terms of their pluripotent state, longevity in culture, X chromosome activation status, epigenetic accuracy, and overall reflection of the “clean slate” at the very beginning of human development. By contrast, reprogramming technologies such as for iPS cell generation are plagued by questions regarding their ability to generate fully pluripotent cells with therapeutic potential, reflecting incomplete reprogramming, as well as the possible transfer of somatic mutations acquired during reprogramming or through a donor's lifetime [117 –119].
Using hESC technology, we were able to generate hESC lines that continue to show hallmarks of pluripotency even after prolonged continuous culture beyond half a year. By contrast, some studies have reported difficulty in continuously culturing hESCs for more than 6 months or 20 generations and in maintaining potency in these cells [89,120]. Furthermore, our hESCs do not show signs of XCI in an undifferentiated euploid state; our protocols, thus, seem to support the derivation of highly potent hESC lines. This is quite remarkable, as, for example, essentially all cells of 9 other euploid, NIH approved, female hESC lines were found to exhibit signs of XCI [121]. In mice, pluripotency factors including Oct4, Nanog, and Sox2 are known to act as XCI-inhibitors by suppressing Xist expression, providing a direct functional link between pluripotency and XCI [122,123]. However, so far, we have not examined other markers of XCI such as the methylation status of the XIST promoter [18] in our cells. We will further examine the reasons that allow us to derive highly potent cell lines without XCI such as by investigating components of our cell culture medium; preliminary results indicate a possible role of the supplied growth factors (data not shown). Other studies have applied a reduced oxygen pressure similar to the female reproductive tract (5%) to prevent XCI in female hESCs [18] or supplied histone modifiers to the hESC culture media [124]. Nevertheless, this could also be achieved with a regular atmospheric oxygen pressure (21%) and without chromatin remodeling as these studies and our work have shown.
Other than the small Y chromosome STR duplication for NYUES11, we have no possible indication of culture-induced genomic instabilities, as they occur especially in hESCs carrying chromosomal abnormalities [90,125 –127]. Tissues that we could derive from our hESCs resembling erythrocytes, melanocytes, intestinal gland cells, skeletal muscle, and beating cardiomyocytes are relevant both for regenerative medicine applications if derived from unaffected embryos and for disease modeling if derived from embryos carrying diseases which affect these tissues, for example, α-thalassemia, neurofibromatosis type 1, familial dysautonomia, Marfan syndrome, and myotonic muscular dystrophy type 1. Our hESCs are, therefore, expected to be of good use to contribute to the general understanding of conditions, facilitate the development of novel therapeutic approaches and drugs, and enable the efficient generation of replacement tissue to treat many currently incurable conditions such as Alzheimer's, cardiac insufficiency, or spinal cord injury. Identity-establishing methods such as DNA fingerprinting will help to keep track of the lines in various settings over time and offer a point of reference for later re-analysis. Further characterization of these and additional new hESC lines such as HLA typing, karyotyping and assessment of global gene expression profiles as well as high-efficiency directed differentiation into target tissue will provide additional versatility for these applications. In the future, these pluripotent hESCs could be differentiated in vitro into organ tissue most affected by a condition such as neurons for Tay-Sachs disease by, for example, addition of specific differentiation factors or withdrawal of pluripotency-maintaining factors.
In summary, our novel hESC lines are of high quality and genetic diversity. They will, thus, provide new tools for basic research, disease modeling, regenerative medicine, drug discovery, and toxicity testing, leading to patient-benefitting applications. This will allow, for example, the growth of replacement tissue for degenerative conditions as a novel treatment strategy, the study of pathological mechanisms for conditions without existing appropriate disease models, and the identification of new drugs for previously incurable diseases with such models. Furthermore, lead compounds can be tested on human cells before clinical trials begin, without relying on controversial animal experiments and short-lived primary cell culture, thereby drastically reducing the risk for late-stage failures. Ultimately, the spectrum of curable diseases is expected to be significantly widened by the use of our cells, while drug development expenses can be utilized in a much more efficient way.
Footnotes
Acknowledgments
The authors would like to thank Alexis Adler for assistance in recruiting donors and culturing embryos, Christine Rice and Young Rock Chung for technical help with experiments, and Ruth Lehmann for advice, encouragement, and critical reading of the article. This work was supported by The Helen L. and Martin S. Kimmel Center for Stem Cell Biology at the NYU School of Medicine.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.