
Abstract
The nonobese diabetic (NOD) mouse is a classical animal model for autoimmune type 1 diabetes (T1D), closely mimicking features of human T1D. Thus, the NOD mouse presents an opportunity to test the effectiveness of induced pluripotent stem cells (iPSCs) as a therapeutic modality for T1D. Here, we demonstrate a proof of concept for cellular therapy using NOD mouse-derived iPSCs (NOD-iPSCs). We generated iPSCs from NOD mouse embryonic fibroblasts or NOD mouse pancreas-derived epithelial cells (NPEs), and applied directed differentiation protocols to differentiate the NOD-iPSCs toward functional pancreatic beta cells. Finally, we investigated whether the NPE-iPSC-derived insulin-producing cells could normalize hyperglycemia in transplanted diabetic mice. The NOD-iPSCs showed typical embryonic stem cell-like characteristics such as expression of markers for pluripotency, in vitro differentiation, teratoma formation, and generation of chimeric mice. We developed a method for stepwise differentiation of NOD-iPSCs into insulin-producing cells, and found that NPE-iPSCs differentiate more readily into insulin-producing cells. The differentiated NPE-iPSCs expressed diverse pancreatic beta cell markers and released insulin in response to glucose and KCl stimulation. Transplantation of the differentiated NPE-iPSCs into diabetic mice resulted in kidney engraftment. The engrafted cells responded to glucose by secreting insulin, thereby normalizing blood glucose levels. We propose that NOD-iPSCs will provide a useful tool for investigating genetic susceptibility to autoimmune diseases and generating a cellular interaction model of T1D, paving the way for the potential application of patient-derived iPSCs in autologous beta cell transplantation for treating diabetes.
Introduction
T
Nonobese diabetic (NOD) mice spontaneously develop autoimmune T1D, which has many similarities to human autoimmune diabetes, and is a valuable model for investigating the pathogenesis of and genetic susceptibility to T1D [20]. A number of researchers have identified strategies for the treatment and prevention of diabetes in NOD mice [21 –33]. For example, anti-CD3 treatment in NOD mice formed the basis for a clinical trial of anti-CD3 monoclonal antibody therapy in human T1D models. Other promising avenues of investigation include finding ways to reverse established autoimmunity and identifying new potential therapeutic targets. Given the similarity to human TID, we chose this model to test the effectiveness of a cell therapy approach.
We established NOD-iPSC lines and applied a chemically defined stepwise method to differentiate the NOD mouse-derived iPSCs (NOD-iPSCs) into functional pancreatic beta-like insulin-producing cells. The differentiated NOD-iPSCs expressed diverse markers of pancreatic beta cells and released insulin in response to glucose and KCl stimulation. Moreover, when transplanted into diabetic mice, the NOD-iPSC-derived insulin-producing cells ameliorated hyperglycemia.
Materials and Methods
Mice
NOD/ShiJcl and NOD/SCID mice were purchased from the Korean Research Institute of Bioscience and Biotechnology. All animal experiments conformed to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of Konkuk University (KU10069 and KU10070).
Preparation of NOD mouse embryonic fibroblasts and NOD pancreas-derived epithelial cells
NOD mouse embryonic fibroblasts (NMs) were isolated from 14-day-old NOD mouse embryos (n=7) [34]. NOD mouse pancreas-derived epithelial cells (NPEs) were isolated from the pancreatic tissue from an 8-week-old NOD mouse, as previously described [35]. Isolated NMs and NPEs were cultured in 60-mm dishes containing the Dulbecco's modified Eagle's medium (DMEM; Sigma, St. Louis, MO), 10% fetal bovine serum (FBS; HyClone, Logan, UT), and 100 U/mL of penicillin/streptomycin (Invitrogen, Carlsbad, CA).
Plasmid preparation and magnet-based nanofection
For generation of exogenous DNA-free iPS cells, magnet-based nanofection was conducted according to the previous reports [17,36]. Briefly, the plasmid DNAs containing Octamer-binding transcription factor 4 (OCT4), SRY-box containing gene 2 (SOX2), Krüppel-like factor 4 (KLF4), and c-MYC were mixed with PolyMAG (chemicell, GmbH, Berlin, Germany). For transfection, cells were seeded into each well of 24-well plates (1×105 cells/well) 1 day before transfection and the cells were transfected by the Magnetofection system.
Lentiviral infection and iPS cell generation
NMs and NPEs were infected with concentrated self-inactivating human immunodeficiency virus type-1-based lentiviral vectors encoding OCT4, SOX2, KLF4, and c-MYC under the control of the elongation factor-1 alpha (EF-1 alpha) promoter. G4-2 ESCs [37,38] and NOD-iPSCs were maintained on feeder layers of mitomycin C-treated mouse embryonic fibroblast (MEF) cells in DMEM supplemented with 20% FBS, 2 mM
Bisulfite DNA-sequencing analysis
Genomic DNA was purified from ESCs, NOD-iPSCs, NPEs, and NMs using the Genomic DNA Prep kit (Qiagen, Hilden, Germany) and 2 μg of DNA was utilized for bisulfite conversion using the EpiTect Bisulfite kit (Qiagen), according to the manufacturer's protocol. Bisulfite-treated DNA was amplified by polymerase chain reaction (PCR), cloned into the pGEMT-easy vectors (Promega, Fitchburg, WI), and then sequenced using T7 forward and SP6 reverse primers. Primer sequences are listed in Supplementary Table S1 (Supplementary Data are available online at
Genotyping analysis
The genotypes of established NOD-iPSCs were determined using microsatellite markers (Mouse Genome Informatics). Of these markers, we selected D11Mit320 and D3Mit21, which distinguish the NOD genotype from the C57BL/6 genotype. Genomic DNA was isolated using the Genomic DNA Prep kit (Qiagen) and specific DNA regions were amplified from the purified DNA using specific primers (Supplementary Table S1). Amplified products were analyzed on a 4% agarose gel.
Flow cytometry analysis
Single-cell suspensions of differentiating NOD-iPSCs and G4-2 ESCs were obtained by dissociating cells using TrypLE (Invitrogen). Differentiated cells were fixed with 4% paraformaldehyde (PFA; Sigma), permeabilized with 0.5% Triton X-100 (Sigma), and immunostained with specific antibodies. The primary and secondary antibodies used were anti-SOX17 (Santa Cruz Biotechnology, Santa Cruz, CA) and Cy 3-conjugated goat anti-rabbit (Jackson ImmunoResearch labs, Inc, PA), respectively. Flow cytometry data were analyzed on 10,000 cells by using a FACScan flow cytometer and Cell Quest software (Becton, Dickinson and Company, Franklin Lakes, NJ). For multicolor flow cytometry analysis, cells were obtained by dissociating cells using TrypLE (Invitrogen), washed twice in PBS, and then stained using the Multicolor Flow Cytometry kit (R&D Systems, Minneapolis, MN), according to the manufacturer's protocol.
Quantitative real-time reverse transcription–polymerase chain reaction
Total RNA was isolated using the TRI reagent (Sigma), according to the manufacturer's protocol. One microgram of total RNA was used for cDNA synthesis (ImProm-II reverse transcriptase; Promega), and cDNA was subjected to PCR amplification with specific primers. Primer sequences are listed in Supplementary Table S1. Quantitative real-time PCR was carried out using the SsoFast EvaGreen Supermix (Bio-Rad, Hercules, CA). Gene expression was quantified only in the linear range of each primer pair. The ΔΔCT method [39] was used to quantify changes in the expression of each specific gene, using the housekeeping gene GAPDH as the normalizer.
Spontaneous differentiation
To induce differentiation into the 3 germ layers, cells were cultured in suspension to form embryoid bodies (EBs) in low-attachment dishes. The cells were subjected to an 8-day induction procedure (4−/4+) that consisted of 4 days of culture as aggregates without 0.1 μM retinoic acid (RA; Sigma), followed by 4 days of culture with RA. EBs were cultured in the differentiation medium (DMEM-GlutaMax-1; Invitrogen) containing 1% nonessential amino acids (NEAA; Sigma), 0.1 mM β-mercaptoethanol, and 10% FBS. EBs were cultured for 2 weeks on plates coated with 0.1% gelatin (Sigma).
Teratoma formation assay
One million NOD-iPSCs were injected intramuscularly into the hind leg of NOD/SCID mice. Teratomas were excised 5 weeks after injection and processed according to standard procedures for paraffin embedding and hematoxylin/eosin staining.
Eight-cell stage injection and embryo transfer
B6D2F1 (C57BL/6×DBA/2) and imprinting control region (ICR) male and female mice were used to produce in vitro fertilized 8-cell embryos. Embryo injections were performed on 8-cell embryos before obvious compaction using an Olympus micromanipulator (Olympus, Center Valley, PA). Eight-cell embryos were held using a holding pipette, and a perforation in the zona pellucida was created by the XYClone laser system (Hamilton Thorne Biosciences, Beverly, MA). Twenty NOD-iPSCs or ESCs were injected into each 8-cell embryo using 20 μM injection needles. Injected embryos were cultured for 24 h and transferred into the uteri of recipient ICR female mice at 2.5 dpc.
Differentiation of NOD-iPSCs into pancreatic beta-like cells
Directed differentiation was conducted with slight modifications of previously described protocols [40,41]. ESCs and NOD-iPSCs were plated onto dishes coated with Matrigel (1:50; BD Biosciences, Franklin Lakes, NJ) and incubated for 3 days with X-Vivo 10 (Cambrex, East Rutherford, NJ) supplemented with 0.1% bovine serum albumin (BSA) (Sigma) containing 50 ng/mL activin A (R&D Systems). Then, the differentiated cells were cultured for 2 days in the F12/Iscove's modified Dulbecco's medium (1:1; Invitrogen) supplemented with 0.5% BSA and 1% insulin-transferrin-selenium (ITS; Stemcell Technologies, Vancouver, Canada) along with 2 μM RA. After RA induction, the cells were cultured in the DMEM (low glucose; Invitrogen) supplemented with 0.5% BSA and 1% ITS, together with 10 ng/mL basic fibroblast growth factor (bFGF; R&D Systems) and 20 ng/mL epidermal growth factor (EGF; Sigma) for 3 days, and for another 3–5 days in the DMEM/F12 medium (Invitrogen) with ITS, 10 ng/mL bFGF and 10 mM nicotinamide (Sigma) for maturation.
Immunofluorescence
For immunofluorescence staining, ESCs, NOD-iPSCs, and differentiated cells were fixed with 4% PFA (Sigma), permeabilized with 0.5% Triton X-100 (Sigma), and immunostained with specific antibodies. To block any nonspecific binding, cells were incubated first in 10% normal goat serum (Vector Laboratories, Burlingame, CA) for 1 h and then incubated with the primary antibody at 4°C overnight. After washing with PBS, cells were incubated with the secondary antibody for 1 h and nuclei were stained with 5 μg/mL DAPI. The primary antibodies used included those against OCT4, stage-specific embryonic antigen-1 (SSEA-1), and paired box gene 6 (PAX6), pancreatic and duodenal homeobox 1 (PDX1), SOX 17, Islet-1 (ISL-1), Somatostatin, beta-tubulin III, and Brachyury (all from Santa Cruz Biotechnology), Nestin (Abcam, Cambridge, United Kingdom), alpha-fetoprotein (AFP; R&D Systems), forkhead box A2 (FOXA2), C-peptide, and Glucagon (Cell Signaling, Danvers, MA), and Insulin (Cell Signaling and Sigma). The secondary antibodies used were Cy 3-conjugated goat anti-mouse or rabbit (Jackson ImmunoResearch labs) and DyLight 649-conjugated goat anti-mouse or rabbit (Jackson ImmunoResearch labs) antibodies. All images were captured on a Carl Zeiss laser scanning (LSM-710) confocal microscope (Carl Zeiss, Oberkochen, Germany).
Insulin release ELISA assay
Primary adult mouse islets were isolated according to a standard procedure as described [42]. Insulin release was measured by incubating the cells in the Krebs-Ringer buffer. After preincubating with the Krebs-Ringer buffer at 37°C for 90 min, the differentiated cells were further incubated at 37°C for 60 min with the Krebs-Ringer buffer containing 2.5 mM
Transplantation of NPE-iPSC-derived insulin-producing cells
Streptozotocin (STZ) was injected intraperitoneally for 3 days at 50 mg/kg into 6- to 8-week-old NOD/SCID mice. When nonfasting blood glucose levels were above 13.9 mM on 2 consecutive days, 5×106 differentiated cells were transplanted into the left subcapsular renal space. Blood glucose levels were measured every 2 days between 9:00 and 10:00 am, under nonfasting conditions, using a portable glucometer (ACCU-CHEK Active; Roche, Basel, Switzerland). To quantify beta cell proliferation, bromodeoxyuridine (BrdU, Sigma) incorporation into beta cells was evaluated 12 h after an intraperitoneal injection of 100 mg/kg BrdU by double immunofluorescence staining for BrdU, insulin, and nestin. The primary antibodies used included those against BrdU (Santa Cruz Biotechnology), insulin (Cell Signaling), and nestin (Abcam, Cambridge, United Kingdom). The secondary antibodies used were Alexa Fluor 488 goat anti-rabbit (Invitrogen) and DyLight 649-conjugated goat anti-mouse (Jackson ImmunoResearch labs) antibodies as described [43]. We conducted left nephrectomy (removal of both left kidney and the graft) at 5 weeks post-transplantation and analyzed by hematoxylin/eosin staining or immunohistochemistry [immunofluorescence or diaminobenzidine (DAB)-nickel reactions].
Statistical analysis
Data were expressed as in terms of mean and standard error. Statistical significance was determined by the unpaired Student's t-test. Statistical analysis was performed on data using Origin software (OriginLab Corporation, Northampton, MA). All results were derived from at least 3 independent experiments.
Results
Establishment and characterization of NOD-iPSC lines
We prepared MEFs or pancreas-derived epithelial cells (PEs) from NOD mice and used quantitative reverse transcription (RT)-PCR to analyze the level of pancreas-specific gene expression, including the expression of hepatocyte nuclear factor 3 beta (HNF 3 beta), PDX1, and PAX6. Pancreas-specific genes were expressed to a greater extent in NPEs than in NMs and control ESCs (G4-2) (Fig. 1A). At first, as we previously established a magnet-based nanofection technique for generation of exogenous DNA-free iPS cells, this transient expression technique was applied for generation of NOD-iPSCs. However, we could not stably establish the NOD-iPSCs without constitutive expression of exogenous factors in the NOD background [44]. Next, we infected the cultured NMs and NPEs with a combination of lentiviruses that encoded OCT4, SOX2, KLF4, and c-MYC under the control of the EF-1 alpha promoter, and morphological transformations of the infected cells were examined. Colonies were selected 14 days after infection, based on their morphological resemblance to mouse ESC colonies (Fig. 1B). NOD-iPSCs formed compact colonies, as observed by phase-contrast microscopy, and were positive for alkaline phosphatase activity (Fig. 1C). Reprogramming efficiency was defined as the number of GFP positive colonies per 10,000 NMs or NPEs seeded. We observed a higher reprogramming efficiency for NPEs (0.08%) than for NMs (0.04%). Genotype analysis of the established NOD-iPSC lines confirmed a pure NOD genetic background (Fig. 1D). Additionally, all the NOD-iPSC lines acquired the characteristic stem cell-cycle signature of ESCs (Supplementary Fig. S1). The expression levels of the endogenous pluripotency markers OCT4, SOX2, KLF4, and c-MYC were similar in NOD-iPSCs and G4-2 ESCs. Moreover, additional ESC markers (Nanog, FGF4, and Cripto) were also detected in NOD-iPSCs, and their expression levels were similar to those seen in ESCs (Fig. 1E and Supplementary Fig. S2). Both NM-iPSCs and NPE-iPSCs were stained with antibodies against OCT4 and SSEA-1, similar to G4-2 ESCs (Fig. 1F and Supplementary Fig. S3). Bisulfite genomic-sequencing analyses revealed similarities between NOD-iPSCs and G4-2 ESCs. The promoter region of the OCT4 gene was highly unmethylated in NOD-iPSCs, whereas CpG dinucleotides in this region were highly methylated in NMs and NPEs (Fig. 1G). Moreover, using fluorescence-activated cell-sorting analysis, we confirmed high-level expression of OCT4, SSEA-1, and SOX2 markers in both NM-iPSCs and NPE-iPSCs (Fig. 1H and Supplementary Fig. S4).
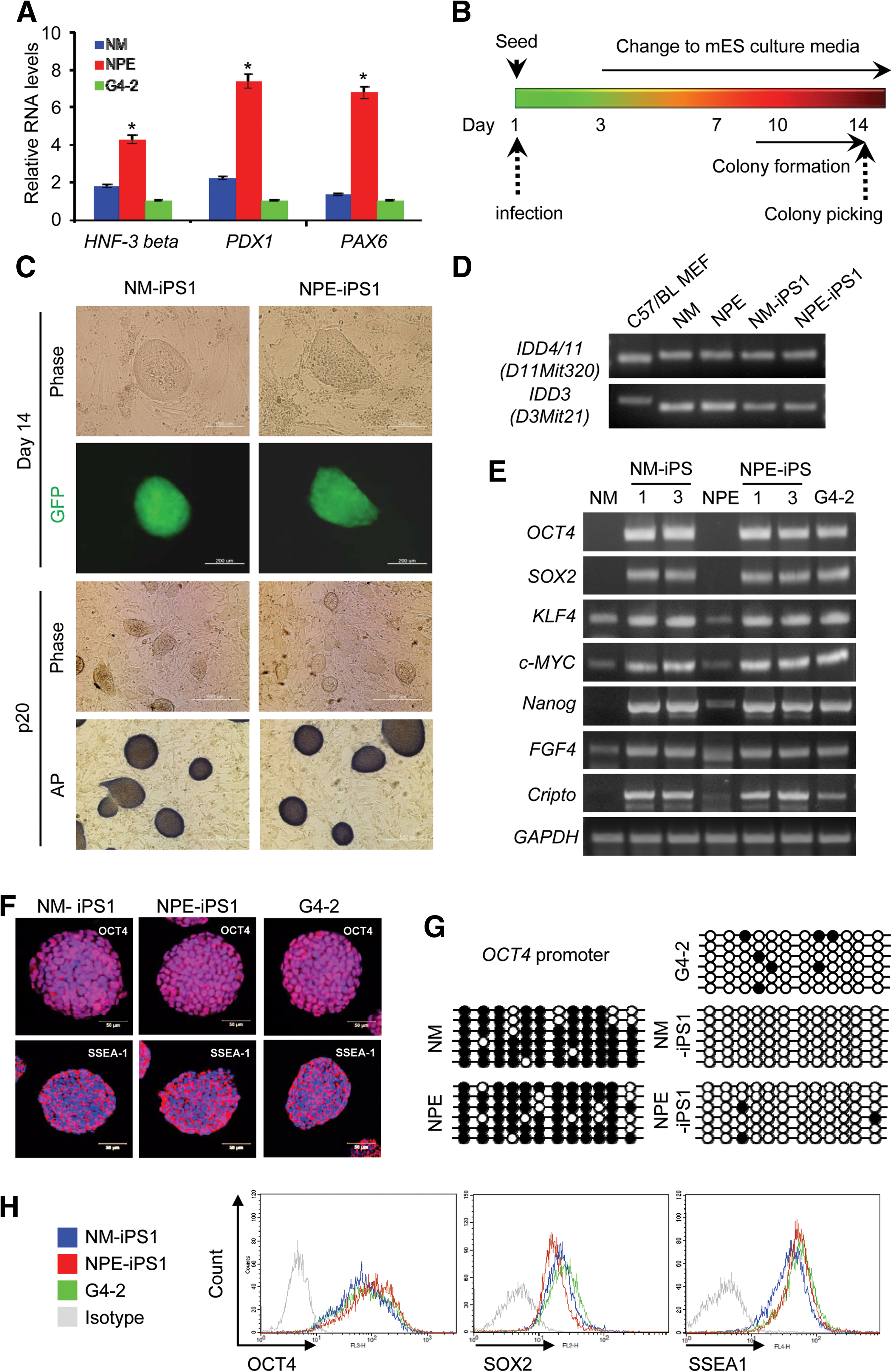
Generation of NOD-iPSCs from a NOD mouse.
NOD-iPSCs spontaneously differentiate into 3-germ-layer cell types
To determine their pluripotency, we studied whether NOD-iPSCs could be spontaneously differentiated into 3-germ-layer cells. EB formation (Fig. 2A) was induced by culturing G4-2 ESCs and NOD-iPSCs in a differentiation medium (20% DMEM without LIF) on low-attachment plates, followed by plating onto gelatin-coated dishes for additional culture. We confirmed the differentiation of NOD-iPSCs into diverse cell types from the 3 germ layers by checking the expression of specific markers for ectodermal (nestin and beta tubulin III), endodermal (AFP and Gata4), and mesodermal (brachyury and BMP4) lineages. RT-PCR (Fig. 2B) and immunocytochemical analysis (Fig. 2C and Supplementary Fig. S5) confirmed that differentiated NOD-iPSCs (both NM-iPSCs and NPE-iPSCs) expressed markers for all 3 germ layers. To examine the in vivo developmental pluripotency of the NOD-iPSCs, we performed a teratoma formation assay. When injected into NOD/SCID mice, both NM-iPSCs and NPE-iPSCs formed teratomas, including derivatives of the endoderm (gut-like epithelium and respiratory epithelium), mesoderm (smooth muscle and cartilage), and ectoderm (nerve fibers and skin) (Fig. 2D
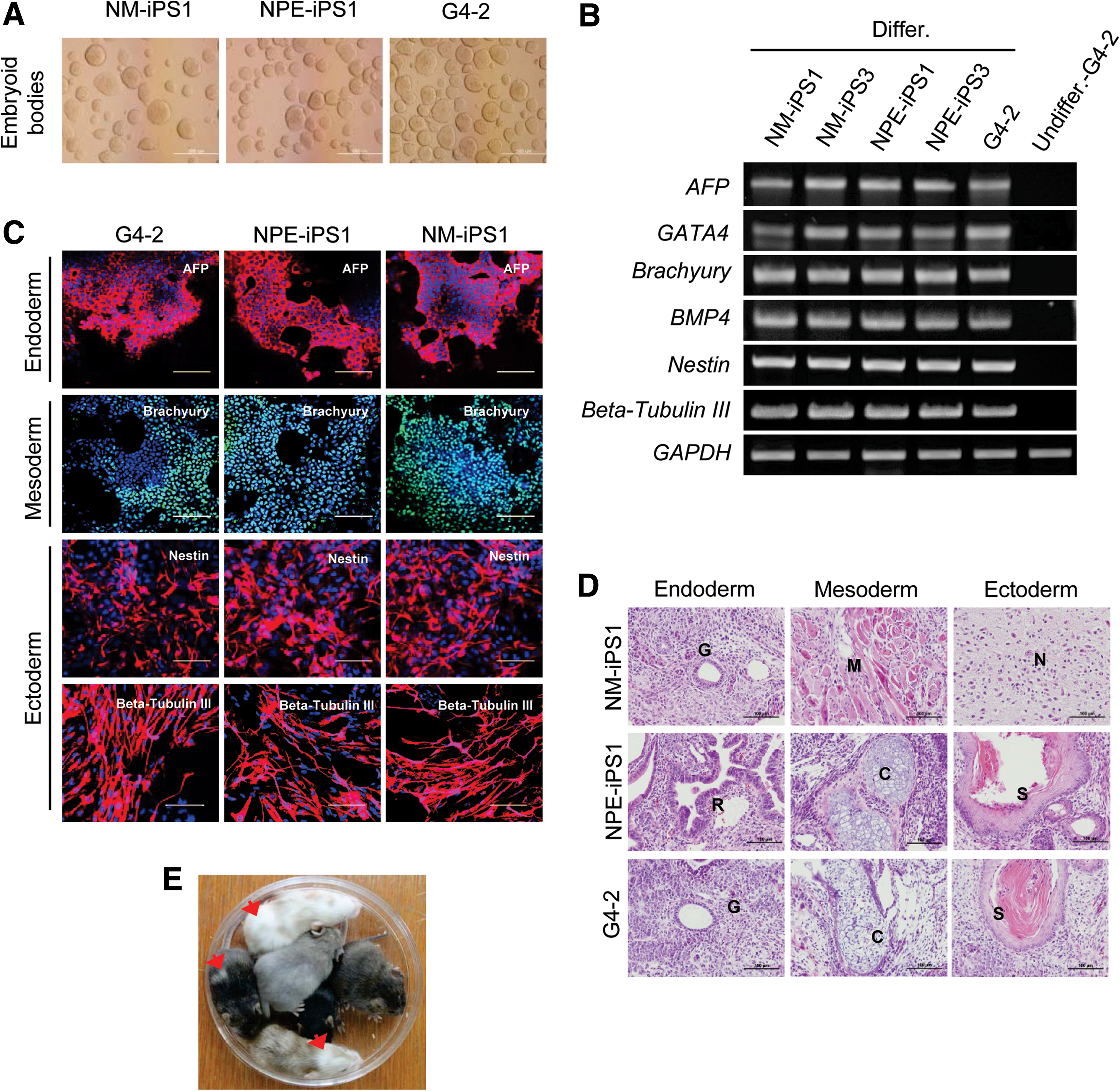
In vitro and in vivo differentiation of NOD-iPSCs into the 3 germ layers.
Highly efficient differentiation of NPE-iPSCs into insulin-producing cells
To confirm the transitions in the specific marker gene expression patterns of each cell (NM-iPSCs, NPE-iPSCs, and G4-2 ESCs) during direct differentiation, quantitative RT-PCR was performed at each step throughout differentiation for genes involved in pancreas development (Fig. 3A, C). After formation of the induced definitive endoderm, the specific marker gene expression patterns in the endoderm were monitored and SOX17 expression peaks were observed at step 1 (Fig. 3C), at a time when the OCT4 expression level rapidly decreased (Fig. 3B). The NPE-iPSC-derived definitive endoderm exhibited higher SOX17 peak expression than did the NM-iPSCs and G4-2 ESCs. Furthermore, the increased SOX17 expression level in the NPE-derived definitive endoderm was confirmed by flow cytometry. NPE-derived definitive endoderm differentiated cells were found to have a higher percentage of SOX17-positive cells, that is, >87.23%, than did NM-iPSCs (72.55%) and G4-2 ESCs (74.75%) in a 3-day culture (Fig. 3D). In step 2, the cells differentiated into PDX1-positive pancreatic progenitors and we were able to detect upregulated PDX1 gene expression on day 6 (Fig. 3C). After the induction process (steps 3 and 4), the expression of pancreatic beta cell-specific genes, ISL-1 and insulin, was detected in the differentiated cells. In particular, our data showed that the expression of pancreatic beta cell-specific genes was 2-fold higher in NPE-iPSC-derived, than in NM-iPSC- and G4-2 ESC-derived differentiated cells. These results suggest that NPE-iPSCs can be more efficiently differentiated into pancreatic lineage cells than NM-iPSCs or G4-2 ESC-derived cells can.

Dynamic expression patterns of pancreatic lineage genes during direct pancreatic differentiation from NOD-iPSCs.
NOD-iPSCs can be induced to differentiate into insulin-producing cells
We used immunocytochemistry and quantitative RT-PCR to characterize the expression of differentiation state-specific genes after each of the 4 steps in our differentiation process. In step 1, consisting of 3 days of treatment with activin A and wortmannin, the endoderm-specific expression markers SOX17, HNF-3 beta, CXCR4, GATA4, and FOXA2 were expressed in the differentiating cells. In step 2, further differentiation toward PDX1-positive pancreatic progenitors was achieved by treating the cells with RA, Noggin, and FGF7 for 3 days. In step 3, to facilitate the proliferation of pancreatic progenitors, differentiated cells in the pancreatic progenitor state were treated with EGF for 4 days. After EGF treatment, the expression of PAX6, NeuroD1, Neurogenin3, and ISL-1 was detected in the differentiated cells (Fig. 4A, B and Supplementary Fig. S6). Insulin-producing cells differentiated from NPE-iPSCs expressed pancreatic lineage-related genes to a greater degree than those differentiated from either NM-iPSCs or G4-2 ESCs (Fig. 4B). In the final stage (step 4), we induced the differentiation of NOD-iPSC-derived pancreatic progenitors into cells of endocrine lineage, which were positive for insulin, C-peptide, and PDX1 (Fig. 5A and Supplementary Fig. S7). We observed the apparent co-expression of insulin/PDX1 or C-peptide/PDX1, and some of the insulin-positive cells were costained for Glucagon or Somatostatin, suggesting that while these cells synthesized insulin, they were still immature [45]. We also confirmed the expression of pancreatic beta cell-specific genes, including IAPP, insulin, and GLUT2, in the differentiated cells. Additionally, we confirmed that the differentiated cells expressed both insulin 1 and insulin 2 in the final stage (Supplementary Fig. S8). We observed higher expression of these genes in cells differentiated from NPE-iPSCs than in those from NM-iPSCs and G4-2 ESCs (Fig. 5B). To determine whether insulin release by NOD-iPSC-derived insulin-producing cells was responsive to glucose or other physiological stimulation, we exposed these cells to low concentrations of glucose, high concentrations of glucose, KCl, or 100 μM carbachol and then used ELISA to analyze the quantity of insulin released into the culture medium. Upon stimulation with high levels of glucose (25 mM), KCl (30 mM), or 100 μM carbachol the amount of insulin released was at least 2-fold the amount released when cells were stimulated with low levels of glucose (2.5 mM; Fig. 5C). We conducted more experiments to compare insulin release from iPSC-derived beta cells and that from natural beta cells using primary islets from NOD/scid mice. Although the amount of released insulin was lower compared to primary islets, this finding indicates that these cells secrete insulin in response to glucose, KCl, and carbachol stimulation, in a manner mimicking that of adult mouse islets (50.15±0.16 vs. 33.82±0.35 ng/mg protein at 25 mM glucose; 62.8±0.3 ng/mg protein at 30 mM KCl; 55.59±0.32 ng/mg protein at 100 μM carbachol, Supplementary Fig. S9). In particular, NPE-iPSC-derived cells released more insulin when stimulated with high levels of glucose stimulation (12.22±0.32 ng/mg protein at 25 mM glucose; 12.56±0.24 ng/mg protein at 30 mM KCl; 12.6±0.35 ng/mg protein at 100 μM carbachol, *P<0.05) than did either NM-iPSC- or G4-2 ESC-derived cells.

Differentiation of NOD-iPSCs into insulin-producing cells by a stepwise direct differentiation protocol.

Differentiation of NOD-iPSCs into pancreatic insulin-producing cells.
Transplantation of NPE-iPSC-derived glucose-responsive insulin-secreting cells reverses hyperglycemia in diabetic mice
To determine whether the induced cells could function in vivo, the ability of NPE-iPSC-derived glucose-responsive insulin-producing cells to reverse hyperglycemia was examined in vivo using NOD/SCID mice, which were rendered diabetic by the STZ treatment. Blood glucose levels that were elevated after STZ administration were significantly lower after transplantation of NPE-iPSC-derived cells (n=8; Fig. 6A), suggesting that these cells can normalize hyperglycemia in diabetic mice. Normalization of blood glucose levels occurred within 2–4 days after transplantation. In contrast, sham-transplanted control diabetic mice (n=3) remained hyperglycemic, with glucose levels above 18.3 mM. After transplantation of NPE-iPSC-derived insulin-producing cells, normoglycemia (7.91±0.26 mM) was maintained up to 35 days after transplantation in the NPE-iPSC group, while the sham-transplanted control group continued to exhibit glucose levels above 18.3 mM at 35 days after transplantation. The body weights of the sham-transplanted control and NPE-iPSC-derived cell–transplanted group were also monitored. Consistent with the previous results, only the mice grafted with NPE-iPSC-derived cells transplanted group showed attenuated weight loss and continued to gain body weight over time (Supplementary Fig. S10). Serum insulin level after transplantation of NPE-iPSC-derived cells was 0.17±0.008 ng/mL, which was significantly higher than that in sham-transplanted control diabetic mice (∼0.05±0.01 ng/mL; Fig. 6B), while lower than that in normal mice (1.98±0.021 ng/mL). The amelioration of blood glucose level in mice by the graft of NPE-iPSC-derived cells was not related to spontaneous regeneration of beta cells because we observed no evidence of proliferating beta cells in STZ-treated mice by double immunofluorescence-using anti-BrdU antibody together with -insulin or -nestin antibody (Supplementary Fig. S11). Forty days after transplantation, we removed the graft-bearing kidney and found that removal of grafts from those mice transplanted with NPE-derived cells resulted in a rapid return to hyperglycemia, again confirming that the graft may be responsible for the restoration of glucose levels (Fig. 6A). Hematoxylin/eosin staining after nephrectomy revealed that the transplanted cells were able to engraft under the kidney capsule (Fig. 7A, B). Moreover, DAB staining revealed insulin- and C-peptide-positive cells in the grafts (Fig. 7C). These results indicate that NPE-iPSC-derived glucose-responsive insulin-producing cells, when transplanted into the left subcapsular renal space, were able to secrete insulin in vivo and to lower blood glucose levels in STZ-treated diabetic mice.

Transplantation of NPE-iPSC-derived insulin-producing cells into STZ-induced diabetic NOD/SCID mice.
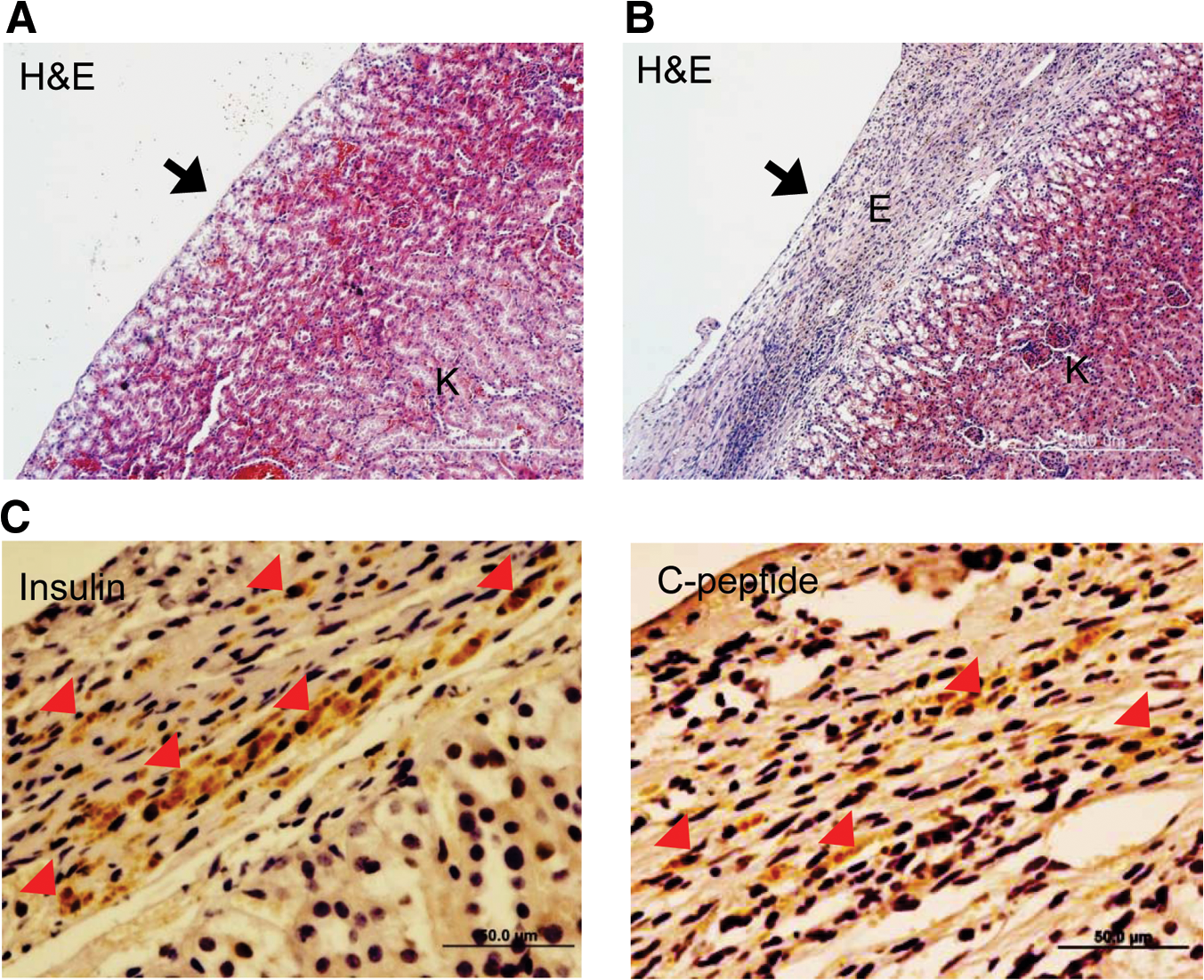
Analysis of a grafted kidney.
Discussion
Recent studies suggest that cell therapy has potential for treating T1D, which is caused by T-cell-mediated autoimmune destruction of pancreatic beta cells in genetically predisposed individuals [1]. Islet transplantation has been regarded as a potential treatment for T1D for the last 2 decades, following a report of excellent clinical outcomes by researchers in Edmonton [4,6]. However, there are 2 major drawbacks in the application of islet transplantation for the treatment of human T1D. First, the regimen of immunosuppression required to prevent rejection of the allotransplant drastically impacts the lifestyle of the patient, making it inappropriate for younger aged diabetics [2 –8]. Second and more importantly, adequate quantities of islet tissue are not available to treat the increasing population of diabetic patients. Thus, one of the most productive areas of stem cell research is cell replacement therapy for TID; however, no one has succeeded in creating a fully functional and mature insulin-producing pancreatic beta cell [46]. Although stem cell-derived beta cells are less efficient than normal beta cells, they are expected to be possibly efficient enough to help a person with diabetes. Our data suggest that as an alternative to the Edmonton protocol, a combination of cell reprogramming and differentiation techniques could be used to generate genetically matched patient-specific pancreatic beta cells. This would create an unlimited source of beta cells and reduce the requirement for immune-suppressive drugs.
NOD mice have been of great value for research in the broad field of T1D [20] and in the development of humanized mice [47]. The NOD mouse strain is considered a nonpermissive strain for ESC derivation [48], because ESC derivation requires specific exogenous factors to maintain the ICM-like pluripotent state [44]. In our study, when the magnet-based nanofection technique was used for generation of exogenous DNA-free iPS cells, we could not stably establish the NOD-iPSCs. However, we were able to efficiently establish NOD-iPSC lines from both NMs and NPEs using a lentiviral expression system. In this system, the OCT4, SOX2, KLF4, and c-MYC genes are expressed under the control of the EF-1 promoter. Our data show that the efficiency of generating iPSCs from NPEs was at least 2-fold that of iPSC generation from NMs. We detected the apparent expression of Nanog and Cripto in NPEs, but not in NMs, suggesting the possibility that the endogenous expression of Nanog and Cripto in NPEs may accelerate reprogramming [49]. Analysis of gene expression patterns revealed that iPSCs established from both NMs and NPEs have pluripotency similar to that of mouse G4-2 ESCs. Additionally, the ability of these cells to spontaneously differentiate into the 3 germ layers was investigated using specific markers, such as endoderm (AFP; hepatocyte and GATA4; zinc-finger transcription factors), mesoderm (brachyury; T-box family of transcription factors and BMP4; a member of the bone morphogenetic protein), and ectoderm (nestin; a type VI intermediate filament protein and beta tubulin III; neuron-specific beta tubulin). These cells were also almost the same as that of control G4-2 ESCs. However, NPE-iPSCs differentiated more efficiently into pancreatic beta-like insulin-producing cells than did NM-iPSCs and G4-2 ESCs. We developed an optimized stepwise differentiation protocol, based on several different direct differentiation methods [40,41], that led to the successful differentiation of NOD-iPSCs into insulin-producing cells. Interestingly, we found differences between the gene expression patterns of differentiated NM-iPSCs and those of NPE-iPSCs (Figs. 4 and 5). Our data confirm that the expression of pancreatic-specific genes in differentiated NPE-iPSCs was higher than in differentiated NM-iPSCs or G4-2 ESCs. This substantiates previous reports that suggested that genetically matched iPSCs retain a transient transcriptional and epigenetic memory of their cellular origin affecting their potential to differentiate into specific cell types [50 –52].
Our data, particularly the transplantation results, suggest that cells differentiated from patient-derived iPSCs may be an effective therapy in the treatment for T1D; however, further studies are needed to address the long-term safety and effects of these disease model-derived iPSCs. In particular, tumor formation may be a problem associated with the graft of differentiated ESCs or iPSCs. While we did not detect teratoma formation in the grafts in our study, long-term data are needed to show that NPE-iPSC-derived cells do not form tumors. We expect that NOD-iPSCs will be useful for investigations of genetic susceptibility to autoimmune diseases, and for generating a cellular-interaction model of autoimmune diseases. NOD mice spontaneously develop autoimmune responses in a variety of tissues, including salivary, lacrimal, thyroid, parathyroid, adrenal, testis, large bowel, and red blood cells, in addition to diabetes [28,53]. NOD mice can also be used as models to experimentally induce autoimmune diseases, such as experimental autoimmune thyroiditis, encephalomyelitis, and systemic lupus erythematosus [54]. Therefore, further research with NOD-iPSCs should provide a way to examine the onset and progression of autoimmune diseases in vitro or in reconstituted animal models. These in vitro and in vivo systems will also be useful for testing preventive and therapeutic strategies. In this investigation, we used immunodeficient NOD/SCID mice rendered diabetic by the STZ treatment. One of the advantages of using iPSC-derived beta cells for cell therapy is the avoidance of alloimmunity. However, in the case of autoimmune T1D, autoimmunity that persists after establishment of the disease could pose serious practical and theoretical problems for the application of iPSC-derived beta cell therapy. Thus, further studies using immunocompetent NOD mice are necessary to show that iPSC-derived beta cells, in conjunction with immunosuppression to inhibit autoimmune destruction of grafted iPSC-derived beta cells, can treat established T1D. In addition to the problem of ongoing autoimmunity, a recent study suggests that there may also be immune rejection of even autologous iPSCs [55]. It is therefore necessary to determine if the immunogenicity of transplanted iPSC-derived cells leads to immune rejection. Our data support the potential use of patient-derived iPSCs in autologous beta cell transplantation for the treatment of diabetes, although additional measures may be necessary to avoid the immunogenicity that is intrinsically associated with the production of iPSCs and their differentiation into beta cells.
In summary, we have successfully established NOD mouse iPSC lines and applied a chemically defined stepwise method to differentiate NOD-iPSCs into functional pancreatic beta cells. NPE-iPSCs more efficiently differentiated into this cell type than the NM-iPSCs and even ESCs, suggesting that epigenetic memory may predispose. The differentiated NOD-iPSCs expressed diverse markers for pancreatic beta cells and released insulin in response to glucose and KCl stimulation. Furthermore, we found that the NOD-iPSC-derived insulin-producing cells could ameliorate hyperglycemia in diabetic mice.
Footnotes
Acknowledgments
We thank Dr. Natalie DeWitt (Institut Pasteur Korea, Korea), Dr. Kyung-Mi Lee (Korea University, Korea), Dr. Andre Veillette (Clinical Research Institute of Montreal, Canada), Dr. Cassian Yee (Dept. Medicine, University of Washington), and Dr. Ofer Mandelboim (Hebrew University, Israel) for helpful suggestions and critical reading of the article. This work was supported by a grant from the Korea Health 21 R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A08-4065) and by a National Research Foundation (NRF) grant funded by the Korea government (MEST) (No. 2010-0020348). We acknowledge the graduate fellowship provided by the Ministry of Education and Human Resources Development through the Brain Korea 21 project, Republic of Korea.
Author Disclosure Statement
None of the authors has any conflict of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.