
Abstract
Ex vivo differentiation systems of natural killer (NK) cells from CD34+ hematopoietic stem cells are of potential importance for adjuvant immunotherapy of cancer. Here, we analyzed ex vivo differentiation of NK cells from cord blood-derived CD34+ stem cells by gene expression profiling, real-time RT-PCR, flow cytometry, and functional analysis. Additionally, we compared the identified characteristics to peripheral blood (PB) CD56bright and CD56dim NK cells. The data show sequential expression of CD56 and the CD94 and NKG2 receptor chains during ex vivo NK cell development, resulting finally in the expression of a range of genes with partial characteristics of CD56bright and CD56dim NK cells from PB. Expression of characteristic NK cell receptors and cytotoxic genes was mainly found within the predominant ex vivo generated population of NKG2A+ NK cells, indicating the importance of NKG2A expression during NK cell differentiation and maturation. Furthermore, despite distinct phenotypic characteristics, the detailed analysis of cytolytic genes expressed within the ex vivo differentiated NK cells revealed a pattern close to CD56dim NK cells. In line with this finding, ex vivo generated NK cells displayed potent cytotoxicity. This supports that the ex vivo differentiation system faithfully reproduces major steps of the differentiation of NK cells from their progenitors, constitutes an excellent model to study NK cell differentiation, and is valuable to generate large-scale NK cells appropriate for immunotherapy.
Introduction
N
Due to their strong ability to target tumor cells, NK cells have been described as promising effectors for adoptive immunotherapy of cancer [7]. So far, NK cells for adoptive transfer have mainly been generated through ex vivo expansion of PBNK cells [8]. Obtaining adequate purity and cell numbers of functional NK cells remains the biggest challenge for their therapeutic use. Recently, we have described a cytokine-based culture method with the capability of generating clinically relevant NK cell products from umbilical cord blood (UCB)-derived hematopoietic stem cells [9,10], obtaining high cell numbers, purity, and functionality. Currently, NK cell products generated by this method are investigated in a phase I clinical trial to treat elderly acute myeloid leukemia patients in a nontransplantation setting (Dutch Trial Register NTR2818) [11,12]. Additionally, this feeder cell-free ex vivo differentiation system might provide a powerful tool to study human NK cell development.
In the present study, we have investigated the gene and protein expression profile as well as functional properties of the ex vivo differentiated human NK cells and compared their characteristics to CD56bright and CD56dim PBNK cells. It is intriguing that NK cells generated by this method cannot unequivocally be classified to either the CD56bright or the CD56dim PBNK cell subpopulations. Despite phenotypic similarities to CD56bright PBNK cells, they remarkably display a pronounced gene expression profile for cytotoxic molecules similar to CD56dim cells and exhibit potent cytotoxicity. Upon activation, they also have the capability of producing interferon (IFN)-γ. These findings support that our NK cell differentiation protocol allows in vitro studies of human NK cell development, and that the generated NK cells constitute promising effectors for adoptive immunotherapy against cancer.
Materials and Methods
Cells and cell lines
Adult PB was derived from the Austrian Red Cross Blood Donation Centre or the Sanquin Blood Bank in the Netherlands. Human UCB samples were obtained from the Department of Obstetrics and Gynecology, Medical University of Vienna, or from the cord blood bank of the Radboud University Nijmegen Medical Center (RUNMC, Nijmegen, The Netherlands). The studies, including procedures for cord blood collection, have been approved by the ethics committees of the respective universities, and blood samples were obtained following an informed consent procedure. Within 6 h after blood collection, mononuclear cells were isolated by density-gradient centrifugation using the LSM 1077 Lymphocyte Separation Medium (PAA Laboratories GmbH, Graz, Austria).
K562 cells (LGC Standards, Wesel, Germany) were cultured in the Iscove's modified Dulbecco's medium (Life Technologies Corporation, Carlsbad, CA). Nalm-6 cells and 721.221 were a gift of Drs. M. Lopez-Botet (UPF, Barcelona) and R. Panzer-Grümayer [St. Anna Children's Cancer Research Institute (CCRI), Vienna, Austria], respectively, and were grown in RPMI-1640 (Sigma-Aldrich, St. Louis, MO) containing 50 U/mL penicillin, 50 μg/mL streptomycin, and 10% fetal calf serum (FCS; Integro, Zaandam, The Netherlands).
Ex vivo expansion and differentiation of CD34+ progenitor cells
UCB cells were labeled with CliniMACS CD34 reagent (Miltenyi Biotech GmbH, Bergisch-Gladbach, Germany), and CD34+ cells were selected by magnetic isolation (Miltenyi MACS Separator), according to the instructions of the manufacturer. CD34+ cells were collected, cell number and purity established by flow cytometry, and the cells used for NK cell generation.
CD34+ UCB cells were transferred into culture plates and expanded and differentiated according to culture method III as described previously [9]. In short, CD34+ cells were expanded for 10 days in Glycostem basal growth medium (GBGM®; Glycostem Therapeutics, Nijmegen, The Netherlands) supplemented with a high dose of the factors stem cell factor (SCF) (27 ng/mL; CellGenix, Freiburg, Germany); interleukin-7 (IL-7); thrombopoetin (TPO) (both 25 ng/mL; Stemcell Technologies, Grenoble, France); and Flt3L (25 ng/mL; CellGenix) and a low dose of the factors granulocyte colony-stimulating factor (G-CSF) (250 pg/mL); granulocyte-macrophage colony-stimulating factor (GM-CSF) (10 pg/mL; both Stemcell Technologies); and IL-6 (50 pg/mL; CellGenix) as displayed in Fig. 1A. Differentiation was induced by replacing TPO by IL-15 (20 ng/mL; CellGenix) on day 10 and Flt3L by IL-2 (1,000 U/mL; Chiron Corporation, Emeryville, CA) on day 14. During the first 14 days of culture, low molecular weight heparin (25 mg/mL; Abbott, Wiesbaden, Germany) was included in the growth medium. Cells were grown up to a total of 29 to 35 days.
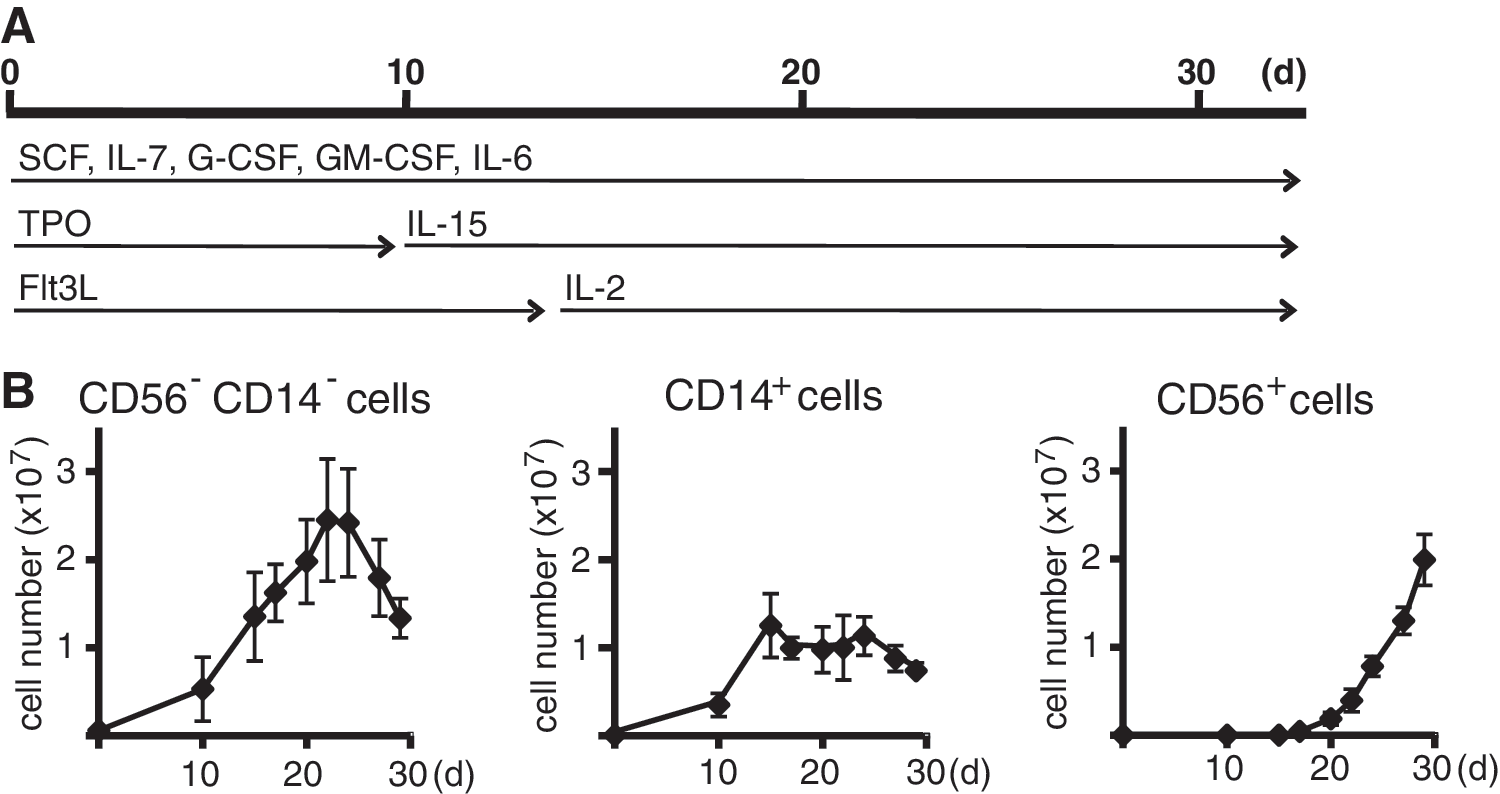
Expansion of progenitors, monocytic cells, and NK cells in the culture system.
Purification and cultivation of PBNK cells
Adult PBNK cells were isolated from PB mononuclear cells by negative magnetic cell sorting via depletion of non-NK cells (NK cell selection kit; Miltenyi Biotec), according to the manufacturer's instructions. For culture, PBNK cells were seeded at 1–2×106 cells per mL GBGM medium or differentiation medium (GBGM medium supplemented, as described for culture after 14 days in ex vivo expansion and differentiation of CD34+ progenitor cells) in 12-well plates (Corning Costar®; Sigma-Aldrich).
RNA preparation and cDNA synthesis
At least 1×105 cells were lyzed with TRIzol (Life Technologies Corporation) and stored at −80°C. Total RNA was extracted according to the protocol provided by Invitrogen, and 1 μg RNA was used to synthesize cDNA with H Minus M-MuLV reverse transcriptase, RNase inhibitor, dNTPs, and oligo-dT primer (all from Fermentas, St. Leon-Rot, Germany), following the manufacturer's protocol.
Real-time RT-PCR analysis
Specific mRNA levels were measured by real-time RT-PCR starting from 100 ng cDNA using a SYBR Green PCR Master Mix and a 7300 Real-time PCR system (Applied Biosystems®; Life Technologies Corporation). Values were normalized to β-actin mRNA as internal standard and analyzed with StepOne software v2.1 (Applied Biosystems®). Oligonucleotide primers were designed using Primer-BLAST software (
Affymetrix microarray analysis
Labeled cDNA was prepared from total RNA, hybridized to GeneChip Human Gene 1.0ST Arrays (Affymetrix, High Wycombe, United Kingdom), and scanned according to the manufacturer's protocols (Affymetrix support site;
Flow cytometry
Cell numbers and expression of cell surface markers were determined by flow cytometry. For immunophenotypical staining, cells from ex vivo NK cell cultures, total mononuclear cells from PB, or negatively selected PBNK cells were used as indicated in legends of figures. Cells were incubated with the appropriate concentration of antibodies for 30 min at 4°C. After washing, cells were resuspended in Coulter® Isoton® II Diluent and analyzed using the Coulter FC500 flow cytometer (Beckman Coulter, Brea, CA). To determine the content of CD34+ cells in the UCB and the purity of the selected CD34+ cells, the following monoclonal antibodies were used: CD45-FITC (J33) and CD34-PE (581) (both from Beckman Coulter). The population of living CD34+ cells was determined by exclusion of 7AAD-positive (Sigma-Aldrich) cells. Analysis was performed according to the most recent ISHAGE protocol (International Society for Hematotherapy and Graft Engineering).
The purity of cells obtained from the differentiation cultures was determined using the following monoclonal antibodies (see also Supplementary Table S2): CD3-FITC (UCHT1; Beckman Coulter); CD56-PE (NCAM16-2; BD Biosciences Pharmingen, San Diego, CA), and anti-CD45-ECD (J33; Beckman Coulter). The population of living CD56+ cells was delineated by exclusion of 7AAD-positive cells. Ten-color analysis was used to determine the phenotype of ex vivo generated and PBNK cells. For this the following monoclonal antibodies were used in appropriate concentration: CD16-FITC (NKP15), CD336 (NKp44)-PE (Z231), CD3-ECD (UCHT1), CD337 (NKp30)-PC5.5 (Z25), CD335 (NKp46)-PE-Cy7 (BAB281), CD314 (NKG2D)-APC (ON72), CD244 (2B4)-APC-Alexa700 (C1.7.1), CD56-APC-Alexa750 (N901), CD161-PB (191B8), NKG2A-PE (Z199), CD158a,h-PE (EB6.B), CD158b1/b2-PE (GL183), CD158e1/e2-PE (Z27), CD158i-PE (FES172), CD85j (HPF1.4) (all provided by Beckman Coulter), and NKG2C-PE (134591; R&D System, Minneapolis, MN). The acquisition was performed on the Navios™ flow cytometer, and the data were further analyzed using Kaluza™ software (both from Beckman Coulter).
Intracellular IFN-γ levels were measured as follows: cells were induced with IL-12 for 12 h or a combination of 50 ng/mL phorbol myristic acetate (PMA) and 1 μM ionomycin (both from Sigma-Aldrich) for 2 h or left untreated. Then, GolgiPlug™ was added for further 4 h. Flow cytometry was performed after permeabilization of the cells using the Cytofix/Cytoderm Fixation/Permeabilization Kit and staining with the FITC-labelled B27 IFN-γ antibody (all from BD Biosciences Pharmingen), according to the manual provided by the manufacturer. An isotype-matched control antibody of BD Biosciences Pharmingen (MOPC-21) was used in 2-fold concentration of the IFN-γ antibody to evaluate specificity of staining.
Preparative cell sorting
In certain cases, cells from the differentiation cultures were sorted into CD14+, CD56+/NKG2A+, and CD56+/NKG2A− cells. For this purpose, cells were stained with the following monoclonal antibodies: anti-CD56-APC (clone NCAM16.2; BD Biosciences Pharmingen), anti-CD14-FITC (clone TÜK4; Miltenyi Biotec), and anti-NKG2A-PE (clone Z199; Beckman Coulter). To separate CD56bright and CD56dim cells from PBNK cells, cells were stained with the anti-CD56-APC mAB. Incubation with the antibodies was usually for 1 h on ice followed by 2 washing steps using PBS supplemented with 2 mM EDTA and 2% FCS. A FACSAria Cell Sorter (BD Biosciences Pharmingen) at the service department of the CCRI was used for fluorescence-activated cell sorting (FACS).
Cytotoxicity assays
Ex vivo differentiated NK cells and PBNK cells were purified by negative magnetic cell sorting, as described under purification and cultivation of PBNK cells. NK cytotoxic activity was measured in triplicates using the human chronic myeloid leukemia cell line K562 in a Europium-release killing assay as described previously [16]. Briefly, target cells were labeled with europium diethylenetriaminopentaacetate (EuDTPA) and subsequently washed, and 2×103 target cells were incubated for 4 h with NK effector cells at various effector to target (E:T) ratios in RPMI-1640 without phenol red (PAA Laboratories) supplemented with 10% FCS. Values for specific release of EuDTPA were determined using the Delfia Enhancement Solution (Perkin Elmer, Waltham, MA) via time-resolved fluorescence. Maximal EuDTPA release was established by cell lysis with 1% Triton X-100. The specific cytotoxicity was calculated as percent specific EuDTPA release=(mean sample−mean spontaneous release)/(mean maximal release−mean spontaneous release)×100.
ADCC against the human lymphoblastoid cell line 721.221 and the pre-B ALL cell line Nalm-6 was measured in triplicates using the Europium-release killing-assay. After labeling of the target cells with EuDTPA, cells were washed and incubated with 10 μg/mL Rituximab (Hoffmann-La Roche Ltd., Basel, Switzerland, kindly provided by the pharmacy of the University Hospital Vienna, Austria) for 1 h at RT or left untreated. After extensive washing, 2×103 target cells were incubated for 4 h with purified NK effector cells at an E:T ratio of 12:1. Specific cytotoxicity was measured and calculated as described above.
Statistics
Results from experiments performed in triplicates are described as mean±standard deviation of the mean (SD). Combined results from at least 3 individual experiments are shown as mean±standard error of the mean (SEM). Statistical analysis was performed using the Student's t-test. A P value of <0.05 was considered as statistically significant.
Results
Time-dependent cellular composition of ex vivo differentiation cultures
Initially, we further characterized the cellular composition of our recently developed cytokine-based expansion and differentiation method [9] for generating NK cells from CD34+ hematopoietic stem cells (Fig. 1A). In this protocol, CD34+ cells are first expanded for 10 days using SCF, Flt3L, IL-7, and TPO. Thereafter, at day 10, TPO is replaced by IL-15 and at day 14, Flt3L by IL-2 to initiate NK cell differentiation and proliferation. Usually, at day 28, the percentage of CD56+ NK cells within the expanded population reached 50% or more and upon extended culture to day 35 over 95% pure, and viable NK cells can be obtained as previously reported [9,10]. A compilation of numbers and purity of NK cells achieved in several experiments is shown in Supplementary Table S3, which emphasizes the high numbers of pure NK cells obtained by this ex vivo NK cell differentiation method. We previously observed that during the differentiation period, monocytic cells can be detected in varying amounts [9]. Here, we have analyzed the accumulation of CD56−/CD14−, CD56−/CD14+, and CD56+/CD14− cells during the differentiation process in more detail (Fig. 1B). Starting with CD34+ cells from one cord blood sample, the expansion of CD56−/CD14− cells peaked at day 22 of culture obtaining values of 2 to 3×107 cells and declined thereafter. Monocytic CD56−/CD14+ cells expanded until day 15 of culture and then reached a plateau phase of approximately 1×107 cells, thereafter their number started to decline again at day 25. CD14−/CD56+ NK cells were first detected at day 17 and accumulated in the displayed experiments to 2×107 cells at day 29/30, resulting in a purity of NK cells of about 50% at this stage of differentiation.
Expression of NK cell-specific genes is initiated during early ex vivo NK cell differentiation
To characterize the upregulation of genes specific for the NK cell lineage in our ex vivo differentiation system, we performed gene expression profiling of cell samples taken between days 10 and 35 of culture. The top 25 genes most strongly upregulated during NK cell differentiation (Table 1 and Supplementary Table S4) were established by comparing the samples taken at the culture end point (day 35) and the initiation of differentiation (day 10). This group of genes exhibited a 29-fold to 158-fold upregulation and included genes for major NK cell receptors such as all members of the CD94/NKG2 receptor family, as well as for proteins relevant for NK cell-mediated cytotoxicity or involved in intracellular signaling. In general, when we analyzed the gene expression profiles at various time points between day 10 and 35, we observed detectable expression for all 25 genes starting between day 17 and day 20, followed by continuous further upregulation until day 35 (see Supplementary Table S4).
Upregulation of gene expression at day 35 is displayed as fold induction relative to day 10 of culture. Genes occurring several times in the original Affimetrix array data set are only given once using their highest value.
Based on their known relevance for NK cell activation and cytolytic function, we selected 17 of the 25 highest induced genes and confirmed their upregulation by real-time RT-PCR using mRNA isolated at several time points during culture (Fig. 2). In addition, although not among the top 25 genes, CD16 (FcRγIII) was included in this analysis because of its important function in ADCC and its differential expression in CD56dim and CD56bright PBNK cells. In Fig. 2A, mRNAs for proteins generally regarded as NK markers such as CD56, CD161/NKRP1, or CD16 are shown together with mRNAs for the highly expressed IL-2 receptor β chain and CD96. We observed detectable induction in their mRNA expression levels at day 15 of culture and continuous upregulation during the further NK cell differentiation process. Figure 2B displays the mRNAs for the NKG2 and CD94 C-type lectin-like NK receptor chains highly upregulated from day 14 onward. In parallel, mRNA-encoding proteins of the cytolytic machinery, including perforin, granzymes, and granulysin, are swiftly upregulated (Fig. 2C). Finally, we observed mRNA induction of several intracellular signaling proteins, including SH2D1B, RASGRP1, and ITK, potentially involved in NK cell differentiation (Fig. 2D).

A wide range of genes characteristic of NK cells is upregulated during ex vivo differentiation. In parts
These data demonstrate that many of the relevant NK cell receptors and intracellular proteins important for NK function are strongly upregulated in the established culture system, and this can be reliably used to study gene expression during NK cell development. For example, CD56 and CD94 are among the most strongly upregulated genes. The corresponding proteins clearly display consecutive appearance at the cell surface (Fig. 2E). At day 19, significant populations with expression of CD56 alone or with expression of CD56 and CD94 can be observed. However, no CD56−CD94+ cells are detectable. When the CD56+CD94− cells were isolated by preparative FACS sorting and cultured separately, consecutive acquisition of CD94 was observed in a large proportion.
Ex vivo generated CD56+NKG2A+ NK cells display a more mature gene expression signature than CD56+NKG2A− NK cells
The most prominent relative upregulation in our NK cell culture system was observed for NKG2A. Since NKG2A can be assumed to be important for the functional maturation of NK cells as a major inhibitory receptor [17,18], we analyzed the correlation of NKG2A expression with other NK cell-related genes upregulated during differentiation. For this purpose, we investigated separately CD14+, CD56+NKG2A−, and CD56+NKG2A+ cells for the same set of genes displayed in Fig. 2. Cultures at day 27 were FACS-sorted into CD14+, CD56+NKG2A−, and CD56+NKG2A+ cells, and the expression levels of the genes were analyzed in these fractions by real-time RT-PCR (Fig. 3). With the exception of CD16, significant expression of all genes was observed only in CD56+ NK cells, but not in CD14+ monocytic cells. CD16 showed higher relative mRNA expression levels in CD14+ monocytic cells as compared to CD56+ NK cells. Regarding the NK cell populations, mRNA expression levels in the CD56+NKG2A− and the CD56+NKG2A+ subsets were similar for CD56, NKG2C, and granzyme A, suggesting that expression of these genes is independent of NKG2A. In contrast, expression levels for all other genes analyzed were significantly higher in the CD56+NKG2A+ subset compared to the CD56+NKG2A− subset. Not only genes for receptors regulating NK cell activity, but also the most strongly upregulated signaling proteins and molecules involved in cytotoxicity were more prominently present in NKG2A+ NK cells when compared to NKG2A− NK cells. This is in line with the notion that acquisition of NKG2A expression is followed by more prominent expression of other markers of mature NK cells [17], indicating that the outgrowing CD56+NKG2A+ NK cell subpopulation is more mature than the majority of the CD56+NKG2A− NK cells in the culture.
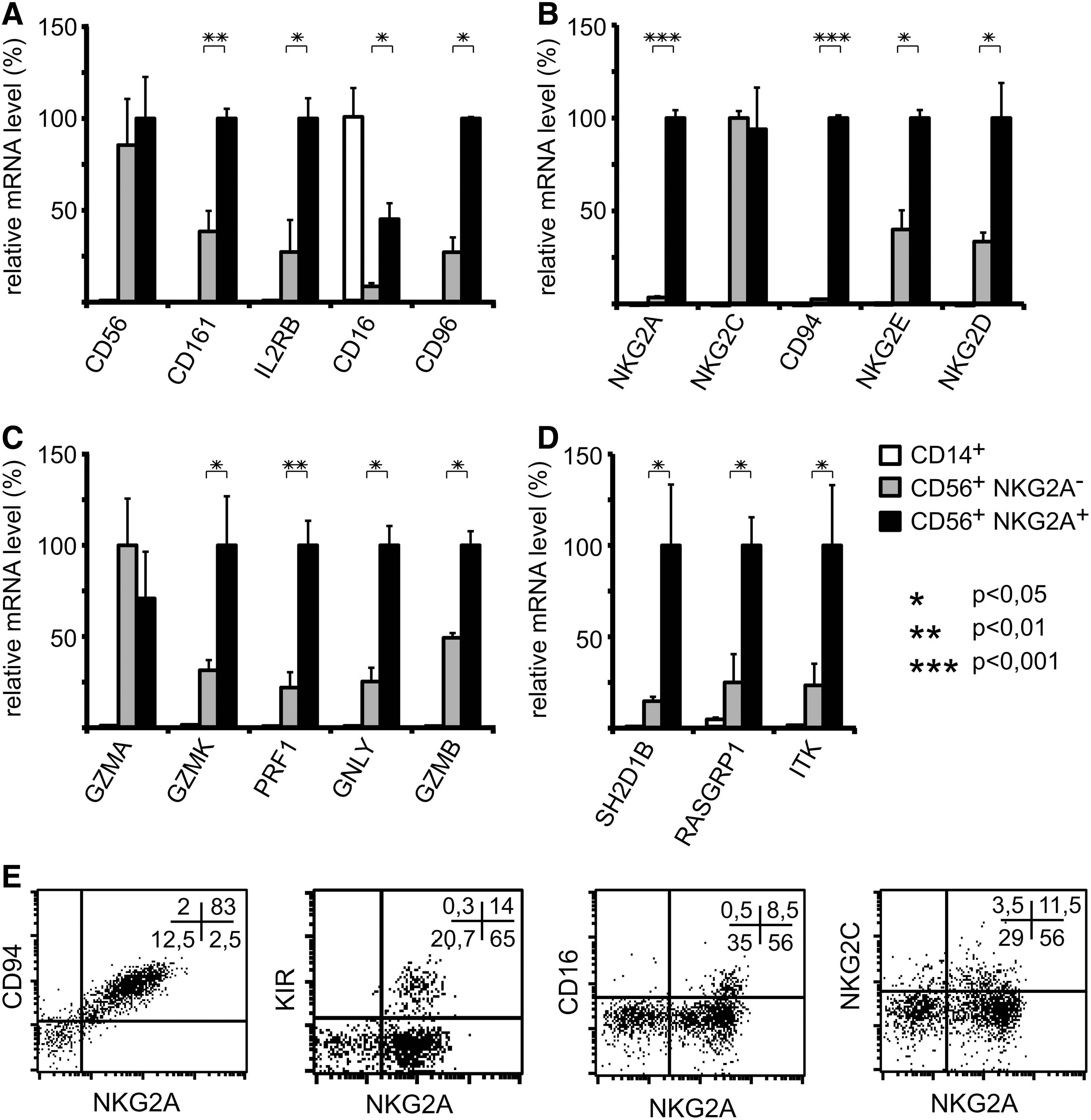
Functional markers of mature NK cells are most prominently expressed in ex vivo generated CD56+NKG2A+ NK cells. In parts
This is further confirmed by the surface expression pattern for KIR and CD16, which are almost exclusively expressed on cells displaying the inhibitory NKG2A/CD94 receptor (Fig. 3E). Expression of the activating NKG2C/CD94 receptor, although observed largely on NKG2A-positive cells, can also be present on cells lacking NKG2A.
Ex vivo generated NK cells display several surface antigen expression pattern close to CD56bright PBNK cells
Because a direct comparison has so far not been performed, we analyzed whether ex vivo differentiated NK cells can be classified to a subset of PBNK cells, which are classically divided into CD56bright or CD56dim NK cells. Known characteristics of these subsets are that CD56bright NK cells exhibit, compared to CD56dim cells, higher CD56 surface levels, diminished KIR expression, except for KIR2DL4, and strongly reduced or even absent surface CD16. Ten-color flow cytometry analysis showed that ex vivo differentiated NK cells (Fig. 4A) display CD56 surface levels even higher than CD56bright PBNK cells (Fig. 4C). This correlates with higher CD56 mRNA expression levels detected by real-time RT-PCR (data not shown). Strongly elevated surface expression on ex vivo differentiated NK cells is further detected for the activating NKp44 receptor (Fig. 4A compared to B). The proportion of cells expressing NKG2A corresponds with PBNK CD56bright cells and is larger than for CD56dim cells (Fig. 4A compared to C and D). Furthermore, ex vivo differentiated NK cells exhibit a percentage of cells with CD16 surface expression rather comparable to CD56bright PBNK cells, which in this case is much lower than for CD56dim cells. On the other hand, KIR surface expression is present on a significant number of ex vivo generated NK cells. In regard of NKRP1/CD161, 2B4/CD244, and NKG2D, comparable surface expression for ex vivo generated NK cells and for both PBNK cell subsets was detected. For NKG2C, we observed a high donor-dependent variability similar for ex vivo generated NK cells and both PBNK cell subsets (data not shown). The proportion of cells double positive for NKG2A and NKG2C was regularly higher in ex vivo generated NK cells than the proportion of cells single positive for NKG2C.
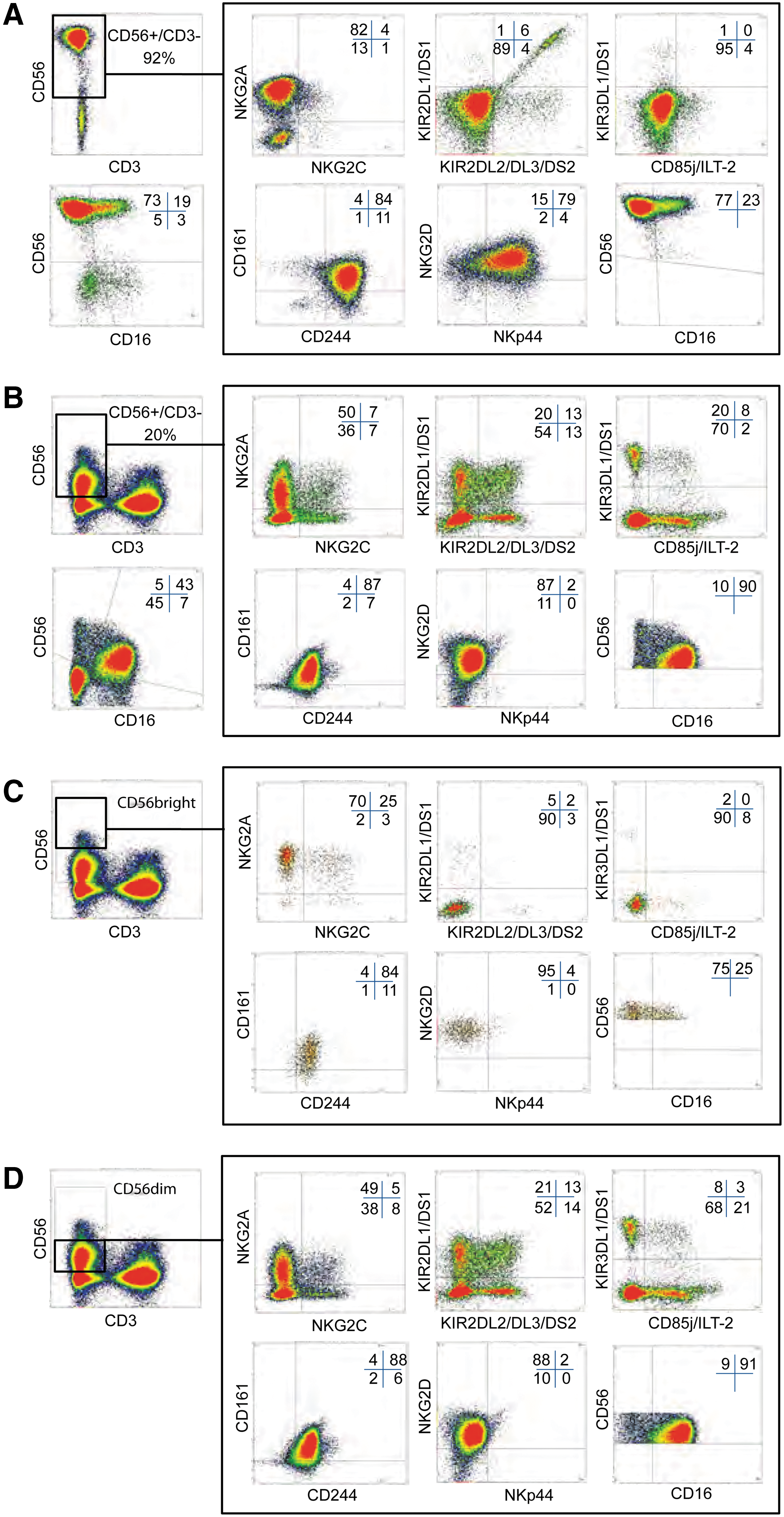
Ex vivo generated NK cells display phenotypic properties of PBNK cells in flow cytometry. Ex vivo generated NK cells in cultures from day 28 and NK cells in PB mononuclear cells were analyzed in parallel by 10-color flow cytometry using a panel of antibodies for characteristic surface markers and NK cell receptors as described in the Materials and Methods section.
Ex vivo differentiated NK cells show a gene expression repertoire of cytotoxic molecules similar to CD56dim PBNK cells and reveal potent cytolytic function
We next investigated the cytotoxic gene expression profile of the cells and whether this could be matched with their cytolytic function. Therefore, we analyzed several granzymes as well as perforin, which are highly induced during ex vivo NK cell differentiation, and compared the corresponding mRNA expression levels to freshly isolated CD56dim and CD56bright PBNK cell subsets (Fig. 5A). In line with previous reports, the cytotoxic CD56dim PBNK cells showed higher mRNA expression levels for granzyme A, granzyme B, and perforin, whereas CD56bright PBNK cells displayed elevated expression of granzyme K. Interestingly, ex vivo differentiated NK cells exhibit, similar to CD56dim PBNK cells, higher mRNA levels of granzyme A, granzyme B, and perforin and lower levels of granzyme K mRNA when compared to CD56bright PBNK cells. The expression of granulysin was comparable for both human PBNK cell subsets and ex vivo generated NK cells.
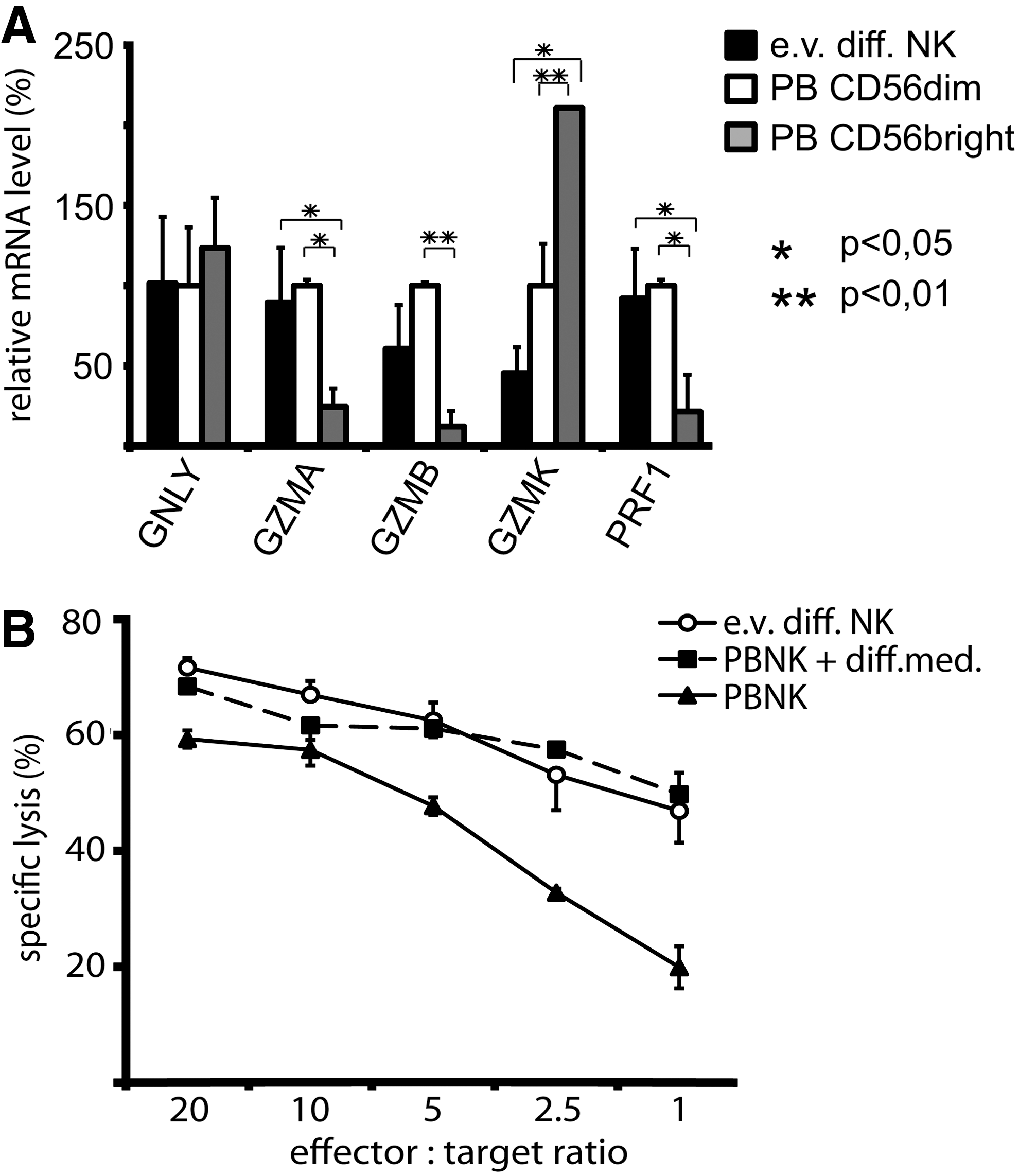
Comparison of cytolytic profiles of ex vivo generated NK cells and PBNK cells.
Based on the findings that ex vivo generated NK cells express a similar repertoire of cytotoxic molecules as CD56dim PBNK cells, we further analyzed their cytotoxic capacity in parallel to PBNK cells that consist of about 90% of CD56dim NK cells. For this purpose, we used purified PBNK, which were kept for 3 days in basal CB6M® medium or in differentiation CB6M® medium. Generally, the cytotoxic efficiency of ex vivo generated NK cells was at least equal to PBNK cells activated by culturing in the differentiation medium (Fig. 5B).
These data display that the ex vivo differentiated NK cells generated with our culture system possess a similar cytolytic molecule expression profile as CD56dim PBNK cells and, in line with their cytolytic gene expression profile, exhibit potent NK cytotoxicity.
Ex vivo differentiated NK cells can produce IFN-γ and display antibody-dependent cellular cytotoxicity
In addition to their cytolytic activity, the secretion of cytokines such as IFN-γ is a main function of NK cells. Therefore, we evaluated the potential of ex vivo differentiated NK cells to produce IFN-γ mRNA and protein in comparison to PBNK. For this purpose, cells were activated for a short period with IL-12 or a combination of PMA and ionomycin. While ex vivo differentiated NK cells without further stimulation had undetectable levels of IFN-γ mRNA, IL-12 stimulation was capable to induce significant levels of mRNA. However, only a fraction of the response of PBNK kept in a basal or differentiation medium was obtained (Fig. 6A). The combination of PMA and ionomycin, used as a positive control, triggered similar high responses for ex vivo differentiated NK cells and PBNK cells. When we tested the capacity of the cells to produce IFN-γ by intracellular staining of the cytokine, the production again was evident for IL-12- and PMA/ionomycin-treated cultures (Fig. 6B and Supplementary Fig. S1). In the case of IL-12 induction, a significant fraction of ex vivo differentiated NK cells displayed IFN-γ production. This response was higher for ex vivo differentiated NK cells than for PBNK without prior cultivation in the differentiation medium, but lower than for PBNK cells kept in the differentiation medium. PMA/ionomycin was able to elicit IFN-γ production in about half of the ex vivo differentiated cells and nearly all PBNK cells. This demonstrates that ex vivo differentiated NK cells are principally prepared to produce IFN-γ and possess an immunoregulatory capacity, but appear less responsive to stimuli such as IL-12 when compared to PBNK.
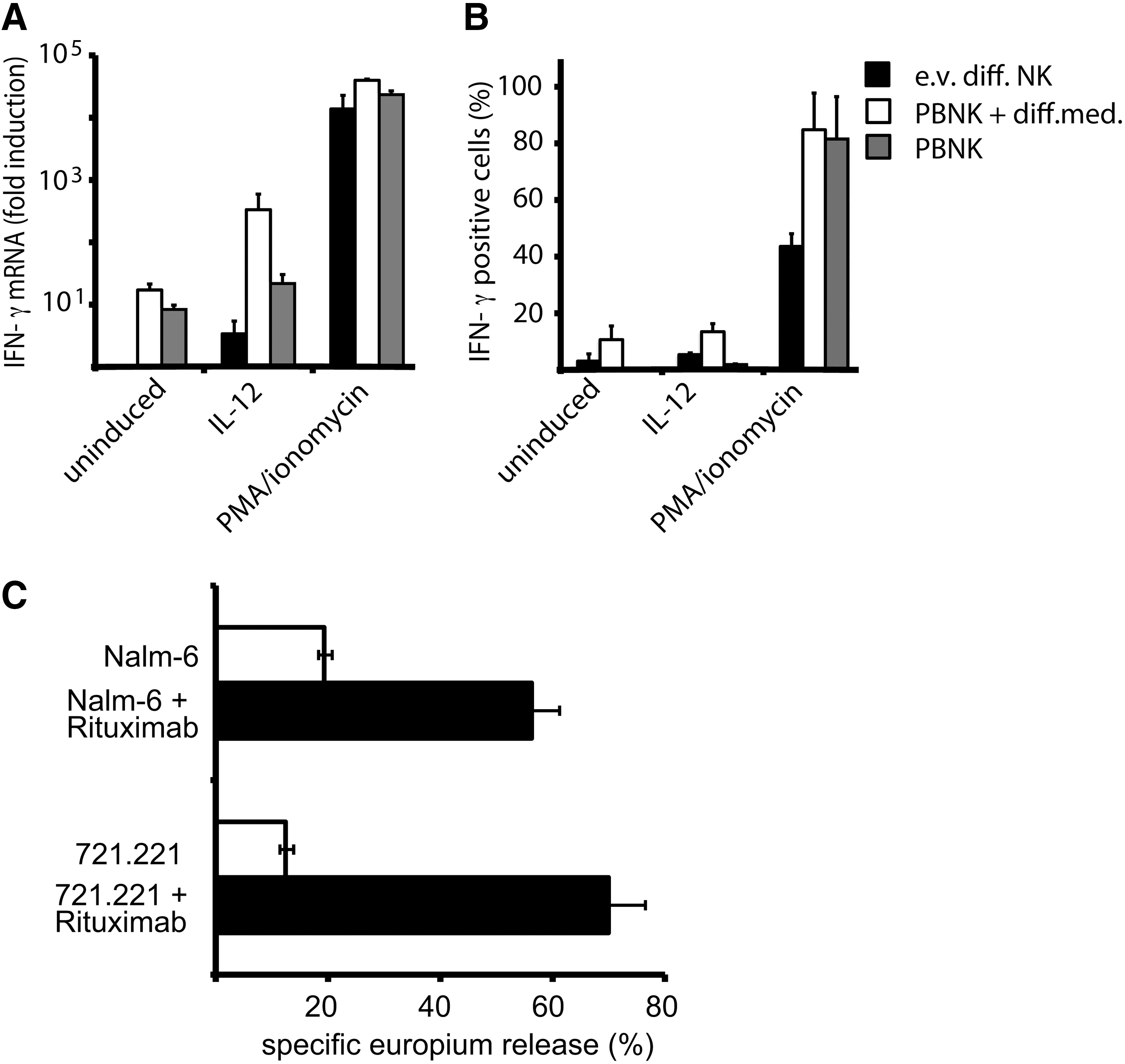
Ex vivo generated NK cells can produce IFN-γ and display potent ADCC.
In regard of therapeutic potential of ex vivo differentiated NK cells, we were further interested to test to which extent these cells would display ADCC. For this purpose, we used the human lymphoblastoid cell line 721.221 and the pre-B ALL cell line Nalm-6 in the absence or presence of Rituximab, a humanized anti-CD20 antibody, which is increasingly used in the therapy of leukemias [19]. Indeed, while in the absence of the specific antibody ex vivo differentiated NK cells displayed significant, but low cytotoxicity toward both leukemia cell lines, the addition of Rituximab mediated strongly increased killing, reaching levels of 50%–70% of the target cells (Fig. 6C). This shows that ex vivo differentiated NK cells are promising effector cells for the enhancement of antibody-based leukemia therapies.
Discussion
Ex vivo differentiation systems for NK cells from UCB stem cells are important from 2 perspectives. They can be used to elucidate basic mechanisms of human NK cell differentiation and can be employed to generate defined NK cell products for the treatment of malignancies such as leukemias [20,21]. A better understanding of the differentiation process will facilitate the production of therapeutic NK cells with specific and improved properties. However, NK cell development has so far been studied mostly in the murine system [22 –24], whereas detailed knowledge about differentiation of human NK cells on the molecular level is still limited. Until recently, in vitro systems to generate human NK cells were depending on mouse bone marrow stromal feeder cells [25,26], which hampered a detailed gene transcription analysis of the NK cell differentiation process. Only few studies have therefore investigated human NK cell development on the molecular level using ex vivo differentiation systems [27]. More recently, a hematopoietic stem cell reconstitution technique in NOD-SCID IL-2Rγnull (hu-NSG) mice was described and provided evidence that human NK cells develop in vivo in these immune-compromised mice and can acquire functional competence after preactivation [28]. However, despite significant progress in the development of differentiation systems, many issues remain unresolved; among others, the contribution of lymphoid [29] and myeloid [30] pathways to NK cell development from CD34+ stem cells is still a controversial issue. Additionally, it would be of interest to further characterize and directly compare ex vivo generated NK to the subsets of NK cells in PB to achieve a better understanding of the immunological and therapeutic properties of the generated cells.
In the current study, we have characterized in more detail a recently reported and extremely efficient system for ex vivo generation of high numbers of mature NK cells from UCB-derived CD34+ hematopoietic stem cells, which gives rise to a largely pure NK cell product [9,10]. Within this stromal cell-free system, NK cells are generated after stimulation with cytokine mixtures promoting NK as well as monocytic cell development. Indeed, monocytic cells are formed within our ex vivo NK cell differentiation system that express CD14 as well as the FcRγIII receptor similar to cultured monocytic cells [31]. However, these monocytic cells accumulate transiently in the culture and are again reduced at later time points when NK cells start to increase quickly (Fig. 1B). In the intermediary phase of our culture system, these monocytic cells might be important for promoting NK cell differentiation in terms of providing secreted factors and/or cellular interactions such as trans-presenting IL-15 via IL-15 receptor expressed on monocytes [32 –34]. In addition, based on the obtained kinetics of accumulation of CD14−CD56− cells, CD14+CD56− cells, and developing and expanding CD56+ NK cells, differentiation of the CD56+ cells from myeloid precursors is not excluded.
To further characterize our NK cell differentiation system, we performed microarray profiling at different stages of differentiation. The obtained data demonstrate that a broad range of genes characteristic for NK cells was reproducibly upregulated. Among those were NK cell receptor genes, including those for NKRP1 and all members of the NKG2 and CD94 receptor family, but also signaling proteins and cytotoxic molecules (see Table 1 and Supplementary Table S4). As expected, the strongest upregulated genes included those for the NK marker protein CD56, the IL-2 receptor β chain, and the CD94 and NKG2 receptor chains (Fig. 2). Most of the highest upregulated genes were exclusively found in the NK cell fraction and not in the monocytic cells also generated in the culture, one notable exception being FcRγIII/CD16, which was present also in the monocytic fraction (Fig. 3).
When analyzing protein expression on the cell surface, it seems that CD56 expression precedes and may be a prerequisite for CD94 expression (Fig. 2E). In the real-time RT-PCR analysis, the highest increase in mRNA levels was found for the inhibitory NKG2A receptor chain, whereas much less mRNAs for the activating NKG2C and NKG2E chains were detected. However, surface expression of NKG2A as well as of NKG2C and NKG2E should be linked with CD94 expression, as these 3 NKG2 chains can be transported to the cell surface only as heterodimers with CD94 [35]. The preferential high expression of NKG2A in ex vivo generated NK cells is also reflected in flow cytometry data (Fig. 4), which showed elevated surface expression of the heterodimeric NKG2A/CD94 receptor when compared to CD56dim PBNK cells, rather close to the level displayed by CD56bright cells.
Importantly, we further found that, with few exceptions, NKG2A+ NK cells generated in this culture exhibited higher levels of most other NK cell receptors and cytotoxic molecules or signaling molecules characteristic for NK cells when compared to NKG2A− cells (Fig. 3). Thus, it seems that NKG2A expression is connected to further maturation of NK cells. The number of NK cells with expression of the NKG2C receptor chain was to a large degree donor-dependent, as also observed for PBNK cells [36], but usually was present on 1 to 10% of the cells, indicating that the activating NKG2C/CD94 receptor was normally expressed. Ex vivo generated NK cells displayed a relatively high proportion of cells double positive for the NKG2A/CD94 and NKG2C/CD94 receptors, quite similar to CD56bright cells (I.Karas and E.Hofer, unpublished observation).
It has been previously shown that the NKG2A/CD94 receptor is the major inhibitory receptor on developing mouse NK cells [18] and on mature cytotoxic human NK cells differentiated in vitro using a stromal-feeder cell line [17]. In these studies, NKG2A expression was correlated with the acquisition of cytotoxic properties. This is in line with our observation that NKG2A+ cells, compared to NKG2A− cells, express not only more important NK cell surface proteins but also higher levels of cytotoxic molecules. In accordance, discrete stages of human NK cell differentiation have also been found in vivo and support the finding of a nonrandom acquisition of NK cell receptors; for example, prior expression of the CD94 receptor chain is followed by the acquisition of NK cell functions [29]. Studies on subpopulations of human PBNK cells, which lack inhibitory receptors, found these cells to be immature and hyporesponsive [37]. In this context, our data emphasize the notion that for the majority of cells, expression of NKG2A/CD94 discriminates developmental stages during ex vivo NK cell differentiation. It is however possible that a minor portion of NK cells may mature after expression of other inhibitory NK receptors.
The present study provides first insights and attempts to integrate ex vivo generated NK cells within the established model of cytotoxic and cytokine-producing NK cell subsets. Based on our data, the comparison of ex vivo differentiated NK cells with freshly isolated CD56bright and CD56dim PBNK does not allow a complete classification of the generated cells to either of these subpopulations. The CD56 expression is even higher than on CD56bright PBNK cells, and NKp44 is also unusually high, corroborating that these proteins are upregulated by cytokines and their high levels may be determined by the cytokines used in the growth medium. NKG2A is highly expressed at a level similar to CD56bright cells. Furthermore, CD16 expression is rather low, again similar to the levels observed for CD56bright cells. However, there is significant expression of KIR (Fig. 4). Importantly, the gene expression profiling highlighted a potentially mature repertoire of cytotoxic molecules expressed by the ex vivo differentiated NK cells. By more closely examining the expression of perforin and various granzymes by real-time RT-PCR, we were able to show that the ex vivo generated NK cells, similar to CD56dim PBNK cells, express higher levels of granzyme A, granzyme B, and perforin and lower levels of granzyme K when compared to CD56bright PBNK cells [38,39]. Therefore, the ex vivo generated NK cells exhibit most important functional prerequisites for strong cytotoxic activity similar to CD56dim PBNK cells. In line with this, we observed that the killing capacity of the ex vivo generated NK cells is at least equal to the activity of PBNK cells activated for several days in a differentiation medium containing 1,000 U/mL IL-2 (Fig. 5).
A second major function of NK cells is the production of inflammatory cytokines and chemokines, including IFN-γ. Although this immunoregulatory function has been conventionally associated with the CD56bright subtype, a recent re-evaluation has shown that also CD56dim cells can produce IFN-γ, especially at early time points of stimulation by IL-12 or target cell binding [5]. Our evaluation of IFN-γ mRNA and protein production in ex vivo NK cells has shown that these cells, in contrast to PBNK cells, without further stimulation do not produce detectable levels of IFN-γ mRNA and protein. When treated with the physiological stimulus IL-12 for 4 to 16 h, significant amounts of IFN-γ mRNA were detected, and a small fraction of the cells produced IFN-γ. This response remained always lower than for PBNK cells preincubated in the differentiation medium, and longer activation times appeared not to further increase the response (data not shown). Full stimulation with PMA/ionomycin led to IFN-γ responses more comparable with PBNK cells, demonstrating that ex vivo NK cells are principally prepared for IFN-γ production and immunoregulatory function, but this response needs more robust activation than for PBNK.
Furthermore, antibody-based reagents are increasingly used in the therapy of leukemia and other forms of cancer [19,40]. We were therefore interested to determine whether ex vivo generated NK cells would display ADCC toward leukemia cell targets in the presence of Rituximab. Indeed, we observed that the low cytotoxic activity displayed against 721.221 and Nalm-6 cells was strongly increased by Rituximab, resulting in 60%–70% lysis of these cells. This shows that the small fraction of ex vivo generated NK cells with expression of CD16 can be efficiently activated by corresponding antibodies to kill antibody-binding target cells. These cells should therefore display a synergistic antitumor efficacy in combination with therapeutic antibodies.
Collectively, these findings strengthen the potential therapeutic application of highly cytotoxic NK cells generated ex vivo from CD34+ UCB cells. Clinical studies have already revealed the principal potential of allogeneic NK cells for adoptive cancer immunotherapy [20,21,41,42]. These prospects seem to be especially promising for treatment of certain leukemias, for which haploidentical or close to haploidentical stem cell transplantation has been shown to be effective. In this regard, our feeder cell-free ex vivo differentiation system provides large-scale yields by high expansion rates of hematopoietic stem cells derived from UCB, which can be appropriately selected from available huge cord blood banks, and a highly pure NK cell product with adequate receptor expression profiles and efficient killing properties comparable to CD56dim PBNK cells. We propose that this system has great potential for exploitation in adjuvant treatment of cancer.
Nomenclature
According to the HUGO Gene Nomenclature Committee Database (HGNC;
Footnotes
Acknowledgments
We acknowledge the help of the midwives of the Department of Obstetrics and Gynecology of the Medical University of Vienna with the collection of cord blood. Furthermore, we thank Drs. Miguel Lopez-Botet and Renate Panzer-Grümayer for supplying 721.221 and Nalm-6 cells; Ing. D. Prinz for competent preparative cell sorting; and all members of the Department for Vascular Biology and Thrombosis Research for help and discussions throughout the work, especially Maria Witkowsky for keeping our laboratory organized. This work was supported by a Marie Curie research training network grant of the EC to E.H. (MRTN-CT-2005-019248). D.L. was a Marie Curie fellow in the network.
Authors Disclosure Statement
For the authors D. Lehmann, M. Osl, K. Lipnik, M. Bilban, B. Schlechta, and E. Hofer, no competing financial interests exist.
The work relates to the GBGM medium for ex vivo generation of NK-cells, which is a commercially available cell culture medium previously developed by Glycostem Therapeutics. Purchase and/or use of the GBGM medium is not restricted by patent rights. Glycostem Therapeutics has acted as sponsor in part of this research. Authors J. Spanholtz and M. Tordoir are employees of Glycostem, scientifically directed by H. Dolstra. Compensation for Dolstra's involvement in this project is paid by Glycostem to the overall research budget of RUNMC. RUNMC and Glycostem cooperate on the basis of a research collaboration agreement that regulates the above interests. Neither Dolstra nor RUNMC has a financial interest in Glycostem. The other authors based at the Medical University of Vienna have no financial or commercial interest connected to Glycostem.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.