
Abstract
Induced pluripotent stem cells (iPSCs) have recently boomed enthusiasm in stem cell therapy, whereas high potential tumorigenesis of iPSCs has become the biggest obstacle for clinic application and the tumorigenic genes in iPSCs have not been well documented. In this investigation, using tools of bioinformatics, we analyzed the all available datasets regarded to iPSCs from 11 differentiated cell lines and revealed 593 iPSC consensus genes. Notably, of the 593 genes, 209 were expressed in human tumor cell lines and cancer tissues, and some of them were expressed in the iPSC-differentiated hepatocytes; remarkably, 5 oncogenes were overexpressed in the iPSCs and an oncogene RAB25 in the iPSC-differentiated cells, suggesting that these iPSC consensus genes are implicated with the risk of tumorigenesis and cancers. This investigation provides useful information for designing new strategies and methods to curtail the expression of oncogenic genes in iPSCs and produce safe iPSC derivatives for stem cell therapy.
Introduction
Several review papers have addressed recent advances in iPSCs [7 –12], and many cutting-edge technologies have recently been used in iPSC research field and produced massive data (Supplementary Experimental Procedures 2.1). Several groups have performed iPSC database mining and analysis in recent years. In brief, Newman and Cooper et al. selected 6 gene expression profile data (GEPD) from human embryonic stem cells (ESCs) and iPSCs from fibroblasts and analyzed consensus gene signature of the stem cells [10], but not included the data from many other types of iPSCs. Wang et al. inspected published GEPD related to ESCs and fibroblast-derived iPSCs and revealed their similarities and differences of gene expression profile, and attempted to establish new protocols for inducing pluripotency in somatic cells with high efficiency [11]. More recently, Ghosh et al. reported 20 potential cancer genes expressed in iPSC-differentiated cells after intersectional analysis between the genes expressed in iPSC-differentiated cell lines in 3 sets of the published GEPD and the genes expressed in 6 human cancer cell lines [6], whereas whether these 20 genes are also expressed in human cancers has not been systemically analyzed in the report. In addition, some researchers analyzed individual (not multiple) GEPD to tell the differences of gene expression profile either between human iPSCs and their derivatives, or between human iPSCs and their original fibroblasts and tried to address the risk of the iPSCs and derivatives [5,13]. Despite that, these analyses were mainly based on the data of fibroblast-derived iPSCs and only reflect a small portion of information regarded iPSCs, and the iPSC consensus genes have not been systemically analyzed yet; in particular, the iPSC tumorigenic genes that implicated with cancers have not been well documented.
Future progress in resolving the origin of this neoplastic potential of iPSCs and devising strategies to maximize expression of their normal stem cell properties while preventing expression of their neoplastic potential will require better understanding of which genes are consensually upregulated and expressed in iPSCs and then identifying which of these genes control expression of normal stem cell function (proliferation followed by terminal differentiation) versus which promote tumorigenesis. To gain new insight into the specific role that genes found to be consensually upregulated and expressed in all iPSCs play in determining the subsequent growth behavior of iPSCs and derivatives, we utilized tools of bioinformatics to collect all of the available GEPD of various iPSCs derived from 11 different types of differentiated cells (Supplementary Experimental Procedures 2.1), coherently analyzed the consensus genes in various iPSCs, and revealed 593 iPSC consensus genes and the implication of these genes with tumorigenesis and cancers.
Materials and Methods
Data collection and overview
In this investigation, we collected the data of human iPSCs, cancer stem cells (CSCs), and ESCs from the gene expression omnibus (GEO) database of Genbank, GeneCards database, and PubMed literature. With the criteria of “induced pluripotent stem cells” and “human” before April, 2011, 54 GEO records covering nearly all available human iPSC-related items were retrieved from the GEO database. After rational exclusion several incomplete datasets, including 14 overlapping GEO records, 16 GEO records of single nucleotide polymorphism, and several GEO records that did not contain the data of both iPSCs and the original parent differentiated cells, 28 solid datasets of 23 GEO records were used for a deep analysis (Supplementary Experimental Procedures 2.1). These included the datasets of iPSC-fibroblasts (Category I: Group A), iPSC human third molar mesenchymal stromal cells (HTM; Category I: Group B), iPSC hepatic differentiated cell (HDC; Category I: Group C), and other iPSCs (Category I: Group D) derived from various differentiated cells, including keratinocytes, hepatocytes, endothelial cells, melanocytes, smooth muscle cells, mononuclear cells, amniotic cells, and various stem/progenitor cells. In addition, noncoding RNA (NcRNA) expression profile in GSE16654 was analyzed to distinguish iPSCs from fibroblasts. Using the same criteria as described previously for iPSC data collection and exclusion, 14 human CSC (Category II, Supplementary Experimental Procedures 2.2), and 26 human ESC GEO datasets (Category III, Supplementary Experimental Procedures 2.3) were collected. In addition to the GEO database, GeneCards database as well as PubMed references relating to genes overexpressed in tumor cells and cancers dated before April, 2011, were also collected.
Data organization and analysis tools
We first divided the data into 3 categories: iPSCs, CSCs, and ESCs. We then analyzed the consensus genes in iPSCs for their implication in cancers using the well-known bioinformatics methods and tools, including ArrayTools integrated in Excel and MySQL (
Analysis of gene expression in GEO datasets related to iPSCs
Generally, we used the established methods and tools to analyze our selected GEPD based on individual dataset, since the computational methods to automatically analyze multiple GEPD sources at the same time are still under development. First, we picked out the genes overexpressed in iPSCs based on comparative analysis of GEPD of iPSCs and their original parent differentiated cells. So, one gene set from GEPD analysis may contain hundreds or thousands of genes overexpressed in iPSCs. Second, in the intersectional analysis only the genes expressed in at least 2 datasets were counted and those genes expressed in multiple datasets were regarded as iPSC consensus genes (Fig. 1, Supplementary Experimental Procedures 2.4).
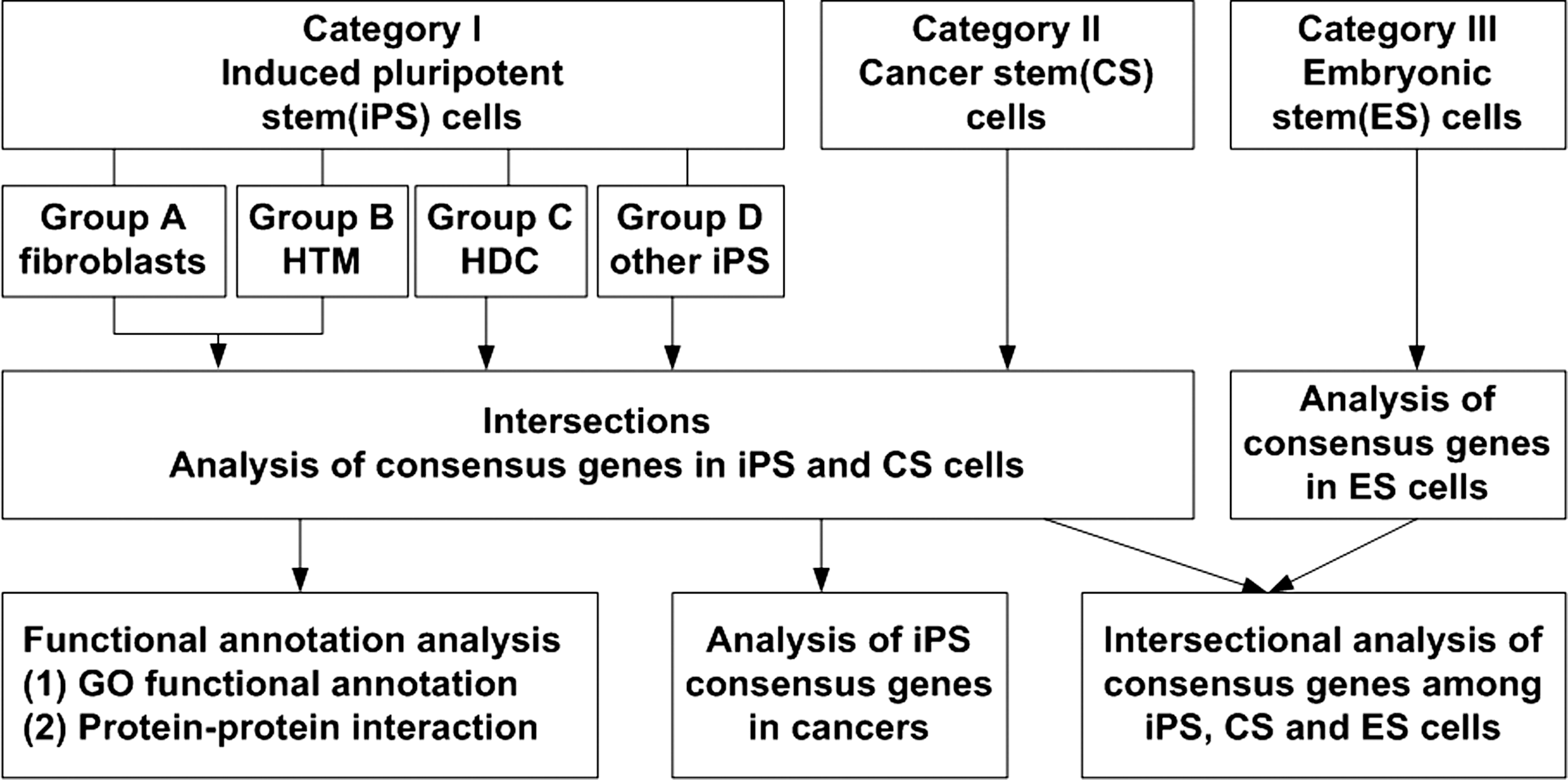
An outline for the analysis of induced pluripotent stem cell (iPSC) consensus genes and their implication with tumorigenesis and cancers.
Functional annotation of consensus genes in iPSCs
GO functional annotation analysis of iPSC consensus genes was carried out by DAVID (The Database for Annotation, Visualization and Integrated Discovery v6.7), and protein–protein interaction by STRING. For GO functional annotation, the classification of biological process and protein localization in different cellular organelles were analyzed by a small-size PHP program (Supplementary Experimental Procedures 2.5).
Analysis of the implication of iPSC consensus genes with tumorigenesis and cancers
To determine whether iPSC consensus genes were expressed in human tumor cells, cancers, and normal ESCs, we analyzed genes expressed in tumor cells, cancer patients, and normal ESCs according to their GEO datasets (top 1,000 genes) and PubMed literature, and performed the intersection among the consensus genes of iPSCs, CSCs, and ESCs in order to distinguish the tumorigenic iPSC consensus genes from normal ESC genes.
Results
Consensus genes overexpressed in iPSCs
Although recent genetic and proteomic studies have revealed thousands of genes that are up- or downregulated in iPSCs from various differentiated cells, the consensus genes overexpressed in a variety of iPSCs have not been systemically analyzed. Using the bioinformatics tools as described in the Methods section above, we noted that several thousand genes were markedly upregulated in iPSCs compared with the original differentiated cells. The expression levels of 335 genes in iPSCs from fibroblasts (Category I: Group A, and Supplementary Experimental Procedures 2.1) and 117 genes in iPSCs from HTM (Category I: Group B) were strikingly increased by more than 100-fold as compared with their original differentiated cells. Among these overexpressed genes, 238 genes in iPSC-fibroblasts and 87 genes in iPSC-HTM are well annotated. These genes were used for further analysis (Fig. 2A, Supplementary Tables S7 and S8 in Supplementary Data 2); the remaining poorly annotated genes were excluded from further analysis due to absence of gene name, ID, and mRNA sequence (Fig. 2A). With a threshold of gene expression level of 10-fold, 3,378 genes in iPSCs from fibroblasts (Category I: Group A) and 837 genes in iPSCs from HTM cells (Category I: Group B) were markedly overexpressed compared with their original differentiated cells. Among these genes, 1,799 genes in iPSC-fibroblasts and 624 genes iPSC-HTM are well annotated (Fig. 2B, Supplementary Tables S10 and S11 in Supplementary Data 2), indicating that the gene expression pattern in iPSCs has globally changed during cell reprogramming.
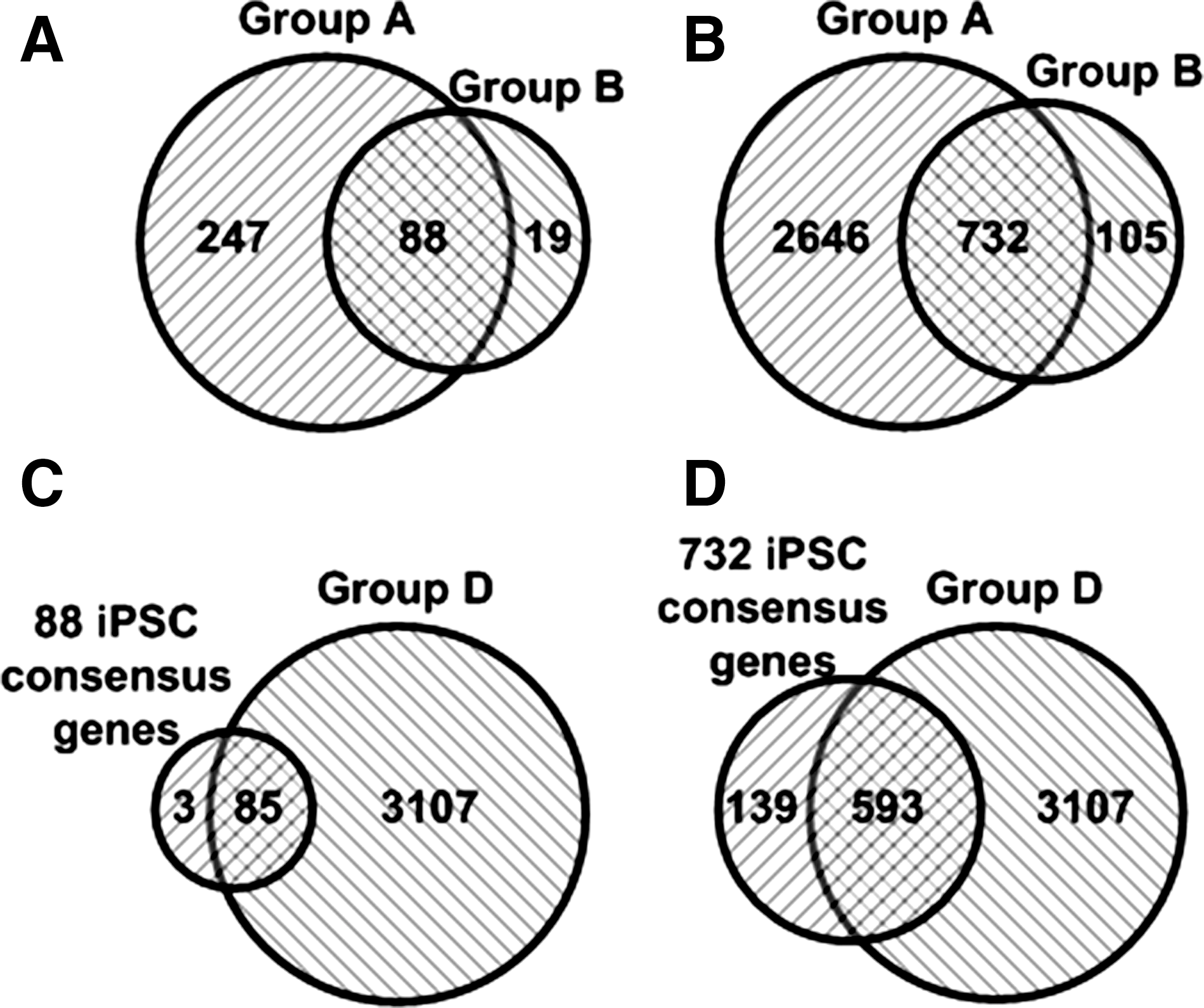
Intersection of genes overexpressed in iPSCs from various differentiated cells. Eighty-eight genes with more than 100-fold overexpression between iPSC-fibroblasts (335 genes) and iPSC-human third molar mesenchymal stromal cells (HTM; 107 genes) were intersected
Next, we performed gene intersection analysis to identify consensus genes in iPSCs from 11 different cells and tissues. Intersection of genes expressed more than 100-fold between iPSC-fibroblasts (335 genes) and iPSC-HTM (107 genes) showed that 88 out of the 107 genes are commonly expressed in both iPSC-fibroblasts and iPSC-HTM cells (Fig. 2A, Supplementary Table S9 in Supplementary Data 2). Intersection of genes with more than 10-fold overexpression between iPSC-fibroblasts (3,378 genes) and iPSC-HTM (837 genes) showed 732 out of the 837 genes commonly expressed in both iPSC-fibroblasts and iPSC-HTM cells (Fig. 2B, Supplementary Table S12 in Supplementary Data 2). Moreover, there are 3,192 genes commonly overexpressed in the iPSCs from other 9 differentiated cell lines (Category I: Group D, Supplementary Table S13 in Supplementary Data 2). Intersection of the 3,192 genes with either 88 genes (more than 100-fold overexpression) or 732 genes (more than 10-fold overexpression) in both iPSC-fibroblasts and iPSC-HTM cells showed that 96.6% (85/88 genes, Fig. 2C) and 81.0% genes (593/732, Fig. 2D) were commonly expressed among these iPSC lines, respectively (Supplementary Tables S14–S16 in Supplementary Data 2). We refer to the 593 genes that are expressed in all groups of iPSCs from various differentiated cells as iPSC consensus genes.
iPSC consensus genes expressed in iPSC-differentiated hepatocytes
Increasing evidence suggests that iPSCs can differentiate into various cell lineages and have a big potential for regenerative medicine. However, whether iPSC-differentiated cells are safe for therapeutic use remains to be established, and information available on gene expression profiles of iPSC-differentiated cells is limited. Here we analyzed the gene expression profile in iPSC-derived HDCs (iHDCs) and parent iPSCs. When 0.3-fold of gene downregulation was set as a threshold, 932 genes were lowly expressed in iPSCs, but highly in its original iHDCs (Fig. 3, Supplementary Tables S21–S27 in Supplementary Data 3). An intersection of the 932 genes overexpressed in iHDCs with either 88 genes (with a threshold of more than 100-fold) or 732 genes (with threshold more than 10-fold) commonly overexpressed in various iPSCs revealed 2 iPSC consensus genes, FREM2 and CDH3, and 27 iPSC consensus genes that were also expressed in the iHDCs (Fig. 3, Supplementary Tables S28–S31 in Supplementary Data 4), respectively. To our surprise, RAB25, a well-characterized oncogenic gene, was expressed in the iHDCs, suggesting that the iPSC derivatives still bear tumorigenic risk. The impact of other 26 iPSC consensus genes in the iHDCs on the tumorigenesis remains to be investigated.

Intersectional analysis of iPSC consensus genes with genes expressed in iPSC-differentiated hepatocytes. Intersectional analysis of 932 genes overexpressed in iPSC-differentiated hepatocytes with either 88 iPSC consensus genes (more than 100-fold expression) (Fig. 2A) or 732 iPSC consensus genes (more than 10-fold expression) (Fig. 2B) showed that 2 iPSC consensus genes in the group
Epigenetic alteration in iPSCs
Accumulated data indicate that the conversion of a differentiated cell to an iPSC involves a series of genetic and epigenetic alterations. Of note, epigenetic changes usually precede genomic changes, resulting in distinct epigenetic alterations in DNA methylation and NcRNAs [17 –19].
Genomic DNA methylation in iPSCs
Although entire genome DNA methylation has been reported for Arabidopsis thaliana [20], whole-genome bisulfate sequencing of DNA methylation in human is still a big challenge. Genomic DNA methylation in individual genes or regions of a chromosome has previously been reported. For example, Deng et al. disclosed methylation of genes in the selected regions of human chromosome 12 and 14; in these small genomic regions, they observed an increase in methylation of 115 genes, and a decrease in methylation of 40 genes in human iPSCs [18]. By intersection of the 593 iPSC consensus genes that we identified in our study (Fig. 2D) with either 115 genes or 40 genes, we found that the methylation of 2 genes (HESX1 and SYN3) was increased, whereas methylation of 3 genes (DNMT3B, POU5F1, and ZIC3) was decreased (Supplementary Table S17 in Supplementary Data 2). The detailed molecular mechanism by which genomic DNA methylation regulates epigenetic alterations in iPSCs remains to be elucidated. It is well recognized that changes in DNA methylation in iPSCs are dynamic during cell reprogramming. Nishino et al. reported that genes with hypomethylation in ESCs were usually overexpressed in human iPSCs [17]. When we performed an intersection between the top 100 hypomethylated genes of 1,260 genes from Nishino's study and the 593 iPSC consensus genes obtained in this investigation, we found that 22 out of the 593 genes were hypomethylated (Supplementary Table S17 in Supplementary Data 2). By contrast, an intersection between the top 100 hypermethylated genes of 1,260 genes reported in Nishino's study and the 593 iPSC consensus genes did not reveal any hypermethylated genes among the iPSC consensus genes. This small sample analysis suggests that DNA hypomethylation may be the predominant epigenetic alteration during cell reprogramming.
Noncoding RNAs
Increasing data have shown that NcRNAs play an important role in cell reprogramming [19]. Several NcRNAs can function as either driver or promoter of cell dedifferentiation into iPSCs, including the hsa-miR-302 family, miR-371/-372/-373, miR-181a, miR-199-3P, and miR-214. Of note, hsa-miR-302 miR-181a, miR-199-3P, and miR-214 have been considered stem cell–specific NcRNAs [21]. In this investigation, we analyzed NcRNAs dataset and spotted that 181 out of 774 NcRNAs were significantly upregulated or downregulated in human iPSCs compared with the original fibroblasts, including 20 NcRNAs overexpressed more than 10-fold, and 47 NcRNAs downregulated by more than 10-fold (Supplementary Tables S18–S20 in Supplementary Data 2). Among the top 20 upregulated NcRNAs expressed in iPSCs, hsa-miR-302 family members are predominant. For example, hsa-miR-302a, hsa-miR-302b, and hsa-miR-302d were overexpressed 541-, 325-, and 567-fold, respectively, as compared with their expression in the original fibroblasts. In addition, it has been reported that miR-302 was overexpressed in normal stem cells [22,23] and regarded as a potential stemness regulator in ESCs [24]. In this context, miR-302 was also highly expressed in CSCs [25], and Lin et al. used mir-302 to reprogram human skin cancer cells into a pluripotent ES cell–like state, indicating that mir-302 plays important roles in cell programming and maintenance of the pluripotent state of iPSCs [26].
Analysis of GO functional annotation and protein–protein interaction of iPSC consensus gene products
GO functional annotation of iPSC consensus genes
Intersectional analysis of gene expression in reprogrammed human cells from various cell types revealed 593 iPSC consensus genes (Fig. 2D). GO functional annotation [14] of these iPSC consensus genes was performed by DAVID [15], and the results were clustered according to the combination of biological process and cellular localization (Table 1, Supplementary Tables S1–S6 in Supplementary Data 1). Not surprisingly, the top cluster of the genes overexpressed in iPSCs is metabolism-related genes, since iPSCs are highly dynamic and active. The second and third clusters of iPSC consensus genes belong to transport and signaling genes, reflecting the critical role for material transport and signal transduction to cell survival and pluripotentiality. The fourth cluster consists of genes responsible for gene transcription. The next several clusters contain genes governing iPSC fate, including cell cycle, proliferation, differentiation, and apoptosis. Other gene clusters are related to immunity, cell adhesion, migration, angiogenesis, and vasculogenesis, which also contribute to the pluripotent functions of iPSCs. Moreover, cellular localization analysis indicates that the majority of iPSC consensus genes are membrane-associated proteins, including the proteins of plasma membrane, extra- or intramembrane, integration to membrane, and nuclear membrane (Table 1, Supplementary Tables S1–S6 in Supplementary Data 1), suggesting that these membrane-associated proteins play central roles in multiple iPSC functions.
Gene count may exceed the total number of genes, since 1 gene may have several functions and will be counted repeatedly in different categories.
ECLAMP, extracellular loosely attached membrane protein; IMP, integral to membrane protein; ICLAMP, intracellular loosely attached membrane protein; ER, endoplasmic reticulum; Golgi, Golgi apparatus.
Protein–protein interaction among proteins encoded by iPSC consensus genes
To gain insight into the molecular mechanism of pluripotentiality of the reprogrammed cells, protein–protein interaction of iPSC consensus gene products was analyzed by using STRING, a database of known and predicted protein interactions. As shown in Fig. 4, STRING analysis revealed protein–protein interactions for 29 genes out of the 85 iPSC consensus genes (Fig. 4, Supplementary Table S32 and Supplementary Fig. S1 in Supplementary Data 5), and 186 genes out of the 593 iPSC consensus genes (Supplementary Table S33 and Supplementary Fig. S2 in Supplementary Data 5). The top 10 of these interacting consensus genes are POU5F1, PROM1, LPAR2, CLDN3, OCLN, LPAR3, CD24, CLDN7, KRT18, and PTPRZ1 that are known to play pivotal roles in cell reprogramming and iPSC function. For example, POU5F1 (OCT4) is critical to the stemness of stem cells and a key master gene in cell reprogramming [1,2]; PROM1 (CD133) is a well-known CSC marker and interacts with 15 of 593 iPSC consensus genes. While further studies are needed to experimentally confirm these protein interactions, this analysis provides a road map for elucidating the mechanism of cell reprogramming and the pluripotent function of iPSCs.
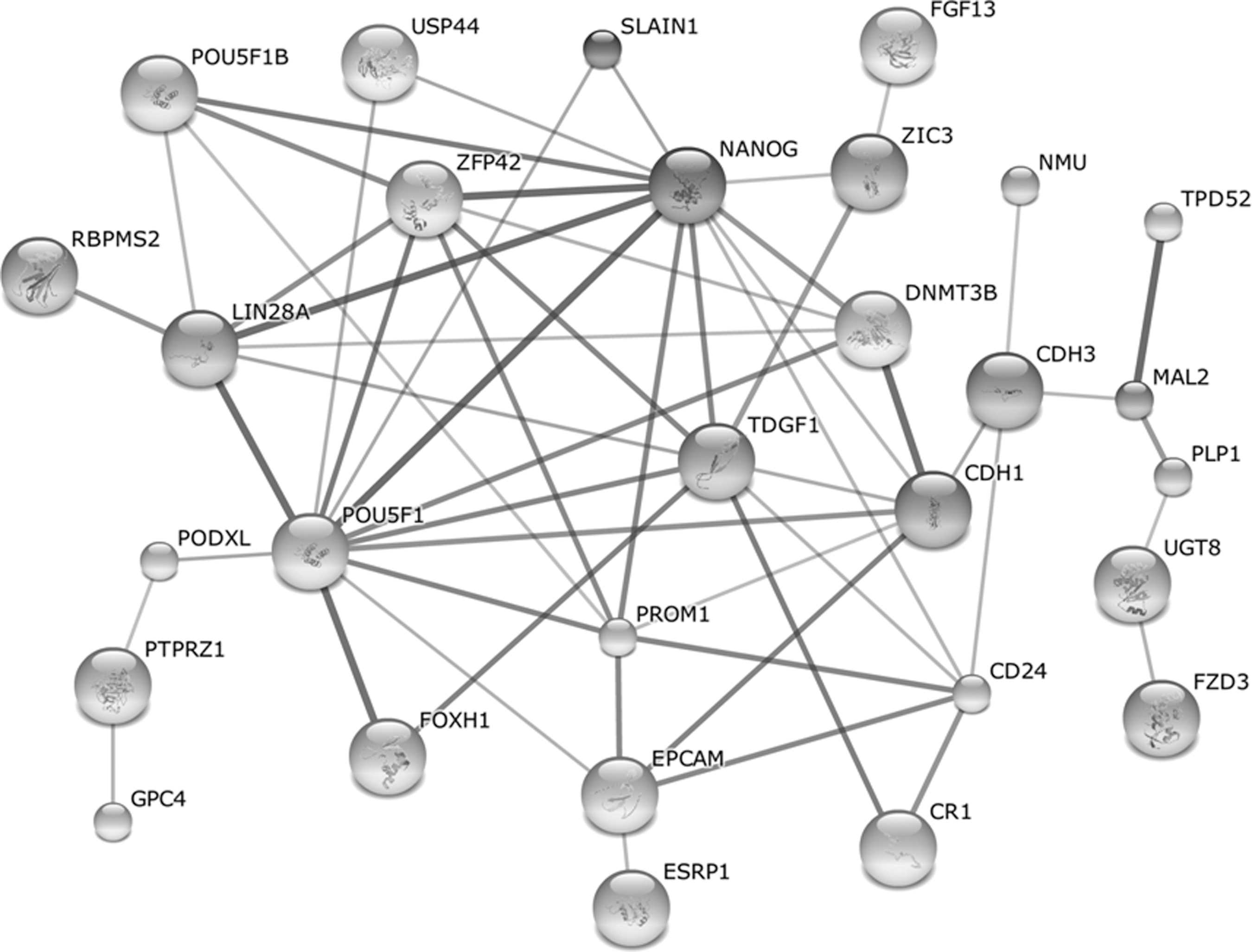
STRING analysis of the protein–protein interaction among the proteins coded by iPSC consensus genes. The protein–protein interaction among 85 iPSC consensus genes was analyzed by STRING. The lines represent positive protein–protein interaction and the thickness of the line stands for the reliability of evidence (See also Supplementary Tables S32 and S33 and Supplementary Figs. S1 and S2 in Supplementary Data 5).
Implication of iPSC consensus genes with tumorigenesis and cancers
Similar to ESCs, iPSCs are pluripotent and have potential application in cell therapy to treat disease. However, iPSCs have been shown to be more tumorigenic than ESCs in the mice, and may thus present an increased risk of tumorigenicity in humans. In this investigation, we analyzed whether iPSC consensus genes are expressed in CSCs, tumor cell lines, and human cancers by mining of GeneCards V3 database (
Expression of iPSC consensus genes in CSCs
Analysis of 14 solid human CSC-related GEO datasets identified 2,344 genes in CSCs that upregulated at least 3-fold as compared with differentiated cancer cells. Intersection between the 593 iPSC consensus genes and the 2,344 genes overexpressed in CSCs indicated that 102 iPSC consensus genes were also expressed in CSCs (Fig. 5A, Supplementary Tables S34–S42 in Supplementary Data 6). In addition, the intersection between 3,378 genes in iPSCs (Category I: Group A) with 10-fold overexpression and 2,344 genes in CSCs identified 462 iPSC genes that were commonly expressed in CSCs (Fig. 5B, Supplementary Tables S34–S42 in Supplementary Data 6). These results imply that a portion of iPSC genes is potentially tumorigenic.
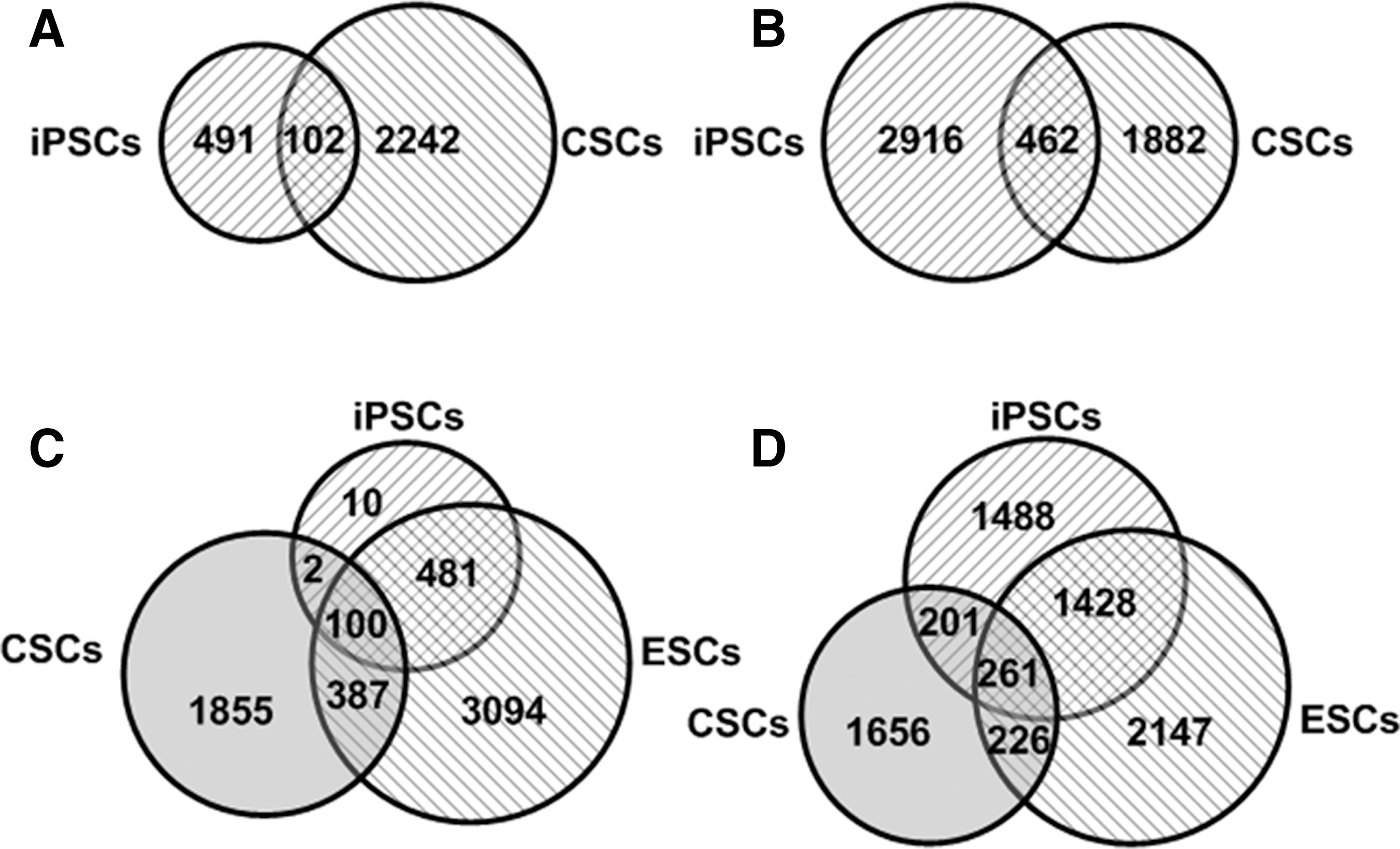
Intersectional analysis of iPSC consensus genes with the genes expressed in cancer stem cells (CSCs) and embryonic stem cells (ESCs). Five hundred ninety-three iPSC consensus genes and 2,344 genes expressed in CSCs show that there are 102 iPSC consensus genes expressed in CSCs
Expression of iPSC consensus genes in human tumor cell lines and cancers
We first searched GeneCards database to see whether iPSC consensus genes were present in human tumor cell lines and cancers. The GNF data derived from DNA microarrays showed that among the 593 iPSC consensus genes, 290 genes were expressed in the 28 tumor cell lines tested. Next, the electronic Northern blot data indicated that 439 iPSC consensus genes were expressed in tumor cell lines. In addition, the SAGE database revealed that 392 iPSC consensus genes were expressed in various tumor cell lines (Supplementary Table S49 in Supplementary Data 8). Intersectional analysis of the iPSC consensus genes expressed in tumor cell lines in GeneCards database (GNF, electronic Northern blot, and SAGE) identified 221 iPSC consensus genes that were commonly expressed in the tumor cell lines examined.
Next, we searched the PubMed literature for how many of the 593 iPSC consensus genes are expressed in various human cancers and noted that 209 out of the 593 genes expressed in a variety of cancers, which were reported in the literatures (Supplementary Table S50 in Supplementary Data 8). Of the 209 genes and 102 iPSC consensus genes expressed in CSCs, 26 tumor-associated genes have been reported to be overexpressed in the cells and tissues from cancer patients (Supplementary Table S52 in Supplementary Data 8), including 1 gene commonly overexpressed in various cancers, CSCs, and iHDCs (RAB25); 6 genes in both various cancers and CSCs (CCNA1, FGFR3, GLI1, KDR, LCK, and RPS6KA1); 1 gene in both various cancers and iHDCs (EFNA1); 1 gene only in CSCs (RRAGD); and 17 genes only in various cancers. Strikingly, 5 out of the 26 genes are well-characterized oncogenic genes (RAC3, RAB25, PIM2, MYBL2, and TET1). Functional analysis of the 26 genes classified them into several catalogues: 1 growth factor (FGF19), 3 growth factor receptors (ACVR2B, FGFR3, and KDR), 9 cell signaling proteins (ARHGEF16, ARHGEF5, GPR158, GPR19, KISS1R, RAB25, RAB39B, RAC3, and RRAGD), 7 protein kinases (BUB1B, CCNA1, EFNA1, LCK, MST4, PIM2, and RPS6KA1), 5 transcription factors (E2F5, GLI1, KLF8, MYBL2, and UTF1), and an epigenetic modifying protein (TET1). Obviously, these growth factors, receptors, signaling proteins, and protein kinases will promote the growth and progression of malignant tumors, and the overexpression of these transcription factors and epigenetic modifying proteins will intensify the global expression of many genes, some of them associated with cancers.
Expression of iPSC consensus genes in iPSC-differentiated hepatocytes
As shown in Fig. 3, 7 iPSC consensus genes were expressed in differentiated hepatic cells, including 23 well-annotated genes. We next analyzed whether the 23 iPSC consensus genes were expressed in human tumor cells and cancers through mining of GeneCards database and PubMed literature. GNF data of GeneCards showed that 12 out of 23 iPSC consensus genes were expressed in various cancer cells, and both electronic Northern blot assay and SAGE data found 21 out of the 23 genes in various cancer cell lines. Moreover, we reviewed PubMed literature and revealed 13 out of the 23 iPSC consensus genes overexpressed in various human tumor tissues and cell lines, especially, an oncogenic gene named RAB25. Notably, expression of 11 out of the 23 genes in various human tumor cell lines or cancer tissues was observed in both GeneCards database and PubMed literature (Supplementary Tables S28–S31 in Supplementary Data 4), including KRT18, VAMP8, PKP2, KRT8, CHMP4C, EFNA1, TNNC1, PRR15, JUP, AFAP1L2, and SLC27A6. These results imply that iPSC-differentiated hepatocytes still contain some of the iPSC consensus genes that are expressed in tumor cells and tissues, and the cells have tumorigenic potential.
Analysis of the genes commonly overexpressed in iPSCs, CSCs, and ESCs
As stated previously, of the 593 iPSC consensus genes, 102 genes were found overexpressed in CSCs (Fig. 5A, Supplementary Table S35 in Supplementary Data 6). To distinguish the tumorigenic genes from normal stem cell genes among the iPSC consensus genes, we performed intersectional analysis among 593 iPSC consensus genes, 2,344 genes expressed in CSCs, and 4,062 genes expressed in ESCs. The intersectional analysis showed that of the 593 iPSC consensus genes, 100 genes were expressed in both ESCs and CSCs; 2 genes, MAT1A and SERPINA5, were expressed only in CSCs; and 481 genes were only expressed in ESCs (Fig. 5C, Supplementary Tables S34–S40 in Supplementary Data 6). On the other hand, intersections among 3,378 genes overexpressed in iPSC-fibroblast datasets (Category I: Group A), 2,234 genes from CSC GEO datasets, and 4,062 genes from ESC GEO datasets identified 201 genes that were expressed in both iPSC and CSCs, but not ESCs; 1,428 genes were commonly expressed in both iPSCs and ESCs, but not in CSCs; and 261 genes were coincidently expressed among iPSCs, CSCs, and ESCs (Fig. 5D, Supplementary Tables S41 and S42 in Supplementary Data 6).
Since 4 Yamanaka factor–induced iPSCs have high tumorigenic potential, researches have recently used small molecules to induce cell reprogramming [27,28]. For example, normal human epidermal keratinocytes (NHEKs) were successfully induced into iPSCs with a combination of OCT4 and small molecules. To see whether iPSCs induced by OCT4 and small molecules have tumorigenic potential, we picked the top 1,000 genes with high-level expression in iPSCs from the GSE25218 dataset that contains GEPD of original NHEKs and iPSCs transduced with OCT4 and a cocktail of small molecules, and performed an intersection of these top 1,000 genes with 593 iPSC consensus genes, 4,062 ESC genes, and 2,344 CSC genes, respectively. The results showed that 472 genes are expressed in both iPSCs and ESCs. But 93 genes were also expressed in both iPSCs and CSCs (Fig. 6A, Supplementary Tables S43–S4 in Supplementary Data 7), and 153 genes were commonly expressed in iPSCs induced by OCT4 and small molecules and iPSCs transduced with the well-known Yamanaka factors—notably, 73 out of the 153 genes were expressed in various cancers, including the well-characterized PIM2 and RAC3 oncogenes (Fig. 6B, Supplementary Tables S47 and S48 in Supplementary Data 7)—suggesting that iPSCs generated by the transduction of OCT4 and a cocktail of small molecules still have tumorigenic potential.
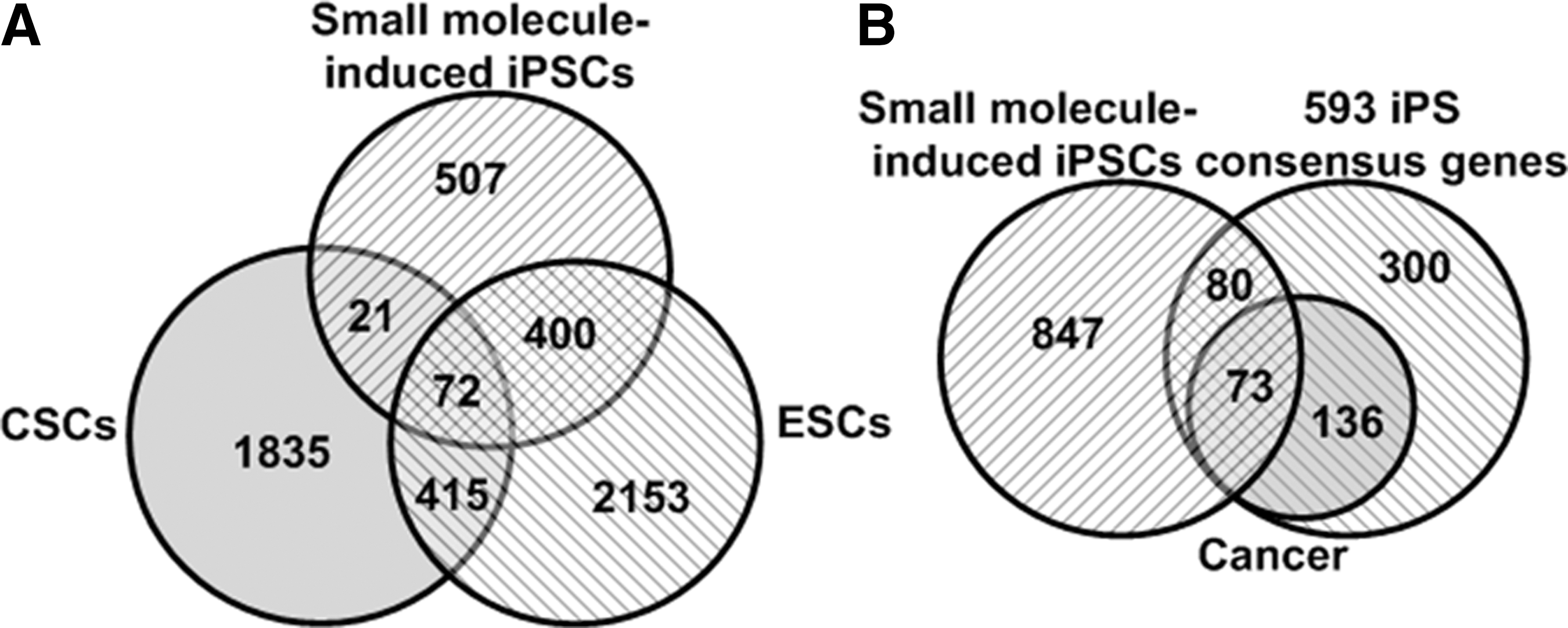
Intersectional analysis of genes expressed in various iPSCs, CSCs, and ESCs. Intersectional analysis among top 1,000 genes expressed in iPSCs induced by OCT4 and a cocktail of small molecules, 4,062 ESC consensus genes, and 2,344 CSC consensus genes was carried out
Discussion
Whereas rapid progress in iPSC research shows great promise, therapeutic application of iPSCs is currently hampered by the potential tumorigenicity of the reprogrammed cells. Our analysis shows that iPSCs express 593 consensus genes; of note, 209 out of the 593 consensus genes were reported coincidently overexpressed in a variety of human cancer cell lines and malignant tumors; remarkably, 26 iPSC consensus genes are associated with cancers, including 5 well-characterized oncogenic genes (RAC3, RAB25, PIM2, MYBL2, and TET1) besides the 5 master reprogramming genes. These data suggest that iPSCs are likely implicated in cancers if they are directly used in stem cell therapy. Consistent with the notion, experimental data have shown that transplantation of iPSCs into mice resulted in lethal teratomas in approximate 25% receptor mice, and that human iPSCs have higher tumorigenic capability than ESCs [5,6].
Although the incrimination of expressing most of the 209 iPSC consensus genes in cancers remains to be experimentally verified, accumulated data have shown that 5 oncogenic iPSC consensus genes play roles in tumorigenesis and cancers (Supplementary Tables S50 and S52 in Supplementary Data 8). In brief, RAC3, an oncogene of the ras family, is expressed in various tumors and contributes to tumor development and the invasive behavior of human glioma, breast carcinoma, and chronic myeloid leukemia (Supplementary References 3.2). RAB25 has been incriminated in the progression and aggressiveness of ovarian and breast cancers, and knockdown of RAB25 expression by RNAi inhibits growth of human epithelial ovarian cancer cells in vitro and tumorigenesis in vivo (Supplementary References 3.3). PIM oncogenic genes are overexpressed in a wide range of tumors of hematological and epithelial origin, and overexpression of PIM enhances tumor cell survival (Supplementary References 3.4). TET1 promotes DNA demethylation at CpG-rich promoters and enhances gene transcription, and TET1-MLL fusion protein causes acute myeloid leukemia (Supplementary References 3.5). MYBL2 is a nuclear protein and involved in cell cycle progression and has been implicated in tumorigenesis by regulating gene expression in neuroblastoma, hepatic carcinoma, and cervical cancer (Supplementary References 3.6). In addition, iPSC-differentiated cells usually contaminate a small amount of undifferentiated iPSCs; hence, iPSC derivatives may have tumorigenic risk. Accordingly, new strategies and approaches will have to be developed in order to silence the tumorigenic genes in iPSCs and derivatives before the cells are utilized in clinic to treat diseases in patients.
It is considered that the expression of the iPSC tumorigenic genes diminished in the iPSC-differentiated cells. However, Ghosh et al. reported that there were 20 potential cancer genes commonly expressed in 6 tumor cell lines and iPSC-differentiated cells, indicating that iPSC derivatives still bear cancer potential [6]; of note, among the 20 genes, c-FOS is obviously related to human cancers, while the contribution of other 19 genes to human malignant tumors remains to be investigated. Our systematic and deep analysis revealed that the iPSC-differentiated hepatocytes still kept expression of 27 iPSC consensus genes, including RAB25, a well-recognized oncogenic gene (Fig. 3, Supplementary Table S30 in Supplementary Data 4). By the way, the 27 genes found in our investigation are totally different from the 20 genes reported by Ghosh et al.; this discrepancy maybe related to the differences of iPSC resource and datasets used between Ghosh et al. and our group. Nevertheless, these data suggest that iPSC-differentiated cells still have potential tumorigenic risk.
Since iPSCs induced by Yamanaka factors have high tumorigenic potential, the first choice to generate safe iPSCs and derivatives should be to produce iPSCs without Yamanaka factors. Several approaches have been explored in recent years. One approach is to induce cell programming using NcRNAs [29 –31] that manipulate gene transcription and translation. However, some NcRNAs, such as miR-302, are also highly expressed in CSCs and associated with tumorigenesis [13]; therefore, the safety of iPSCs induced by NcRNAs remains to be investigated. To this content, measurement of the expression levels of the tumorigenic iPSC consensus genes in NcRNA-induced iPSCs and derivatives are helpful to evaluate the safety of the cells.
Another approach is to induce cell reprogramming with small molecules [27,28]. Unfortunately, the iPSCs induced by a cocktail of small molecules and OCT4 expressed many genes coincidently expressed in a variety of cancer cells (Fig. 5), suggesting that generation of iPSCs by transduction of OCT4 and the small molecules still have tumorigenic potential. Hitherto, it should avoid using Yamanaka factors in generation of therapeutic iPSCs and derivatives. More research works are needed to study how to use small molecules to effectively induce cell reprogramming and to generate safe iPSCs.
The third option is to directly use reprogramming coding mRNAs or proteins to induce differentiated cells into iPSCs. Theoretically, this option has advantage over the classic cell reprogramming protocol [1,2] since the reprogramming coding mRNAs or proteins have no genetic integration and may produce safe iPSCs [1,2,32,33], but the efficiency of generation of iPSCs with this option is much lower than the classic cell reprogramming protocol.
Collectively, revealing iPSC consensus genes and understanding their implication with tumorigensis and cancers provide useful information for designing novel strategy and setting up new methods to generate enough safe iPSCs for stem cell therapy.
Footnotes
Acknowledgments
This study was supported by grants from the Chinese National Natural Science Foundation #30971138, Suzhou city international collaboration #SWH0926, and Suzhou city scientific research #SWG0904, #SS201004, and #SS201138; and by a project funded by the priority academic program development of Jiangsu Higher Education Institutions (PAPD).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.