
Abstract
Human-induced pluripotent stem cell-derived neural progenitors (hiPSC-NPs) have the ability to self-renew and differentiate into glial and neuronal lineages, which makes them an invaluable source in cell replacement therapy for neurological diseases. Therefore, their enhanced proliferation and neuronal differentiation are pivotal features that can be used in repairing neurological injuries. One of the main regulators of neural development is Wnt signaling, which results in the inhibition of glycogen synthase kinase 3 (GSK-3). Here, we assess the impact of GSK-3 inhibition by the small molecule CHIR99021 on the expansion and differentiation of hiPSC-NPs in an adherent condition and a defined medium. Cell proliferation analyses have revealed that inhibition of GSK-3 in the presence of epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) increased the proliferation of hiPSC-NPs across 10 passages. The inhibition of β-catenin signaling by XAV and NOTCH signaling by DAPT reversed CHIR impact on hiPSC-NPs proliferation. The target genes of β-catenin, C-MYC and CYCLIN D1 as well as NOTCH target genes, HES1 and HES5 were upregulated. The treatment of NPs by CHIR in the absence of bFGF and EGF resulted in an increase of neuronal differentiation rather than proliferation by stabilization of β-catenin regardless of the NOTCH pathway. Thus, GSK-3 inhibition has been shown to promote proliferation of the NPs by activating β-catenin and NOTCH-related cell cycle genes in the presence of bFGF and EGF. Additionally, during GSK-3 inhibition, an absence of these growth factors allows for the switch to neuronal differentiation with a bias toward a dopaminergic fate. This may provide desired cells that can be used in therapeutic applications and offer insights into the etiology of some neurological disorders.
Introduction
H
In this regard, Wnt signaling is one of the main NP regulators [8] that predominantly leads to glycogen synthase kinase 3 (GSK-3) inhibition by the canonical pathway [9]. Wnt signaling mediates self-renewal in mouse NPs in vitro [10,11] and accelerates the proliferation of hippocampal NPs by shortening the cell cycle [12]. Activation of the canonical Wnt pathway causes an increase in expansion of NPs and suppression of neuronal differentiation, which has been shown in the early stages of chick spinal cord and mouse forebrain development, as well as in human embryonic stem cells [13 –16]. Conversely, Wnt3a can increase neurogenesis during human ReNcell cell line differentiation and is independent of the Wnt/β-catenin transcriptional activity [17,18].
Recently, accumulative evidence suggests that small molecules can replace growth factors [19 –23]. Small molecules are low molecular weight chemically synthesized molecules that freely diffuse from the cell membrane. These molecules can reversibly alter the function(s) of a single protein (or multiple proteins) by exerting exquisite temporal control in the absence of genetic modification(s). Therefore, small molecules can target intracellular NP signaling pathways, increase proliferation, and direct differentiation [14,24,25].
To date, there is no report that discusses the influence of GSK-3 inhibition on expansion and differentiation of hiPSC-derived NPs. Additionally, a detailed understanding of the molecular mechanisms and signaling pathways involved in the safe and efficient differentiation of hiPSCs into specific neural lineage cells is required.
Therefore, in this study, we have investigated the effect of GSK-3 inhibition by the small molecule CHIR99021 on the expansion and differentiation of hiPSC-NPs in the presence and absence of growth factors, respectively. According to our data, GSK-3 inhibition increased the proliferation of NPs in the presence of basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF). In the absence of growth factors, GSK-3 inhibition enhanced neuronal differentiation of NPs. We have shown that GSK-3 inhibition increases the proliferation and neurogenesis of human NPs via β-catenin stabilization. Additionally, cross talk of NOTCH signaling and β-catenin are involved in enhanced proliferation of NPs. We have also shown that GSK-3 inhibition can direct hiPSC-NPs differentiation into dopaminergic neurons.
Materials and Methods
Human NPs expansion and differentiation
hiPSC-NPs were derived and maintained as previously described [3] from a hiPSC8 line [26]. Briefly, we induced feeder-free hiPSCs that were grown for 7 days into the neural ectoderm by all-trans retinoic acid (2 mM; Sigma-Aldrich) and noggin (250 ng/mL; R&D) in the Dulbecco's modified Eagle's medium: Ham's F-12 (DMEM-F12, Invitrogen, 21331-20) supplemented with 5% knockout serum replacement (KOSR; Gibco, 10828-028), 1% nonessential amino acids (NEAAs; Invitrogen, 11140-035),
The impact of GSK-3 inhibition on proliferation of hiPSC-NPs was studied in the absence (control group) and presence of the GSK-3 inhibitor, CHIR99021 (3 μM; Stemgent, 04-0004), for the first day after passaging. To determine the signaling pathway for proliferation in hiPSC-NPs, for 3 days we used the DAPT small molecule (1 μM; Sigma-Aldrich, D5942) to inhibit the NOTCH pathway and another small molecule, XAV939 (1 μM; Sigma-Aldrich, X3004) [27] to inhibit the β-catenin pathway. We also used pifithrin α (10 μM; Stemgent, 04-0038) to prevent apoptosis through p53 inhibition [28] (Invitrogen).
Spontaneous differentiation was performed in the differentiation medium in the absence of growth factors, which included the neurobasal medium (Invitrogen, 21103-049)/DMEM-F12 (1:1), 1% B27 (Invitrogen, 17504-044), 5% KOSR, 1% N2 supplement,
MTT assay and PKH26 labeling
To assess proliferation, we used the MTT assay, which is based on reduction of the tetrazolium salt, MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma-Aldrich, M2128], by actively growing cells to produce a blue formazan product.
MTT was diluted in the DMEM-F12 at a ratio of 1:5. We incubated the hiPSC-NPs that were in six-well plates with 500 μL of MTT solution at a temperature of 37°C for at least 2 h. Next, the MTT solution was removed and 500 μL DMSO added to create a purple solution. Absorbance at 540 nm was read by an ELISA reader (Elx800; BioTek).
For PKH26 labeling, hiPSC-NPs were seeded in six-well plates. After 2 h, the medium was replaced by a new medium supplemented with PKH26 (1:1,000; Sigma-Aldrich, MIN26), and the cells were incubated for 15 min. At this step, some of the plates were used as controls and analyzed by BD-FACS Calibur flow cytometer (Becton Dickinson). Subsequently, the cells were washed, a new medium without PKH26 was added, and cells were allowed to grow in the presence or absence of CHIR. After 3 days, the cells were dissociated into single cells by the application of trypsin/EDTA for 1 min. The amount of PKH26 fluorescence was detected by BD-FACS Calibur flow cytometer (Becton Dickinson) and analyzed by Mod Fit software version 3.
Gene expression analysis by quantitative reverse transcriptase-polymerase chain reaction
To determine gene expression at the mRNA level, we extracted total RNA by using the TRIzol reagent (Invitrogen, 15596-018). A total of 5 μg of RNA was treated with DNase I (Takara, 2270A), and 1 μg of treated RNA was used as a template for cDNA synthesis (cDNA Synthesis Kit; Fermentas, KI632), according to the manufacturer's instructions. Synthesized cDNA (25 ng) was mixed with the 1× Power SYBR® Green PCR Master Mix (ABI, Prism, 4368702) and specific primers were added, to a final volume of 20 μL. The Applied Biosystems 7900 instrument was used for real-time polymerase chain reaction (PCR). Relative gene expression was analyzed by the comparative Ct method, 2−ΔΔCT [29]. Supplementary Table S1 (Supplementary Data are available online at
Immunoflourescence and flow cytometry analysis
For immunostaining, cells were fixed by 4% paraformaldehyde (Sigma-Aldrich, P6148) for 15 min, then their membranes were permeabilized by 0.2% Triton X-100 (Sigma-Aldrich, T8532) and blocked with 10% host serum in 1% bovine serum albumin (Sigma-Aldrich, A3311). Cells were incubated at 37°C with the following primary antibodies: mouse anti-NESTIN (1:200; Santa Cruz, sc-71665), mouse anti-MAP2 (1:200; Sigma-Aldrich, M1406), mouse anti-GFAP (1:400; Sigma-Aldrich, G3893), and rabbit anti-PITX3 (1:200; Sigma-Aldrich, AV38544), which were diluted in blocking solution for 1 h. After washing 3 times with 0.1% Tween 20 (Sigma-Aldrich, P7949) in PBS, cells were incubated at 37°C with the following secondary antibodies: goat anti-mouse FITC conjugated (1:200; Santa Cruz Biotechnology, sc-2010) and goat anti-rabbit Texas red conjugated (1:200; Santa Cruz Biotechnology, sc-2780) for 45 min. Nuclei were counterstained with DAPI (1:1,000; Sigma-Aldrich, D8417) and analyzed with a fluorescence microscope (Olympus, IX71).
For flow cytometry, cells were fixed by methanol/acetone (3:7) at 4°C, and then blocked with 10% serum of animal whose secondary antibody was derived for 1 h. Cells were incubated with human anti-Ki67 (1:10; ABD, HCA006) overnight at 4°C. Ki67 is a marker present during all the active phases of the proliferating cell's cycle [30]. Next, cells were incubated with goat anti-human FITC conjugated (1:200; Santa Cruz Biotechnology, SC-2456) for 1 h at room temperature. Cells directly incubated with the secondary antibody were used as controls. The FACS area flow cytometer was used for the flow cytometry analysis. Acquired data were analyzed by WinMDI software.
Western blot analysis
Total proteins were extracted by the Trizol® reagent, according to the manufacturer's instructions. For each sample, 10 μg of protein was separated by 12% SDS-PAGE (90 V for 1.5 h) using the Mini-PROTEAN 3 electrophoresis cell system (Bio-Rad). Proteins were then transferred to a PVDF membrane (Bio-Rad) by the semi-dry blotting method (Bio-Rad) and the Dunn carbonate transfer buffer that consisted of NaCHO3 (10 mM), Na2CO3 (3 mM), and 20% methanol. Membranes were blocked for 1.5 h by a 2% blocking solution (Sigma-Aldrich, T8793) and then incubated overnight at 4°C with the primary antibodies, mouse anti-GAPDH (1:5,000; Abcam, ab9484), anti-TH (1:10,000; Sigma-Aldrich, T1299), and rabbit anti-LMX1a (1:1,000; Abcam, ab31006). Membranes were subsequently incubated with the peroxidase-conjugated secondary antibodies, anti-mouse IgG (1:5,000; Sigma-Aldrich, A9044) and anti-rabbit IgG (1:5,000; Sigma-Aldrich, A9169) for 2 h at room temperature. Finally, blots were visualized by ECL detection reagents (Sigma-Aldrich, CPS-1-120). Subsequently, the films were scanned with a densitometer (GS-800; Bio-Rad).
Statistical analysis
All experiments were conducted in at least 3 independent cultures. All data were expressed as mean±standard error of mean and analyzed with one-way ANOVA followed by the Tukey's post hoc test for multiple comparisons. P values less than 0.05 were considered significant. The results of quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) were analyzed by the independent t-test.
Results
GSK-3 inhibition increased the proliferation of hiPSC-NPs
In the initial step, we were interested in determining if GSK-3 inhibition by CHIR would affect proliferation in cultures of previously established hiPSC-NPs [3]. Therefore, NPs were seeded at the same density in the control and CHIR groups. Phase-contrast images showed that when cells were exposed to CHIR during the first 24 h after passaging in the presence of bFGF and EGF, an increase in proliferation was noted (Fig. 1A). However, cell death occurred when the NPs were exposed to CHIR for more than 24 h (data not shown). CHIR-treated cells reached confluency after 3 days, whereas control hiPSC-NPs became confluent after 6 days.

GSK-3 inhibition in the proliferation of hiPSC-NPs.
The MTT analysis at days 1 and 3 after passaging revealed that hiPSC-NPs had higher proliferation in the presence of CHIR when compared to the control group (at least, P<0.05, Fig. 1B).
The flow cytometry analysis of Ki67-labeled cells demonstrated significantly higher Ki67-positive cells in the CHIR-treated cells compared to the control (P<0.05, Fig. 1C).
Additionally, we performed PKH26 staining to determine the dilutions of PKH26 through cell division (Fig. 1D, E). Immediately after PKH labeling, the parent generation was demarcated by mean fluorescence intensity, and the number of cellular divisions determined by decreased fluorescence in subsequent generations (Fig. 1D). Flow cytometry of labeled cells indicated that PKH26 fluorescence fell progressively with each cell division, and GSK-3 inhibition reduced the cell cycle time. There were more cells at generation 5 than control cells (at least P<0.05, Fig. 1E).
To exclude the increased cell number that was related to the inhibition of apoptosis, we used pifithrin α. This is a small molecule that inhibits p53, therefore preventing apoptosis.
According to the MTT analysis, there was no significant difference between pifithrin α and the control (Fig. 1F). Therefore, the increased cell numbers were related to cell proliferation, not reduction by cell death.
To assess the long-term passage effect of CHIR on hiPSC-NPs, we continued to expose the cells to CHIR during the first day after culture for a total of 10 passages. The cells in the CHIR group were passaged every 3 days, whereas those in the control group were passaged every 6 days. The MTT assay of CHIR-treated cells across 10 passages showed more proliferation for CHIR-treated cells compared to the control group (P<0.0001, Fig. 2A). The gene expression analysis for the NP genes, NESTIN, SOX1, and PAX6 after 10 passages showed increased expressions of all 3 genes in the CHIR-treated group. Decreased expression of GFAP was noted in the CHIR-treated cells (Fig. 2B).
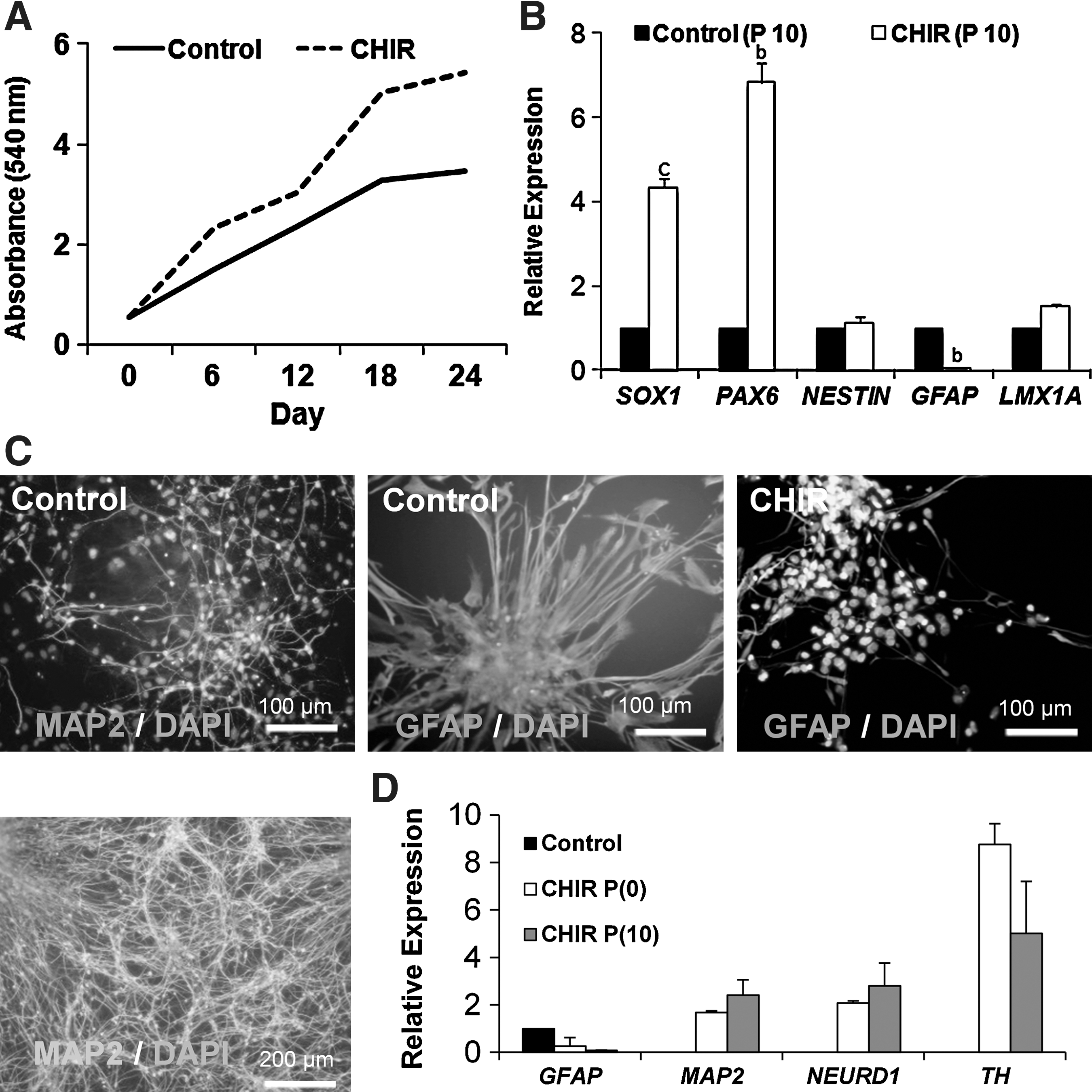
Characterization of hiPSC-NPs proliferation and differentiation capacity upon CHIR treatment.
To assess the differentiation potential of CHIR-treated hiPSC-NPs, cells were differentiated at passage 10 by growth factor withdrawal. After 14 days, hiPSC-NPs of the control group spontaneously differentiated into neurons and astrocytes, a property consistent with normal multipotent NPs (Fig. 2C). Additionally, hiPSC-NPs that received CHIR for 10 passages were CHIR-dependent for their differentiation. These cells produced more neurons rather than glial cells at a higher efficiency when compared to the control group (Fig. 2C). The real-time RT-PCR analysis of spontaneously differentiated NPs indicated that expressions of GFAP, MAP2, NEUROD1, and TH influenced significantly in the presence of CHIR in the differentiation medium compared with the control group (no CHIR during differentiation). However, the potential of differentiated cells that were passaged 0 or 10 times in the presence of CHIR were not significantly different (Fig. 2D).
Possible pathway of hiPSC-NPs proliferation upon GSK-3 inhibition
GSK-3 is a multifunctional protein that regulates cross talk with different pathways such as β-catenin and NOTCH signaling. GSK-3 is known to have an inhibitory effect on NOTCH signaling. Phosphorylation of threonine 2068, 2070, or 2074 in the ankyrin repeats at the c-terminal of NOTCH by GSK-3 inhibits transcription of the Cross-talk of GSK-3 [31 –34]. GSK-3 phosphorylates β-catenin that is to be marked for degradation [35,36].
Cross-talk of GSK-3 inhibition of β-catenin and NOTCH signaling in the proliferation of NPs was checked by XAV939 (a β-catenin inhibitor) and DAPT (a NOTCH-signaling inhibitor) in the NP medium for 3 days. The MTT assay showed that, after 3 days, addition of CHIR did not compensate cell expansion in the presence of XAV939 and DAPT when compared with the control group (at least P<0.05, Fig. 3A).

GSK-3 inhibition and its cross-talk with NOTCH and β-catenin signaling.
It was expected that CHIR might control the β-catenin and NOTCH-signaling target genes during the first 24 h of GSK-3 inhibition. Expression data indicated that mRNA of the C-MYC and CYCLIND1 genes (target genes of β-catenin) [37] increased 12 h after GSK-3 inhibition (P<0.05 for C-MYC, P<0.01 for CYCLIND1, Fig. 3B). Inhibition of β-catenin by XAV significantly blocked their expressions compared to CHIR (P<0.01, Fig. 3B). No changes were detected in the expressions of C-MYC and CYCLIND1 24 h after GSK-3 inhibition (Fig. 3B).
The gene expression analysis for NOTCH target genes showed that 30 min after CHIR treatment, expression of HES5 became elevated and continued until 1 h after GSK-3 inhibition (P<0.05, Fig. 3C). However, its expression decreased upon further treatment with CHIR. When the cells inhibited for NOTCH (by DAPT) and GSK-3 (by CHIR), expression of HES5 decreased at 30 min (P<0.01), 1 h (P<0.05), 12 and 24 h (at least P<0.05) after treatment. The expression of HES1 increased after GSK-3 inhibition (P<0.05). NOTCH inhibition did not decrease HES1 expression when both NOTCH and GSK-3 were inhibited; however, when NOTCH was exclusively inhibited its expression remained constant. HES1 expression decreased at 12 and 24 h (at least P<0.05) after treatment. These findings showed that GSK-3 inhibition promoted hiPSC-NP proliferation through the activation of the β-catenin and NOTCH-signaling pathways.
Possible pathway for hiPSC-NPs differentiation upon GSK-3 inhibition
hiPSC-NPs were allowed to differentiate for 14 days in the absence of bFGF and EGF, but with the addition of CHIR during this time. The expressions of MAP2, GFAP, NEUROD1, and NGN3 were measured by qRT-PCR and/or immunofluorescence staining (Figs. 2D and 4). In the presence of CHIR, the number of MAP2-positive cells increased (90%) compared to the control (29%; P<0.01, Fig. 4A, B), while the number of GFAP-positive cells decreased (7%) compared with the control (62%; P<0.05, Fig. 4A, B). The qRT-PCR analysis showed a significantly increased expression of MAP2, whereas the astrocyte marker, GFAP, decreased upon differentiation (P<0.001, Fig. 2D). Additionally, expression of the β-catenin target genes, NGN3 and NEUROD1, that were implicated in neurogenesis expressed significantly after CHIR treatment (P<0.01 Fig. 2D, data for NG3 not shown).

β-catenin and NOTCH signaling and their cross-talk in the neurogenesis of hiPSC-hNPs.
To explore the potential mechanisms of neurogenesis by GSK-3 inhibition, DAPT and XAV were applied for NOTCH and β-catenin inhibition, respectively. As with the proliferation assay, hiPSC-NPs were differentiated in 6 different groups. In the DAPT- and CHIR+DAPT-treated cells, expressions of MAP2 and GFAP did not significantly change during differentiation compared to the CHIR group (Fig. 4A, B). The application of XAV completely blocked neurogenesis as no MAP2-positive cells were detected (P<0.01). The application of CHIR+XAV did not improve neurogenesis (Fig. 4A, B), while NPs continued their glial differentiation by 71% in the presence of XAV or XAV+CHIR (P<0.05, Fig. 4A, B). Our results showed that GSK-3 inhibition by CHIR increased neurogenesis through β-catenin stabilization, independent of NOTCH signaling.
Evaluation of cell proliferation by the MTT assay indicated that when hiPSC-NPs were treated with CHIR for differentiation, the total number of differentiated cells remained unchanged compared to the control group (Fig. 4C).
The application of another GSK-3 inhibitor, SB216763, led to enhanced neurogenesis and reduced giologenesis (Fig. 4D).
GSK-3 inhibition may bias cells toward a dopaminergic fate
To investigate if the Wnt-signaling pathway can modulate DA neuronal development, we treated hiPSC-NPs with CHIR during differentiation. Expressions of LMX1A and LMX1B, both DA precursor markers, significantly changed at day 7, but their expressions downregulated upon maturation. Expressions of the mature dopaminergic neuron markers TH, NURR1, EN1, and NGN2 were upregulated (at least P<0.05) at day 14 postinitiation of spontaneous differentiation in the presence of CHIR (Fig. 5A).
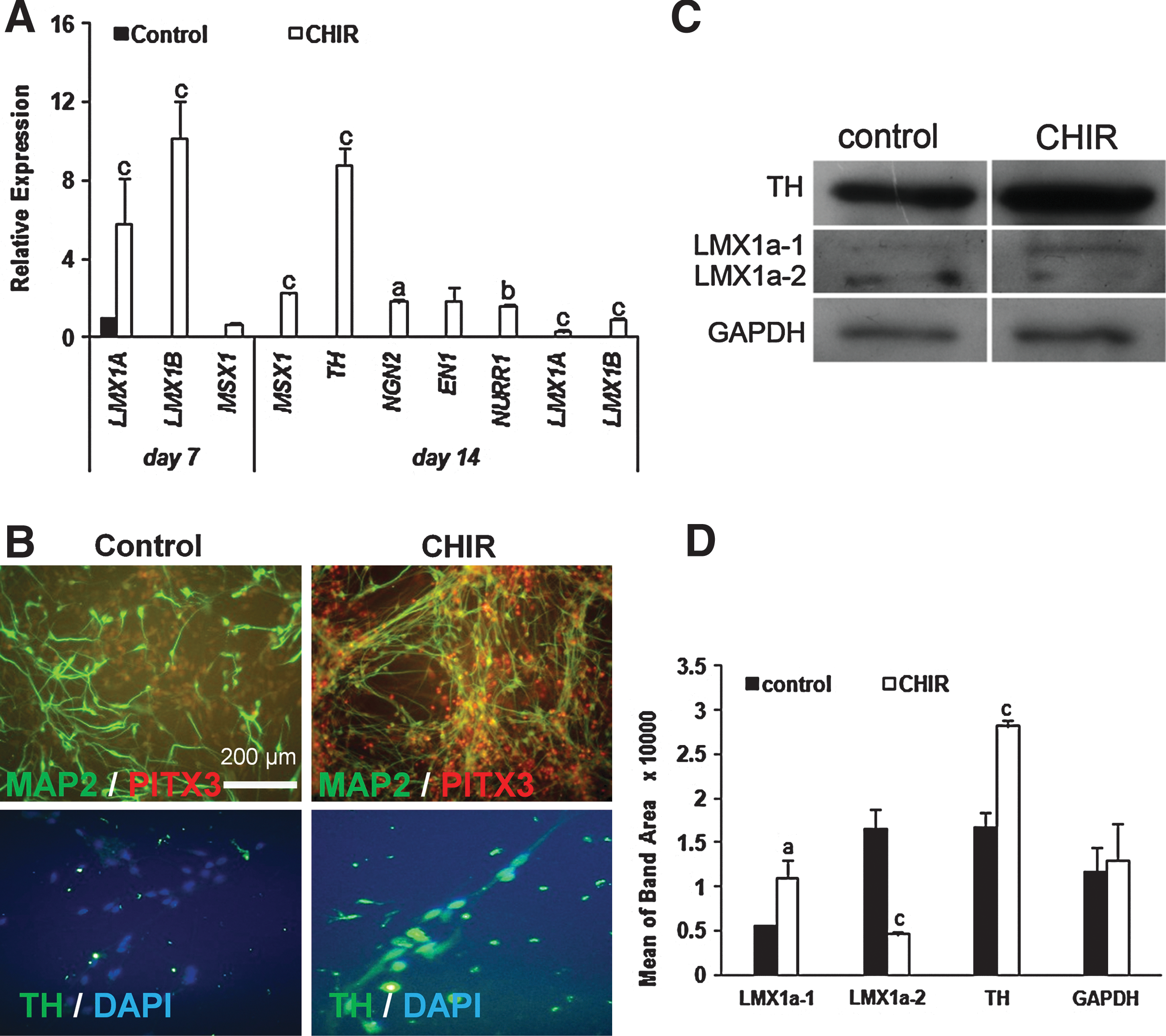
GSK-3 inhibition and neuronal differentiation toward dopaminergic neurons.
To further assess the dopaminergic fate of differentiating cells, we performed immunostaining and western blot. Although a few PITX3-positive cells (<1%), which are a specific transcription factor for dopaminergic neurons [38], have been observed in the control group, the addition of CHIR to differentiating NP cultures increased the number of double-positive MAP2/PITX3 cells to 98% (Fig. 5B). These cells were also TH-positive (Fig. 5B). Interestingly, we found that treatment of differentiating precursors with CHIR resulted in upregulation of the TH protein level as detected by western blot (Fig. 5C, D).
Discussion
The proliferation and differentiation of hiPSC-NPs into a definitive neuronal subtype, such as DA neurons, is of great interest because of their potential use in regenerative medicine to treat disorders such as Parkinson's disease. Understanding the signaling events during hiPSC-NPs proliferation and differentiation is essential for the generation of more efficient protocols with which to establish desired neurons from hiPSC-NPs. There are many reports that have shown the importance of Wnt signaling for different stages of proliferation and differentiation. The role of Wnt signaling (which results in the inhibition of GSK-3), however, in the proliferation and differentiation of hiPSC-NPs has not yet been reported. In this study, we have found that GSK-3 inhibition by small molecule CHIR greatly promoted proliferation of hiPSC-NPs in the presence of bFGF and EGF, and improved neuronal and DA subtype differentiation of hiPSC-NPs. However, key issues such as production, cell-permeability, reversibility of inhibition, and specificity of binding aimed at overcoming safety concerns should be addressed before using small molecules to generate cells for regenerative medicine.
GSK-3 inhibition increased the proliferation of hiPSC-NPs through activation of β-catenin and NOTCH signaling
GSK-3 inhibition plays an important role in adult stem/progenitor cells and in neural development [39 –45]. Continuous activation of GSK-3 has been found in the brains of patients with Alzheimer disease [46] and psychiatric disorders [9]. It has also been demonstrated that NPs of mice with a GSK-3 deletion show hyperproliferation [43]. Therefore, GSK-3 has attracted a substantial amount of attention for developmental biology and therapy.
Here, we found that GSK-3 inhibition by CHIR (3 μM) during the first day after passaging increased the proliferation of hiPSC-NPs in the presence of bFGF and EGF.
Our findings on enhanced proliferation of hiPSC-NPs agreed with recent in vitro studies, which demonstrated enhanced influence of SB-216763 [14,25] or R3303544 [4o7] GSK-3 inhibitors on the proliferation of the hNPs cell line ReNcell VM or murine subventricular zone-derived NPs. In contrast, it has been demonstrated that the GSK-3 inhibitor SB-216763 did not significantly affect proliferation in murine neonatal NPs [48,49]. IM-12 decreased cell growth of the human NP line, ReNcell VM [17]. Additionally, it has been shown that inhibition of GSK-3 by 6-bromoindirubin-3′-oxime or CHIR99021 reduced the proliferation of adult human olfactory epithelium NPs [50]. Differing results may be a result of different GSK-3 inhibitors, the origin of the NPs, or cross-talk between different ligands such as bFGF and EGF in the medium [13,45]. Here, we have used bFGF along with CHIR during maintenance of NPs. bFGF increases NPs proliferation and concurrently inhibits their differentiation. It has been reported that bFGF signaling through phosphatidylinositol 3 kinase (PI3K) activation inactivates GSK-3, resulting in the accumulation of β-catenin in a manner different from that in the Wnt canonical pathway [14].
To explore the mechanisms by which GSK-3 inhibition increased proliferation, we used XAV to inhibit β-catenin signaling and DAPT to inhibit NOTCH signaling. We found that the impact of CHIR on hiPSC-NPs proliferation reversed in the presence of XAV and DAPT. These results showed that inhibition of GSK-3 at this stage resulted in β-catenin stabilization and NOTCH signaling. The downregulation of C-MYC and CYCLIND1 12 h after GSK-3 inhibition, and HES5 and HES1 induction after 30 and 90 min, respectively, confirmed cross-talks among GSK-3, β-catenin, and NOTCH-signaling pathways in the proliferation of NPs.
It has been previously described that ventral midbrain precursors can respond to GSK-3 inhibitors, kenpaullone, and indirubine-3-monoxime (I3M), by stabilization of β-catenin [51]. Accumulation of β-catenin in the nucleus resulted in proliferation by activating LEF/TCF transcription factors and concurrent repression of neuronal differentiation by potentiating the NOTCH1-RBP-Jkappa-signaling pathway [14,34,48]. β-catenin and the NOTCH1 intracellular domain make a molecular complex with the promoter region of the antineurogenic Hes1 and Hes6 genes, permitting their expression [14]. Also, it has been demonstrated that lithium chloride treatment or Wnt-1 overexpression resulting in GSK-3 inhibition leads to the upregulation of the Hes1 promoter [34]. This signaling interplay is particularly essential for NPs expansion, because misexpression of dominant-active GSK-3β completely suppresses the NP self-renewal, increasing their differentiation into neurons. Thus, the GSK-3β/β-catenin signaling axis is regulated by FGF and Wnt signals by linking the cell proliferation to the inhibition of differentiation [14].
GSK-3 inhibition increased neurogenesis independent of NOTCH signaling
This study demonstrated that the inhibition of GSK-3 enhanced neuronal differentiation and blocked gliogenesis of hiPSC-NPs in the absence of bFGF and EGF to 90% MAP2-positive cells. It has been observed that Wnt signaling increased neuronal differentiation of mouse cortical NPs [45,52]. Similarly, GSK-3 inhibition in cultures of embryonic rat ventral mesencephalon cells led to increased neuronal differentiation [51], differentiated adult rat hippocampus-derived neurospheres in vitro [49], and increased neurogenesis of the hNPs cell line ReNcell VM up to 5.7% compared to 2.6% in control cells [25]. Also, GSK-3 inhibitors promoted neurite outgrowth [53]. Strong knockdown of GSK-3 significantly decreased axonal growth in dissociated neuronal cultures [54].
In our study there was decreased expression of GFAP, an astrocyte marker. These data agreed with a recent report on neurospheres derived from neonatal mouse forebrains [55].
The exact mechanism by which GSK-3 inhibitors increased neurogenesis and their effect on the fate of neurons is unknown. To explore the cellular mechanism by which GSK-3 inhibition promotes neurogenesis, we have inhibited β-catenin and NOTCH signaling. Immunostaining data revealed that GSK-3 or NOTCH inhibition increased neurogenesis. Treatment of cells with both GSK-3 and NOTCH inhibitors also showed similar results. Therefore, neurogenesis occurred independent of NOTCH signaling. However, the application of XAV suppressed neurogenesis and induced gliogenesis, which indicated the importance of β-catenin in this regard. It has been demonstrated that β-catenin stabilization by CHIR increases expression of the proneuronal genes, NEUEOD1 and NGN3 [45,56]. NeuroD1 is essential for CNS development, particularly in the hippocampus and cerebellum [57,58]. Overexpression of NeuroD1 is sufficient to induce neuronal differentiation of adult hippocampal neurons [59]. Ngn3 expression in rodents regulates glial differentiation of spinal cord glial precursors [60] and promotes early neurogenesis of the chick spinal cord [37].
GSK-3 inhibition may change the propensity of differentiating NPs toward a dopaminergic fate
It has been reported that GSK-3β inhibitors, I3M and kenpaullone, stabilize β-catenin, and that overexpression of β-catenin in ventral mesencephalic precursors results in increased DA differentiation [51]. Additionally, Wnts are key regulators of proliferation and differentiation of DA precursors during ventral midbrain neurogenesis [61]. In this regard, we have found that GSK-3 inhibition directs neuronal differentiation of hiPSC-NPs to a dopaminergic fate. Also, we have detected DA precursors at day 7 of differentiation due to their high expressions of LMX1A and LMX1B.
Immunostaining for PITX3 as a specific transcription factor for midbrain dopaminergic neurons has shown the presence of ∼90% PITX3-positive cells in hiPSC-NPs that differentiated upon GSK-3 inhibition. Additionally, TH expression at the protein level and mRNA expressions of TH, NURR, MSX1, EN1, and NGN2 increased in the CHIR group compared to the control group at day 14 after differentiation. It has been reported that the Wnt/β-catenin pathway inhibitor Dickkopf1 (Dkk1) increased both neuroectodermal differentiation and the number of mouse ESC-derived DA neurons [44].
Recent studies have also demonstrated that iPSC-NPs can be differentiated in cultures to generate neurons, which produce significant functional improvement when transplanted in rodent models of Parkinson's disease [62,63], monkey spinal cord injury [64], and damaged swine neural retina [65]. More recently, the Studer's group have shown that midbrain floor-plate precursors and DA neurons could be derived from embryonic stem cells days after exposure to small molecule activators of sonic hedgehog and canonical Wnt signaling [66]. Additionally, long-term engraftment in Parkinsonian mice, rats, and monkey models indicate robust survival of midbrain DA neurons, complete restoration of amphetamine-induced rotation behavior, and improvements in tests of forelimb use and akinesia [66].
Conclusions
Taken together, our data indicated that β-catenin stabilization by GSK-3 inhibition increased proliferation of hiPSC-NPs by increasing the expression of cell cycle-promoting genes such as C-MYC and CYCLIND1. Neurogenesis was promoted by increased expressions of proneural genes such as NEUROD1 and NGN3. Additionally, in the presence of CHIR in the differentiation medium, the neuronal differentiating cells showed a bias toward a dopaminergic fate.
Therefore, a promising strategy to increase neuronal yield in hiPSC-NP cultures would consist of a 2-step process that inhibits GSK-3. Initially, hiPSC-NPs would be expanded in conditions that maximize their proliferation by using the expansion medium in the presence of bFGF and EGF. In the second culture step, they would be induced toward neuronal lineage and maturation in the presence of a GSK-3 inhibitor in the differentiation medium in the absence of bFGF and EGF. Our strategy may improve the application of pluripotent stem cell-derived NPs and neuronal cells for cell replacement therapy in neurological disorders such as Parkinson's disease.
Footnotes
Acknowledgments
This study was funded by grants provided from the Royan Institute and the Iranian Council of Stem Cell Research and Technology.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.