
Abstract
Recently, reprogramming of somatic cells from a differentiated to pluripotent state by overexpression of specific external transcription factors has been accomplished. It has been widely speculated that an undifferentiated state may make donor cells more efficient for nuclear transfer. To test this hypothesis, we derived induced pluripotent stem cells (iPS cells) from several somatic cell lines: mouse embryonic fibroblast (MEF), adult tail tip fibroblast (TTF), and brain neural stem cells (NSCs). Three dimensional (3D)-fluorescent in situ hybridization (FISH) and quantitative-FISH (Q-FISH) were then used to evaluate constitutive (pericentric and telomeric) heterochromatin organization in these iPS cells and in their parental differentiated cells. Here, we show that important nuclear remodeling and telomeres rejuvenation occur in these iPS cells regardless of their parental origin. When we used these cells as donors for nuclear transfer, we produced live-born cloned mice at much higher rates with the iPS-induced cells than with the parental cell lines. Interestingly, we noticed that developmental potential after nuclear transfer could be correlated with telomere length of the donor cells. Altogether, our findings suggest that constitutive heterochromatin organization from differentiated somatic cells can be reprogrammed to the pluripotent state by induction of iPS cells, which in turn support nuclear transfer procedure quite efficiently.
Introduction
S
In 1928, Emil Heitz first classified chromatin into euchromatin and heterochromatin, according to the degree of DNA condensation. One type of heterochromatin, called constitutive heterochromatin, is easily formed at genomic regions that bear highly repetitive DNA, such as telomeres, centromeres, or pericentric sequences. Constitutive heterochromatin is mostly transcriptionally silent and maintains high DNA density throughout cell cycles [5]. It has also been proposed to have crucial functions in the inheritance of cell type identity and in the fidelity of chromosome segregation [6,7]. Within the nucleus of interphase cells, the pericentric heterochromatin from different chromosomes gathers into foci, which form bright regions intensely stained by DNA dyes. These subnuclear structures, which especially exist in nuclei of mouse cells, are called chromocenters [8].
Interestingly, there is increasing evidence in favor of heterochromatin as cell fate determinant. Heterochromatin is indeed qualitatively different and more opened in pluripotent cells, such as ES cells and iPS cells, than in terminally differentiated cells and tissues. This decondensation is achieved mainly through histones hyperacetylation. It is believed that this specific chromatin structure can increase nuclease accessibility and the transcriptional activity required to prevent differentiation and promote self-renewal [9,10].
Conversely, constitutive heterochromatin structure seems to adopt a distinct structure in pluripotent cells, crucial for the regulation of pluripotency gene expression. In interphase nuclei, constitutive heterochromatin foci are fewer and less clustered together. The nonhistone chromatin-bound protein heterochromatin protein 1 is more mobile and binds more loosely in ES cells as compared to their differentiated progeny [11]. In totipotent embryos, we have shown that large-scale reorganization of pericentric heterochromatin occurs just after fertilization and concomitantly to major embryonic genome activation [12].
Remarkably, after nuclear transfer, heterochromatin remodeling is less efficient with somatic donor cells than with ES cells [13,14]; and it is known that ES and iPS cells can generate cloned mice that develop to full term with higher efficiency [15,16]. In addition, histone deacetylase inhibitors such as trichostatin A and m-carboxycinnamic acid bishydroxamide, which can affect chromatin acetylation, are known to improve cloning efficiency after somatic cell nuclear transfer [17,18]. In this case, we observed a correlation with improved heterochromatin remodeling [14].
Altogether, these results suggest that remodeling of constitutive heterochromatin may play a major role in cloning efficiency, and that inefficient reprogramming of these regions upon nuclear transfer of differentiated cells may be deleterious.
To test this hypothesis, we derived iPS cells from several somatic cell lines with distinct differentiation states: MEF-iPS, tail tip fibroblasts (TTF-iPS), and adult neural stem cells (NSC-iPS) [19]. These cells were used, as well as their parental cells, for nuclear transfer and we tested their ability to generate cloned embryos/pups.
We then analyzed the 3-dimensional (3D) organization of pericentric heterochromatin in these iPS cells and correlated its reprogramming with the outcome of the nuclear transfer experiments. We also examined telomeres, these typical constitutive heterochromatin regions are located at the end of chromosomes that also seem important for cleavage and development of cloned embryos [20]. Using quantitative fluorescent in situ hybridization (Q-FISH), we examined the relationship between the donor cell telomere length and cloning efficiency upon nuclear transfer of these cells. Remarkably, we show that telomere length can be a good predictor of cloned embryos outcome, both in vitro and in vivo.
Materials and Methods
All animal studies adhered to procedures consistent with the Beijing animal protection laws.
MEF cells, TTF cells, and NSCs
MEF cells were prepared from 13.5 dpc B6D2F1 male mouse fetuses. The fetuses were killed by decapitation and internal organs were removed. TTF cells were prepared by cutting the tail tip from an adult B6D2F1 male mouse. The remaining fetus tissue and tail tip were cut into small pieces (1–2 mm), which were incubated with rotation in the Dulbecco's modified Eagle medium (DMEM) medium contained 0.05% trypsin-ethylenediaminetetraacetic acid for 10 min. The fibroblast suspension was filtered after adding the same volume of the DMEM medium containing 10% fetal calf serum. Then the fibroblast suspension was seeded into tissue culture dishes. Fibroblasts were allowed to grow confluency. For NSCs, B6D2F1 males were sacrificed and brains were extracted. The subventricular zone of the brain was dissected and minced with fine scissors in phosphate-buffered saline. NSCs were cultured as described previously [19].
Preparation of iPS cell and ES cells
iPS cells were generated from MEFs, TTFs, and NSCs with the same genetic background C57BL/6×DBA/2. After transfection to introduce the 4 Yamanaka factors, the pluripotency of these iPS cells were assessed. Several colonies were obtained from the 3 different cell types and yielded stable cell lines. Three iPS cell lines with ability to generate tetraploid complementated live-born mice were chosen for further assessment [3]. ES cells were generated from the same genetic background mice (C57BL/6×DBA/2) as described [3]. All pluripotent stem cells were cultured in the DMEM supplemented with 15% fetal bovine serum, 1,000 U leukemia inhibitory factor, and 1× non essential amino acids.
3D FISH and Q-FISH
The oligonucleotide probes for hybridization were prepared by amplifying major satellite regions from mouse genomic DNA using the 5′-CATAT TCCAGGTCCTTCAGTGTGC-3′ and 5′-CACTTTAGGACGTGAAATATGGCG-3′ primers. These probes were then labeled with the Bioprime Array CGH genomic labeling system (Invitrogen). For the telomeric probe, we used a mixmer from Christophe ESCUDE tTaGgGtTaGgGtTaGgG [3′]Biotine. Probes were labeled with Cyanine 3 fluorescent molecules.
After a number of washing steps in the hybridization buffer, cells and DNA probes were denatured at 90°C, and then they were hybridized together at 37°C. After 24 h of hybridization, DNA was counterstained with Yopro I or propidium iodide for 20 min at 37°C before adding Citifluor (Citifluor Products) and coverslips on EPOXY slide (No. X1XER303B; Fisher) to keep a stable distance between the specimen and the coverslips. Cells are then kept in an original shape, without flattening, before scanning.
Microscopy and image analysis
Confocal microscopy was performed with a Zeiss LSM 510 confocal laser scanning microscope equipped with an oil immersion objective (Plan Apochromatic 63× n.a.1.4) with lasers at 488- and 535-nm wavelengths. Entire nuclei of cells were scanned with a distance between light optical sections of 0.36 μm. With these parameters, each telomere signal can be individually distinguished and quantified in 3D by the ForciPicker algorithm from Image software.
Donor cells
ES cell and iPS cells were synchronized in metaphase as previously described [16]. Subconfluent cultures were passaged the day before cloning and diluted twice. The culture medium was replaced with a culture medium containing 0.05 μg/mL demecolcin. After 3 h of incubation, the mitotic cells appeared as loosely attached cells and were collected by shaking the flasks. The cells were resuspended in the Chatot, Ziomek, Bavister (CZB)-HEPES medium on ice before use.
MEF cells, TTF cells, and NSCs were collected from the dish with trypsin treatment and a single-cell suspension was obtained by pipetting before nuclear transfer.
Nuclear transfer
Female B6D2F1 (C57BL/6×DBA/2) mice (8–10 weeks old) were superovulated with 10 IU pregnant mare serum gonadotropin injection followed by 10 IU human chorionic gonadotropin (hCG) 48 h later. Matured oocytes were collected at 13 h after the hCG injection. Micromanipulation was performed as described [16] using the one-step method to reconstruct cloned embryos. When iPS or ES cells were used as donor cells, the reconstructed oocytes were cultured in the CZB medium for 1 h and then activated for 3 h in the calcium-free CZB medium containing 10 mM Sr2+. When MEFs, TTFs, and NSCs were used as donor cells, the reconstructed embryos were activated for 5 h in an activation medium containing 5 μg/mL cytochalasin B. Cloned embryos were cultured in the CZB culture medium at 37°C under 5% CO2 in air. Cloned embryos of 2-cell, 4-cell, or blastocysts were transferred into the oviducts of 0.5-day-postcoitum or uterine horns of 2.5-day-postcoitum pseudopregnant Crl:CD1 (ICR) females mated with vasectomized males. At embryonic day 19.5 (E19.5), live mouse pups were delivered by cesarean sections.
Statistical analysis
Development of reconstructed embryos at different stages and telomere length from different cell lines were compared between groups by one-way analysis of variances with software SPSS 13.0.
Results
Remodeling of somatic cell nuclear structure after induction of pluripotency
Three iPS cell lines (with the same B6D2F1 genetic background) were derived from MEFs, adult tail tip cells (TTFs), and NSCs by transduction of the 4 Yamanaka factors. These iPS cells were morphologically similar to ES cells and expressed pluripotent markers (data not shown). As induction of pluripotency is believed to remodel the somatic cell nuclear structure [21], we next questioned whether pericentric heterochromatin would be remodeled in the nuclei of these iPS cells in comparison to their parental cells. 3D FISH with mouse major satellite probes was therefore performed to assess the distribution of pericentric regions in these cells. By comparing the 3D FISH images obtained from MEF-iPS and MEF cells (Fig. 1), we found that the pericentric heterochromatin formed large distinct patches in MEF cells and not in MEF-iPS. In MEF cells (Fig. 1A), these patches had clear boundaries and were regularly dispersed in the nuclei of MEF cells, just like chromocenters in mouse somatic cells [8]. In MEF-iPS (Fig. 1B), pericentric heterochromatin presented a loosely and dispersed structure; FISH signal boundaries became blurred and difficult to define. Moreover, quantification with ImageJ software showed that pericentric heterochromatin of iPS cells occupies a larger volume in these nuclei than in MEF cells. We obtained similar results with the 2 other cells lines (TTF-iPS and NSC-iPS, data not shown), suggesting that the pericentric heterochromatin compact structure is lost upon pluripotency induction. Interestingly, such an open chromatin structure predicts the hypertranscriptional activity of the genome as often observed in pluripotent cells [22,23].
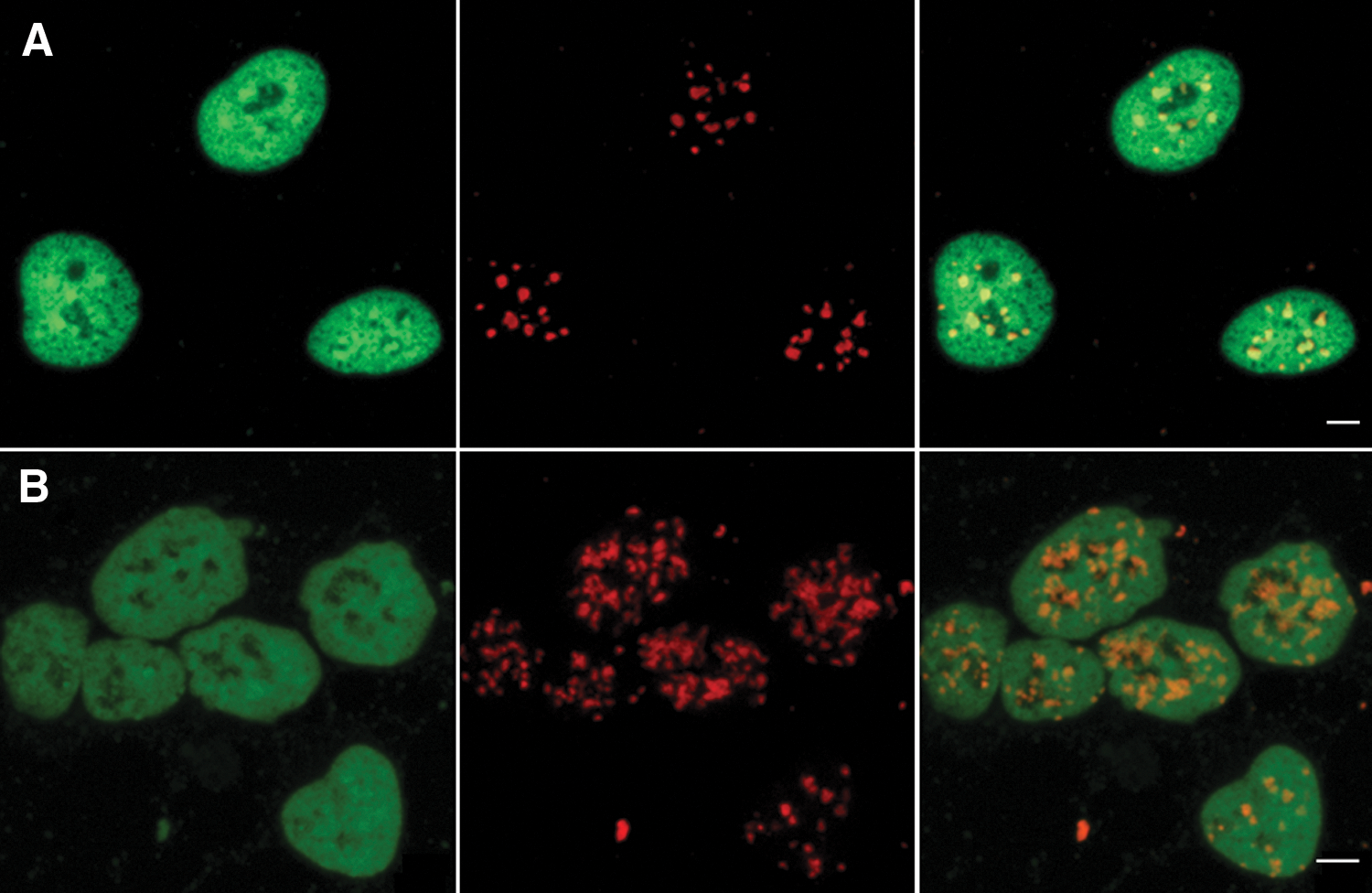
Differential distribution of pericentric heterochromatin in MEF and MEF-iPS cells.
Telomeric length of iPS cells and their parental cells
We next analyzed telomere length in the 3 different iPS cell lines previously generated and in their parental cell lines with specific telomeric DNA probes. Confocal images obviously showed that telomeric FISH signals in pluripotent stem cell lines were higher than in somatic cell lines (Fig. 2). We then measured the telomere length by digital fluorescence microscopy and quantification of telomere fluorescence intensity using a 3D foci picker program (Q-FISH). This technique—which requires only a small number of cells—provides information on the telomere length and localization of individual chromosomes as well [24]. Individual telomere FISH signals from confocal Z-series (Fig. 3A) were segmented and represented by unique colors (Fig. 3B). More than 30 telomeres from 3 replicates were stochastically chosen in different cells and measured for each cell line.

Comparison of 3D-FISH signal in different iPS and parental cell nuclei hybridized with Cy3-telomeric probe (red). Single Z-sections of confocal stacks are presented here.
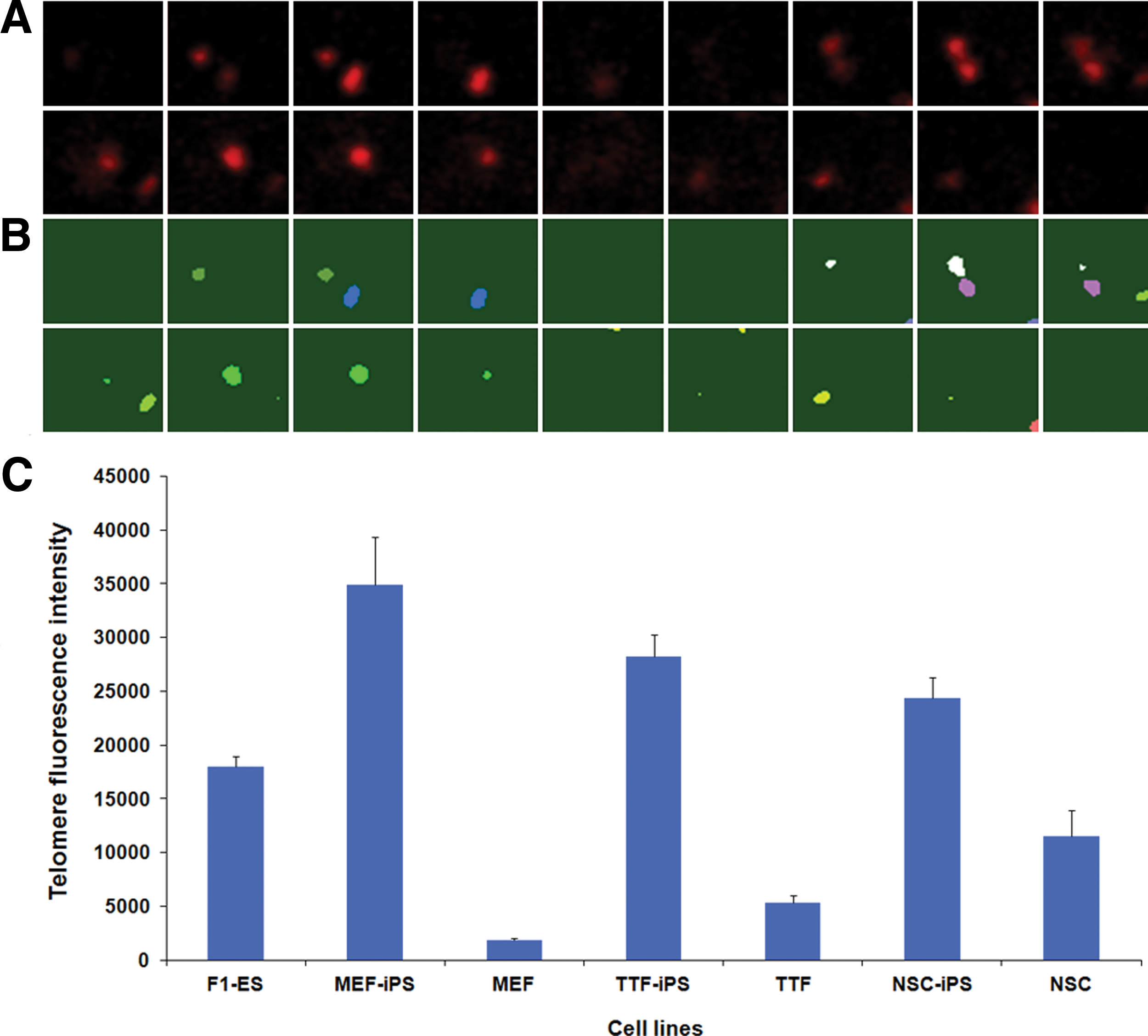
Assessment of telomere length in iPS cells and in their parental cell lines by quantitative-FISH.
The statistical analysis (Fig. 3C) showed similar telomere length when comparing TTF cells with MEF cells (P=0.283) and to some extent NSCs (P=0.057). However, all the somatic cell lines had significantly shorter telomeres than ES cells (P<0.05). After induction of pluripotency, telomere lengths in iPS cell lines were greatly increased relative to their parental somatic cell lines (P<0.001): telomeres of MEF-iPS, TTF-iPS, and NSC-iPS cells were ∼19 times, 5 times, and 2 times longer than in their parental cells, respectively. Notably, all the iPS cell lines had markedly increased telomere length compared to ES cells (P<0.05). These results show that induction of pluripotency can greatly increase telomere lengths and cause changes in chromatin structure independent of parental cell origin.
Preimplantation development of cloned embryos derived from iPS and their parental cells
To gain insights into the function of nuclear structure during reprogramming, iPS cells and their parental cell lines were used as donor cells for nuclear transfer. More than 200 cloned embryos (from at least 3 separate experiments) were generated for each cell line and cultured in vitro until the blastocyst stage. Embryonic development of these cloned embryos was observed and compared at different stages (Table 1).
F1-ES versus MEF are statistically different, P<0.05.
MEF-iPS are statistically different from all other donor cells, P<0.05.
ES, embryonic stem; MEF, mouse embryonic fibroblast; iPS, induced pluripotent stem cells; TTF, tail tip fibroblast; NSC, neurons stem cells; NA, not applicable.
As expected, in vitro development of cloned embryos derived from somatic cells (TTF and MEF) was lower than with stem cells (ES or NSC). No TTF cloned embryos (n=266) and only 3.5% MEF cloned embryos (n=412) reached the 4-cell stage in these experiments. On the other hand, as judged by the ratio of embryos able to develop to the blastocyst stage, preimplantation embryonic development was dramatically improved by induction of pluripotency. Cloned embryos derived from all 3 iPS cell lines had a higher rate of development to blastocyst than embryos cloned, in parallel, with their parental somatic cells (Table 1). Surprisingly, we obtained even better rates with MEF-iPS than with ES cells. Indeed, 46.1% of MEF-iPS cloned embryos (n=332) were able to reach blastocyst, which is significantly higher than the 27.1% obtained with ES cells (n=681).
Postimplantation development of cloned embryos derived from iPS and their parental cells
To test their capacity for postimplantation development, cloned embryos derived from these 7 cell lines were transferred into the uterus of surrogate female mice. We compared the implantation sites, cloned placentas, and offspring survival of all these clones (Table 2). After considerable trials of nuclear transfer with these 3 iPS cell lines, their parental somatic cells, and ES cells, a total of 29 live cloned pups (black-coated as the original B6D2F1 male donor cell mice) were delivered after 19.5 days gestation by CD-1 mice (white-coated) (Fig. 4). As expected, no cloned placentas or live offsprings were generated from MEF and TTF cell lines. For ES cells, 5 cloned live pups (0.4%) from 1,151 reconstructed embryos were generated. In total, 13 MEF-iPS-cloned mice, 4 TTF-iPS-cloned mice, and 6 NSC-iPS-cloned mice survived. This represents a cloning efficiency of 1.3%, 0.5%, and 0.7%, respectively. Most of the cloned pups were not overweight at birth, and the single full-term cloned mouse derived from an NSC nucleus had a lower weight than the other cloned mice. However, their placentas weighed 2- to 3-fold more than normal placentas (Table 3). To demonstrate the fertility of these cloned mice, they were mated to CD-1 females at adulthood. New progeny with uniform brown coat color were generated from all cloned mice (Fig. 4F).
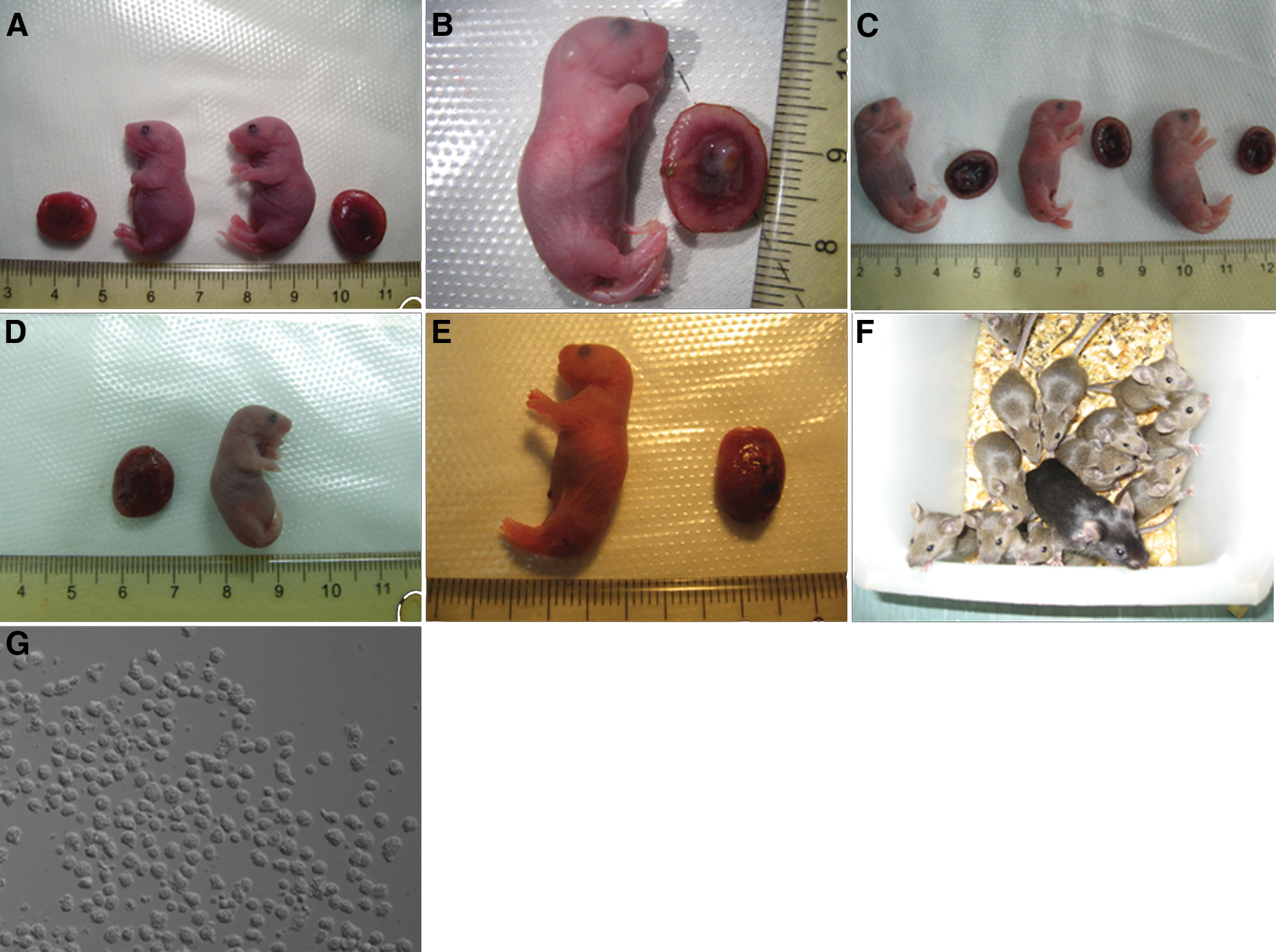
Mice cloned by NT of different cell lines, and their placentas. Donor cell origin:
Only 2 pups could be analyzed.
SEM, standard error of the mean.
Correlation between donor cells and developmental potential of cloned embryos
The Q-FISH we performed on the 7 cell lines used as donor cells showed very different telomere length (Fig. 3): MEF-iPS cells at passage 20 had the relative longest telomeres, whereas MEF cells at passage 3 had the shortest telomeres. We also noticed that TTF-iPS and NSC-iPS telomeres were much shorter than the telomeres of MEF-iPS.
Remarkably, cloned embryos derived from MEF-iPS cells had the best full-term developmental potential in vivo (1.3%, n=13 live cloned pups), whereas production of cloned pups from TTF-iPS and NSC-iPS cells was less efficiency (0.5%, n=4 and 0.7%, n=6, respectively). Moreover, none of the MEF cells generated a cloned pup.
Because of these results, we next decided to test whether the telomere length of donor cells could be used to predict the developmental fate of cloned animals. We normalized telomere lengths, blastocyst ratio, implantation sites ratio, and cloned pups ratio of iPS cell lines and their parental cells against the parameters obtained for ES cells (Fig. 5). It then clearly appeared that the telomere length follows the same trend as the in vivo and in vitro developmental potential of cloned embryos for each donor cell line. This suggests that donor cells telomere length might be fundamentally associated with developmental potential after nuclear transfer.
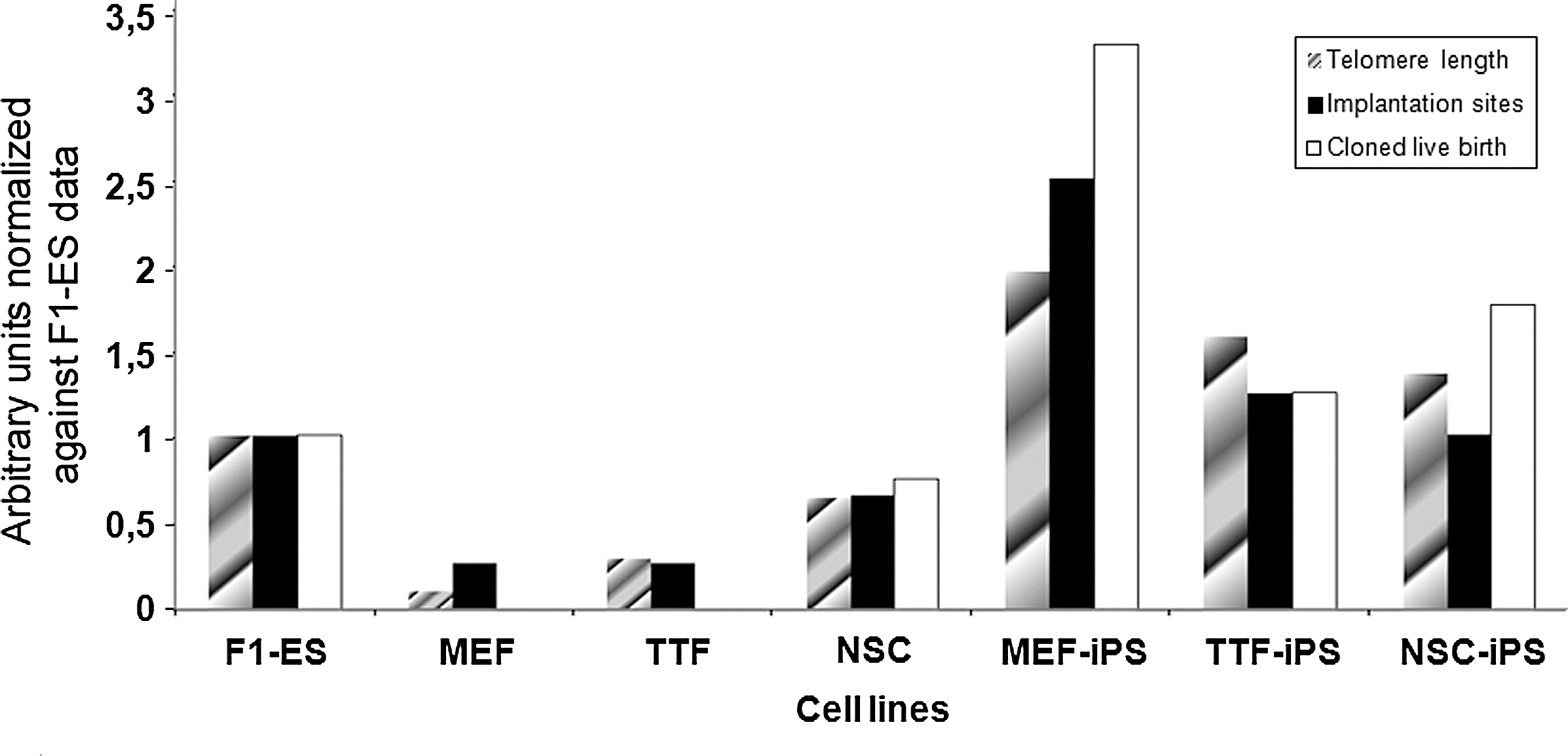
A relative analysis of telomere length and embryonic development parameters of cloned embryos. Cloned embryos were derived from the 3 iPS cell lines and their parental cell lines. Normalization was performed against F1-ES cell data.
Discussion
The one-step injection method has been used frequently for animal cloning. Although we produced more than 1,000 MEF cloned embryos and 396 TTF cloned embryos in this study, no surviving cloned fetuses were obtained. It is well known that these cells are difficult to use as a donor cell source for cloning. Only 1 group has obtained cloned mice using adult tail tip cells with the same low implantation efficiency as in the present study: 3.1% in our experiments, n=4/130, versus 4.1%, n=8/195 by Ogura et al. [25]. Several groups also stated that cloned mice from highly differentiated cells can be obtained by serial NT [26,27], including tail tip cells from senescent mice, which could not be successfully cloned directly [28]. Several reports have suggested that the use of undifferentiated donor nuclei can enhance generation of cloned animals [29]. Particularly, ES cell nuclei can give rise to cloned mice with distinctly higher efficiency than somatic cell nuclei, thanks to their plastic chromatin structure [16,30]. Here, we report for the first time that MEF and TTF cells can be successfully reprogrammed and return to pluripotency. We found that derivation of iPS cells from these poor donor cells clearly improves their potential to give rise to healthy fertile mice by nuclear transfer. However, it should be mentioned that manipulation of MEF and TTF cells is more difficult than with stem cells, especially as these cells have larger size and more rigid membranes. Reprogramming them into iPS cells that can be more easily used injected thus may contribute to higher developmental potential following nuclear transfer.
ES cell populations are characterized by a dynamic decondensed higher order chromatin structure, most probably through histones hyperacetylation [31]. After nuclear transfer, this chromatin decondensed state may allow access of different remodeling factors from the ooplasm, and thus increase the rate of reprogramming. On the other hand, the more condensed state of somatic donor nuclei is difficult to reprogram upon nuclear transfer. Constitutive heterochromatin (as well as other genomic sequences) must be completely remodeled into a totipotent nuclear organization during the first embryonic cell cycles and abnormalities have often been observed in such cloned embryos [13,14]. One could hypothesize that the delay between nuclear transfer and embryonic activation is too short to allow efficient reprogramming. Here, we report that preliminary derivation of somatic cells into iPS can remodel the somatic nuclei chromatin into an ES-like intermediate state before their introduction into enucleated oocytes. Consistent with this result, all iPS cell used lines in this study could be successfully reprogrammed to a totipotent state and generate full-term cloned mice, independently of their parental origin.
Previous reports have shown that telomere of iPS cells are elongated mostly through the activity of telomerase and cell divisions after iPS induction [32,33]. It has also been shown that the telomere length can affect stem cell function in Terc+/− mice, which possess short telomeres [34].Consistent with these results, we have shown that iPS cell lines can elongate their telomeres during induction of pluripotency independently of their parental cell source. However, the efficiency of this telomere rejuvenation is dependent on the differentiation state and the original telomere length in the parental cells: donor cells such as MEF and TTF, with relatively short telomeres and determined differentiation state, exhibit higher efficiency in elongating than telomere than NSC. It has already been shown that the telomere length can be used as a marker to evaluate pluripotency of stem cells and iPS cells [35]. Similarly, our results suggest that the telomere length of donor cells may predict developmental outcome of cloned embryos after nuclear transfer. We also showed that, iPS cells can generate cloned fetuses with similar and even higher efficiency than ES cells. These findings are consistent with the association of telomere lengths with true developmental pluripotency [35].
Altogether, our findings suggest that constitutive heterochromatin (telomeric and pericentric) organization from differentiated somatic cells can be reprogrammed to the pluripotent state by induction of iPS cells, which in turn support the nuclear transfer procedure quite efficiently. We conclude that production of cloned animals through a 2-step procedure combining iPS derivation and nuclear transfer is feasible, independently of the somatic cell origin.
Footnotes
Acknowledgments
We thank Christophe ESCUDE (Museum National d'Histoire Naturelle, Paris, France) for providing telomere probes and Tiphaine AGUIRRE-LAVIN (INRA, Jouy en Josas, France) for her help with FISH procedures. We are grateful to Mark E. GILL (Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland) and Walid MAALOUF (Nurture, QMC, University of Nottingham, United Kingdom) for critical reading of the article rigidity. This work was supported by INRA Jeune Equipe funding, the MRI at INRA, the French embassy in Beijing, the Ministry of Science and Technology of China No. 2009CB941003 and National Natural Science Foundation of China No. 90919060.
Author Disclosure Statement
The authors have nothing to disclose. No competing financial interests exist.