
Abstract
Autophagy is a complex “self-eating” process and could be utilized for cell survival under stresses. Statins, which could reduce apoptosis in mesenchymal stem cells (MSCs) during both ischemia and hypoxia/serum deprivation (H/SD), have been proved to induce autophagy in some cell lines. We have previously shown that atorvastatin (ATV) could regulate AMP-activated protein kinase (AMPK), a positive modulator of autophagy, in MSCs. Thus, we hypothesized that autophagy activation through AMPK and its downstream molecule mammalian target of rapamycin (mTOR) may be a novel mechanism of ATV to protect MSCs from apoptosis during H/SD. Here, we demonstrated that H/SD induced autophagy in MSCs significantly as identified by increasing acidic vesicular organelle–positive cells, type II of light chain 3 (LC3-II) expression, and autophagosome formation. The levels of H/SD-induced apoptosis were increased by autophagy inhibitor 3-methyladenine (3-MA) while decreased by rapamycin, an autophagic inducer. ATV further enhanced the autophagic activity observed in MSCs exposed to H/SD. Treatment with 3-MA attenuated ATV-induced autophagy and abrogated the protective effects of ATV on MSC apoptosis, while rapamycin failed to cause additional effects on either autophagy or apoptosis compared with ATV alone. The phosphorylation of AMPK was upregulated whereas the phosphorylation of mTOR was downregulated in ATV-treated MSCs, which were both attenuated by AMPK inhibitor compound C. Further, treatment with compound C reduced the ATV-induced autophagy in MSCs under H/SD. These data suggest that autophagy plays a protective role in H/SD-induced apoptosis of MSCs, and ATV could effectively activate autophagy via AMPK/mTOR pathway to enhance MSC survival during H/SD.
Introduction
S
Autophagy is a complex process to degrade intracellular components by forming autophagosomes, which then fusion with lysosomes to form autolysosomes [6]. Its role in cell survival and death has become a hot topic and been studied deeply in recent years. There is evidence showing that autophagy could both serve as a pro-survival and pro-death way, which may be dependent on the different cell types and conditions employed in studies [7,8]. For example, under ischemia or H/SD condition, autophagy could reduce apoptosis in myocytes and neutron by providing energy, or eliminating reactive oxygen species and damaged organelles [9 –11]. On the other hand, in some cell types, H/SD-induced autophagy has been proved to be a promoter of apoptosis, leading to cell death, or autophagy itself could induce autophagic cell death as type II programmed cell death, if the dysregulated autophagy becomes too extensive and prolonged [12]. However, the functional role of autophagy in H/SD-induced apoptosis in MSCs has not been fully elicited.
Statins are a kind of cholesterol-lowering drugs with multiple biological activities. They can not only attenuate the inflammation and oxidative stress to provide a better microenvironment for implanted MSCs after AMI, but also increase the anti-apoptotic capacity of MSCs itself under H/SD condition, of which, both make statins a convincing candidate to deal with the “low survival” problem of MSCs mentioned previously [3,13 –15]. Several signaling pathways have been implicated in this protective role of statins, including AMP-activated protein kinase (AMPK), Janus kinase/signal transducers and activators of the transcription, phosphatidylinositol 3-kinase (PI3K)/Akt, and mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK/ERK1/2) pathways [13,14,16]. More recently, autophagy induction has been noticed as a novel way by which statins regulate cell survival and death [17 –20]. Still, the autophagic regulation of statins in MSCs has not been elucidated.
Noticeably, AMPK, which could be activated by statins to protect MSCs from apoptosis, is also an important regulator of autophagy [21]. Under H/SD or other energy shortage situation, AMPK could sense the cellular energy change and be activated by a decreased ATP/AMP ratio, thus indirectly suppressing the activation of mammalian target of rapamycin (mTOR), one of its major downstream target [21]. And that interruption of mTOR signaling could stimulate autophagy [9,22]. Thus, AMPK/mTOR pathway is thought to be a positive regulator of autophagy in hypoxia, starvation, or other energy stress [21].
Therefore, we designed this study to explore the functional role of autophagy in MSC apoptosis during H/SD. And we hypothesized that atorvastatin (ATV), one of the most prescribed statins, could utilize autophagy activation via AMPK/mTOR pathway to protect MSCs from apoptosis under H/SD.
Materials and Methods
Cell culture
Isolation and culture of adult rat MSCs were performed as previously described [5]. Briefly, bone marrow was harvested from the tibia and femur of Sprague-Dawley rats (60–80 g, male) and seeded into cell culture flasks with Iscove's modified Dulbecco's medium (Invitrogen) containing 10% FBS (Invitrogen) and 1% penicillin-streptomycin (Invitrogen) at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. When the cells reached 80% confluence, they were detached using 0.25% trypsin-EDTA (Invitrogen) and subcultured at the ratio of 1:2. Using this expansion protocol, only the adherent cells were selected. Nonadherent hematopoietic cells decreased with consecutive passages. All cells used in the experiment were passage 3.
Cell treatment
The MSCs were washed with phosphate-buffered saline (PBS), exposed to different concentrations of ATV (kindly provided by Pfizer) (control, 0.001 μM, 0.01 μM, 0.1 μM, 1 μM, and 10 μM) in serum-free medium, and then incubated in a sealed, hypoxic GENbox jar fitted with a catalyst (BioMérieux) to scavenge free oxygen for desired time. Oxygen tension in the medium was measured using anaer indicator (BioMérieux). Autophagy inhibitor 3-methyladenine (3-MA) (Sigma-Aldrich) of 5 mM, autophagy promoter rapamycin (Cell Signal Technology) of 10 nM, or AMPK inhibitor compound C (Calbiochem) of 10 mM was added to further investigate the role and mechanism of autophagy in MSC apoptosis.
Measurement of autophagy and apoptosis by flow cytometry
Cell autophagy was assessed by detecting acidic vesicular organelles (AVO) using acridine orange (AO) (Sigma-Aldrich) according to published procedures [23,24]. Briefly, cells were stained with 1 μg/mL AO for 15 min, removed from the plate with trypsin-EDTA (Invitrogen), and collected in PBS. In the AO-stained cells, the cytoplasm fluoresce bright green, whereas the AVO, including lysosomes and autolysosomes, fluoresce bright red. Green (510–530 nm) and red (650 nm) fluorescence emission from 104 cells illuminated with blue (488 nm) excitation light was measured on flow cytometer (Becton-Dickinson) using Cell Quest software.
Cell apoptosis was assessed using Annexin V-FITC/PI Kit (Biosea Biotechnology). Briefly, cells were collected and resuspended in 200 μL medium buffer. About 10 μL of Annexin V solution was added to the cell suspending solution and incubated for 15 min avoiding light at room temperature. Then, 300 μL medium buffer and 5 μL of propidium iodide (PI) were added and the cell suspension was analyzed by a flow cytometric machine immediately. Annexin V-/PI- represented viable cells, Annexin V+/PI- cells represented early apoptotic cells, and PI+ cells represented necrotic or apoptotic cells in terminal stages. About 104 cells were acquired on a flow cytometer (Becton-Dickinson) and analyzed with Cell Quest software.
Detection of autophagosome by transmission electron microscopy
After treated for desired time, cells were detached from the plates and fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer at 4°C for 2 h. After washed with PBS, cells were postfixed with 1% osmic acid at 4°C for 1.5 h. Cells were then dehydrated by a serial gradient ethanol and then embedded in Embed-812 medium (Electron Microscopy Sciences). Ultrathin sections were cut using an Ultrotome (Leica, Reichert Ultracuts) on uncoated copper grids and stained with 0.2% lead citrate/1% uranyl acetate. Images were recorded under a transmission electron microscope (JEM1230; JEOL).
Western blotting
Equal amounts of protein (30 μg/lane) were separated by electrophoresis for western blotting analysis. The proteins were transferred to nitrocellulose membranes (Whatman) using a semi-dry electroblotting apparatus (Bio-Rad), and the membranes were blocked for 1 h at room temperature in 5% skim milk. The membranes were then incubated overnight at 4°C with primary antibodies. The primary antibodies used were as follows: phosphorylated (Thr172) and total AMPK, phosphorylated (serine 2,448) and total mTOR, beclin1, light chain 3 (LC3) of microtubule-associated protein 1, cytochrome C, and β-actin from Cell Signal Technology; bcl-2 and bax from Santa Cruz Biotechnology. After washing, the membranes were incubated for 1 h at room temperature in blocking solution containing peroxidase-conjugated secondary antibodies. Then the membranes were washed and processed for analysis using a Chemiluminescence Detection Kit (Pierce) as described by the manufacturer. Target signals were normalized to the β-actin signal and analyzed semi-quantitatively with Quantity One system.
Statistical analysis
Data were analyzed using SPSS 15.0 software. All values were presented as mean±SD. Differences among groups were tested by one-way analysis of variance. Two-sided P-values were used and P<0.05 was considered significantly different.
Results
Autophagy and apoptosis were both induced in MSCs under H/SD
To investigate H/SD-induced autophagy in MSCs, we detected the levels of the type II of LC3 (LC3-II), a biochemical marker of autophagy, by western blotting. Under normal condition, LC3 protein exists in the cytosol as type I (LC3-I); when autophagy is activated, LC3-I can be recruited to autophagosome membrane and converted to LC3-II [25]. As shown in Fig. 1A, compared with the normal group, the LC3-II/LC3-I ratio was significantly increased after H/SD treatment (3 to 48 h), peaking at 3 h (1.30±0.17 vs. 0.74±0.10, P=0.001) and plateauing within 6 h (1.28±0.18 vs. 0.74±0.10, P=0.002) thereafter, indicating that autophagy was induced in MSCs under H/SD.
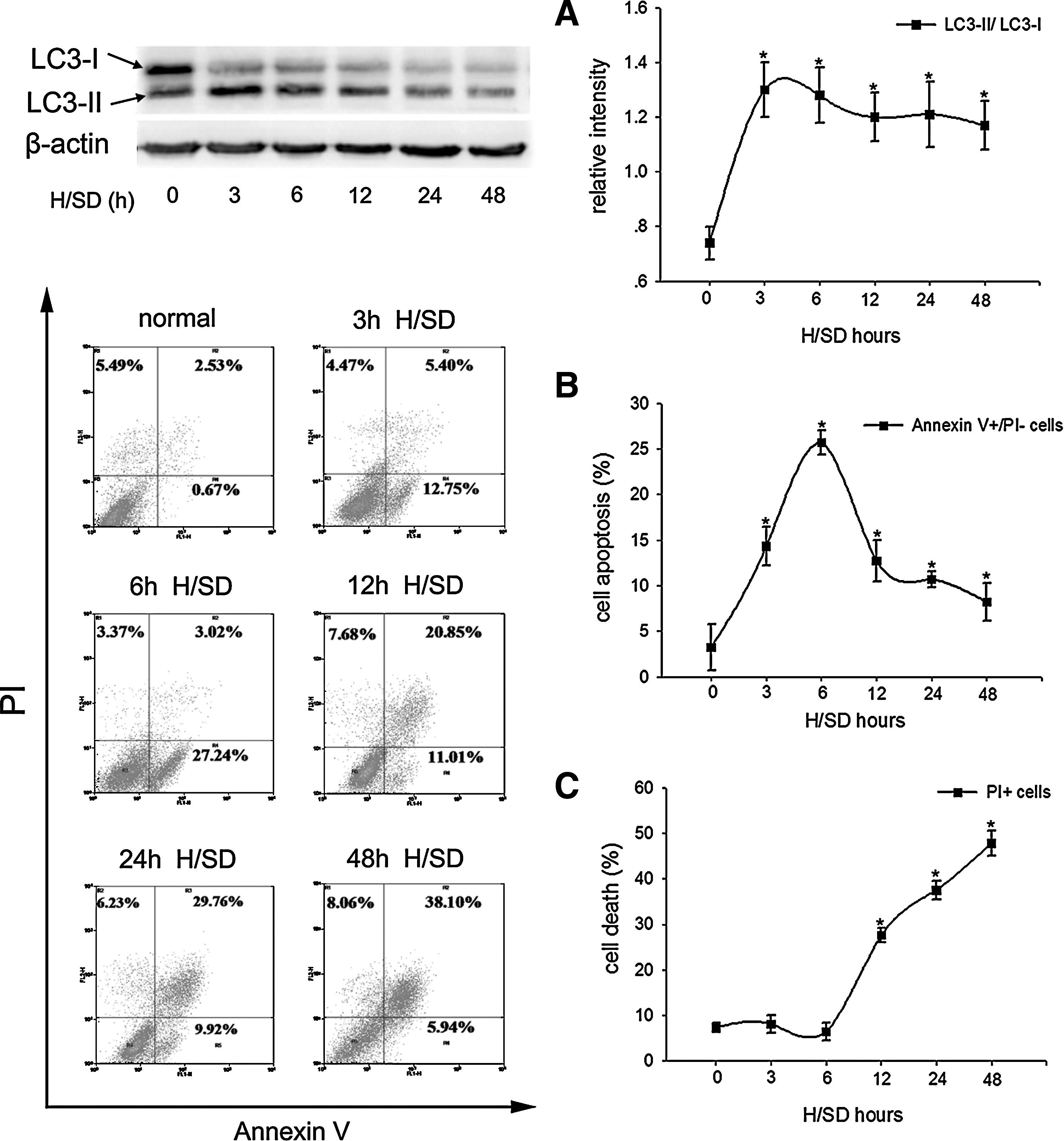
Autophagy and apoptosis were both induced in MSCs under H/SD (3 to 48 h) condition.
We next examined the apoptosis of MSCs under H/SD (3 to 48 h). In agreement with previous studies [5,13], we also found that the early apoptotic rate peaked at 6 h in MSCs exposed to H/SD (25.71±1.34 vs. 3.24±2.50, P<0.001) (Fig. 1B). However, following a longer treatment with H/SD, the population of PI+ cells, which represented the necrotic or apoptotic cells in terminal stages, was increased significantly (Fig. 1C). These data suggested that apoptosis was also induced in MSCs under H/SD condition.
Autophagy protected MSCs from apoptosis under H/SD condition
To investigate the association of autophagy and apoptosis, MSCs were exposed to H/SD for 6 h with or without autophagy inhibitor 3-MA and autophagy promoter rapamycin. As expected, the autophagic activity in MSCs was induced significantly in the H/SD control group compared with the normal group, showed by increased rate of AVO-positive cells (4.59%±0.47% vs. 3.06%±0.60%, P=0.013) (Fig. 2A), higher LC3-II/LC3-I ratio (1.23±0.23 vs. 0.54±0.20, P=0.040) (Fig. 3A), and more autophagosome formation (Fig. 3B). 3-MA suppressed the autophagic activity induced by H/SD in MSCs; meanwhile, compared with the H/SD control group, 3-MA-treated MSCs showed higher rate of apoptosis (26.02%±3.15% vs. 19.44%±3.23%, P=0.023), higher expression of pro-apoptotic proteins bax (1.51±0.15 vs. 1.18±0.14, P=0.009) and cytochrome C (1.43±0.10 vs. 1.21±0.09, P=0.013), and lower expression of anti-apoptotic protein bcl-2 (0.78±0.06 vs. 1.11±0.11, P=0.007) (Fig. 3C, D). Conversely, enhancing MSC autophagy by rapamycin significantly attenuated the levels of apoptosis in MSCs exposed to H/SD (Fig. 3C, D). In addition, we found that the population of necrotic or apoptotic cells in terminal stages induced by longer H/SD exposure (48 h) was also enhanced by 3-MA (50.93±3.30 vs. 41.77±4.40, P=0.007) while reduced by rapamycin (31.80±2.57 vs. 41.77±4.40, P=0.004) significantly (Supplementary Fig. 1B; Supplementary Data are available online at

MSCs were treated with H/SD for 6 h and autophagy was tested by AVO-positive MSCs (labeled in the circle) using a fluorescent dye AO through flow cytometric assay.
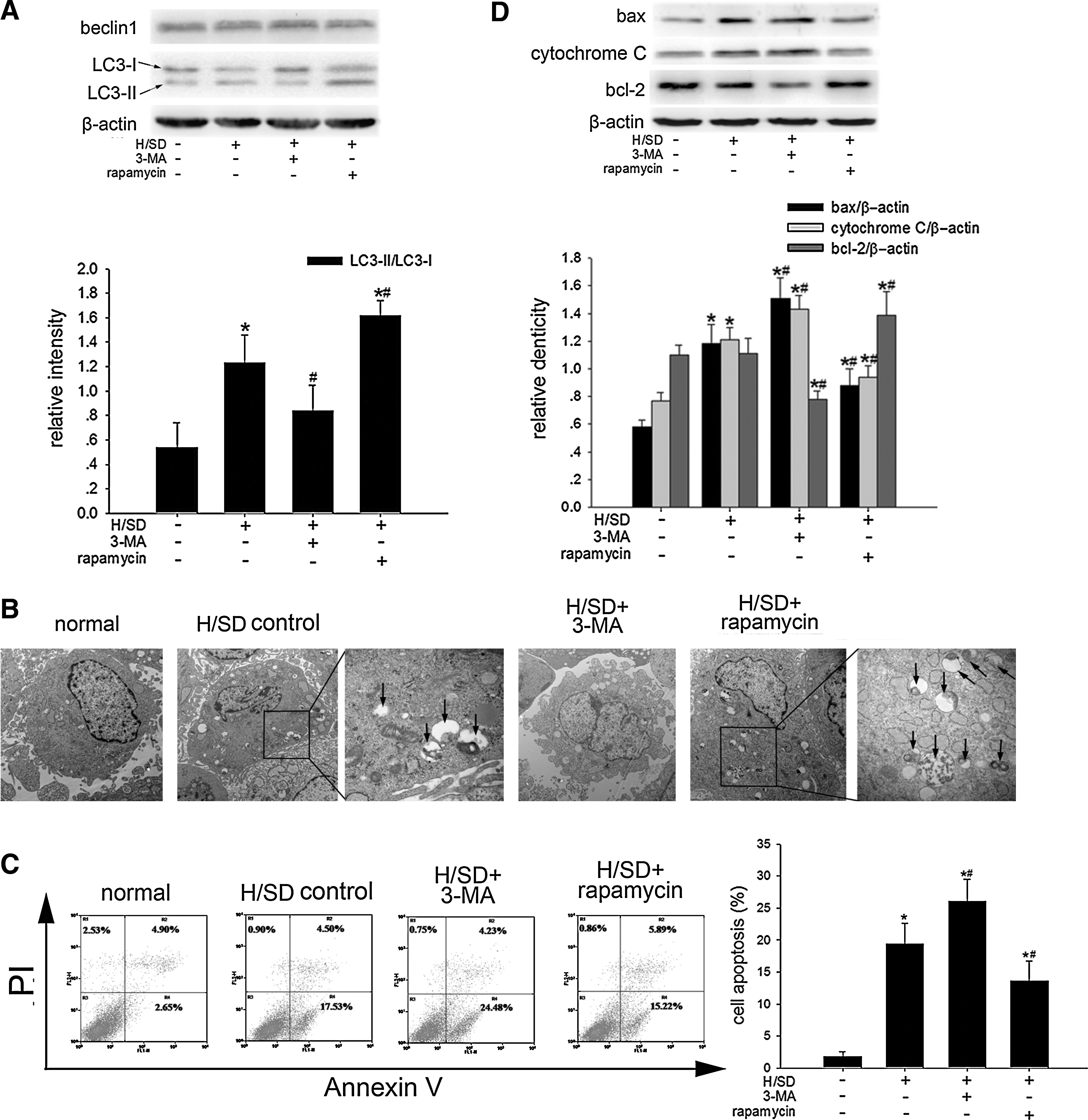
Autophagy as a protective mechanism in MSC apoptosis under H/SD for 6 h.
ATV promoted autophagy in MSCs under H/SD
It has been reported that statins could induce autophagy in several cell lines in certain conditions [17 –20]. To test whether statins were able to modulate autophagy in MSCs, we treated MSCs with various concentrations of ATV (0.001 μM, 0.01 μM, 0.1 μM, 1 μM, and 10 μM) and then exposed them to H/SD for 6 h. As shown in Fig. 2B, AVO-positive MSCs identified by flow cytometer elevated in ATV-treated groups compared with the control group under H/SD, especially in the 1 μM concentration-treated group (6.36%±0.83% vs. 4.67%±0.63%, P=0.007). And we also found that 1 μM ATV-treated MSCs showed higher LC3-II/LC3-I ratio than the control ones (1.39±0.11 vs. 0.89±0.15, P<0.001), although the expression of autophagic marker beclin1 had no difference among groups (Fig. 4A). 3-MA significantly decreased the number of AVO-positive MSCs, LC3-II/LC3-I ratio, and autophagosomes observed in 1 μM ATV-treated MSCs under H/SD, while rapamycin did not further increase any of them (Figs. 2C and 4B, C). Moreover, 1 μM ATV induced LC3-II expression in MSCs during longer H/SD exposure (48 h) as well (Supplementary Fig. 2A). These results suggested that ATV enhanced the autophagic activity in MSCs exposed to H/SD, which might be mediated by the same way as rapamycin did.

Autophagy activation in MSCs following ATV treatment under H/SD for 6 h.
Autophagy was involved in the protection of ATV against apoptosis in H/SD-treated MSCs
In consistence with previous reports [13,14], we confirmed that ATV could protect MSCs against H/SD-induced apoptosis by flow cytometric assay and western blotting in the present study (Fig. 5A, B). And interestingly, the apoptotic rate was found to be negatively correlated with the rate of AVO-positive MSCs in ATV-treated groups under H/SD (Pearson correlation: −0.59, P=0.010). To further test the functional role of autophagy in the protective effects of ATV, we exposed MSCs to 1 μM ATV with 3-MA or rapamycin under H/SD for 6 h, and then assessed the rate of apoptosis. As shown in Fig. 6A, B, during 6 h of H/SD, 3-MA attenuated the anti-apoptotic capacity of 1 μM ATV-treated MSCs as identified by higher apoptotic rate (21.07%±2.13% vs. 14.67%±3.75%, P=0.017), increased expression of bax (1.12±0.15 vs. 0.86±0.15, P=0.043) and cytochrome C (1.08±0.05 vs. 0.81±0.04, P=0.004), and decreased expression of bcl-2 (0.80±0.04 vs. 1.26±0.11, P<0.001). On the other hand, rapamycin failed to have any effects on apoptotic activity in the 1 μM ATV-treated MSCs (Fig. 6A, B). Besides, 1 μM ATV reduced the population of necrotic or apoptotic cells in terminal stages in MSCs exposed to H/SD for 48 h, which was also counteracted by 3-MA (Supplementary Fig. 2C). These results indicated that autophagy induced by ATV might be a possible mechanism for its protection against H/SD-induced apoptosis in MSCs.
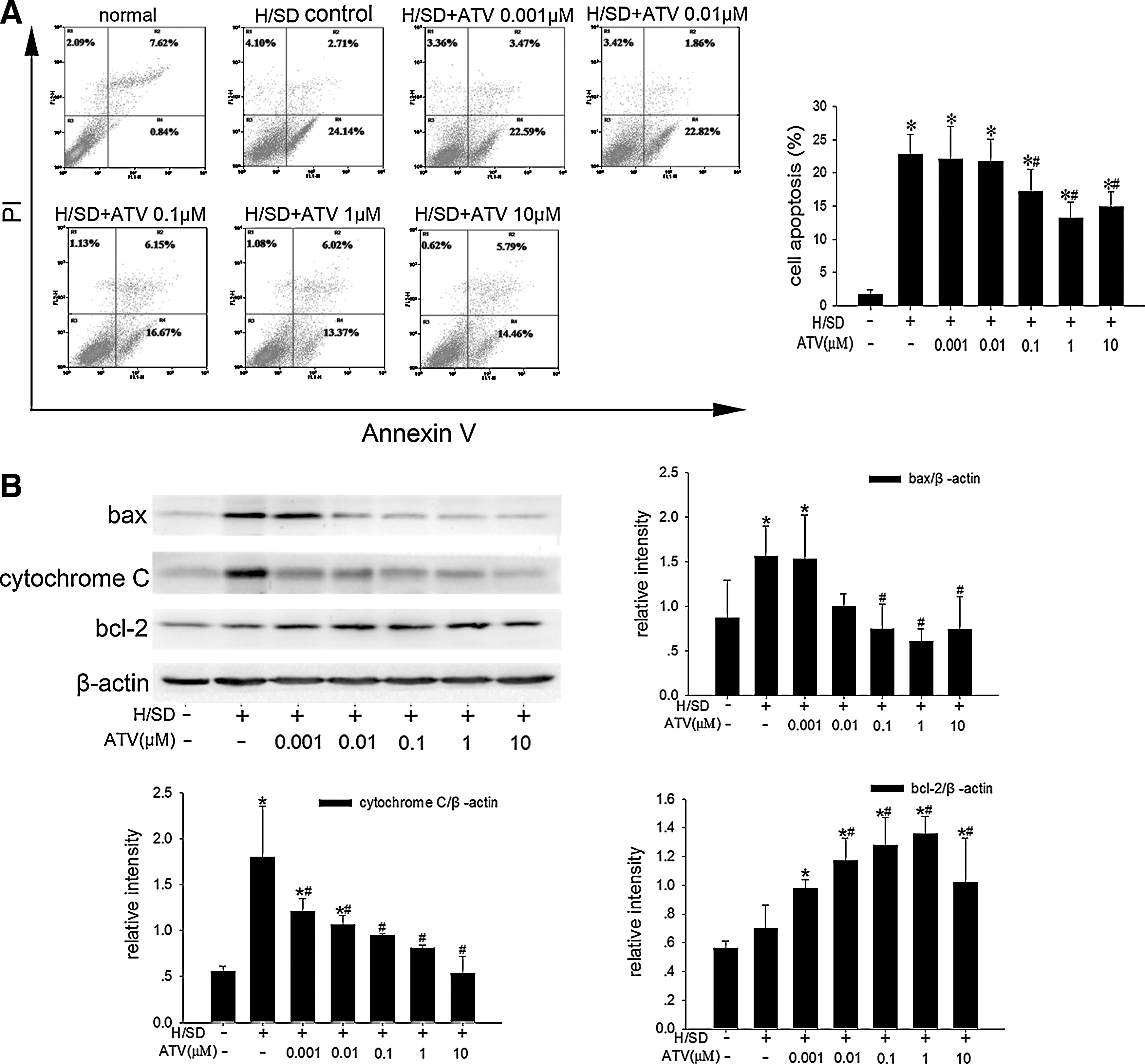
The apoptosis was reduced by ATV in a dose-dependent manner under 6 h of H/SD, as identified by flow cytometric assay
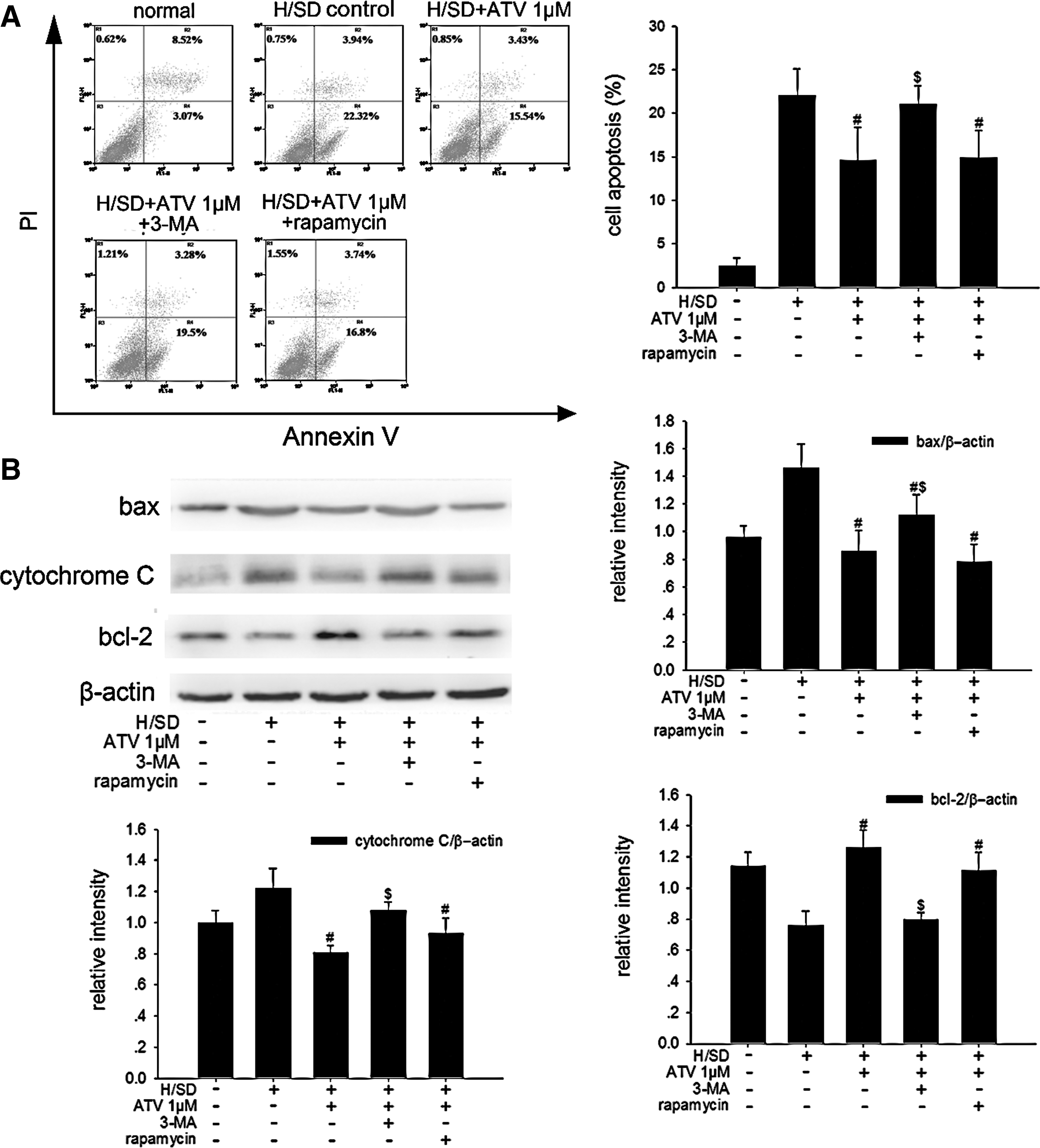
Inhibition autophagy abrogated the effects of ATV on apoptosis reduction in MSCs under 6 h of H/SD.
Autophagy induced by ATV was dependent on AMPK/mTOR pathway
There is abundant evidence showing that AMPK/mTOR pathway plays an important part in regulating autophagy [21,26]. To investigate whether AMPK/mTOR signaling also mediated the autophagy stimulated by ATV, AMPK inhibitor compound C was used and then the levels of autophagy were assessed in the ATV-treated MSCs under H/SD for 6 h. The results revealed that the phosphorylation of AMPK was upregulated (1.57±0.13 vs. 0.86±0.11, P<0.001) while the phosphorylation of mTOR was downregulated (0.60±0.29 vs. 1.29±0.19, P=0.002) in the 1 μM ATV-treated MSCs compared with the control ones under H/SD, although the expression of AMPK and mTOR did not change dramatically among groups (Fig. 7A). Compound C abrogated the effects of 1 μM ATV on both AMPK phosphorylation (0.87±0.21 vs. 1.57±0.18, P=0.001) and mTOR phosphorylation (1.22±0.25 vs. 0.72±0.21, P=0.039) (Fig. 7B). Further, treatment with compound C reduced the autophagy in the 1 μM ATV-treated MSCs that was showed by decreasing rate of AVO-positive MSCs (3.53%±0.56% vs. 6.35%±0.65%, P<0.001), LC3-II/LC3-I ratio (0.61±0.23 vs. 1.33±0.17, P<0.001), and autophagosome formation (Figs. 2D and 8A, B), suggesting that AMPK/mTOR pathway played a role in the stimulative effects of ATV on MSC autophagy under H/SD.
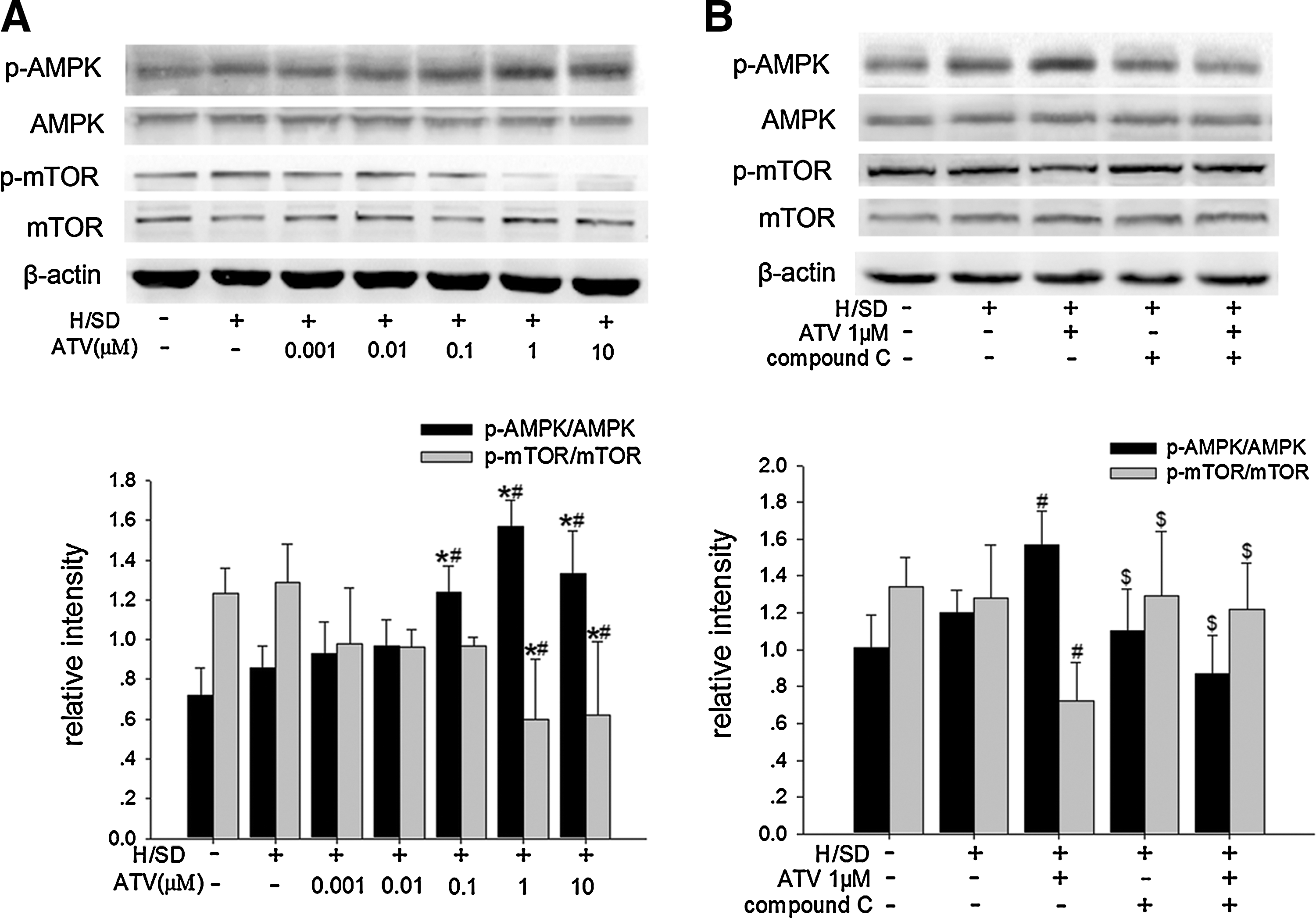
AMPK/mTOR pathway examined by western blotting in MSCs following ATV treatment under H/SD for 6 h.

Autophagy activation by ATV was dependent on AMPK/mTOR pathway in MSCs under H/SD for 6 h. AMPK inhibitor compound C counteracted the effects of ATV on LC3-II expression
Discussion
In the present study, the results showed that the autophagy reached its plateau within 6 h thereafter and the apoptosis peaked at 6 h in MSCs treated with H/SD, so we mainly adopted H/SD for 6 h as the time point to observe the relationship between autophagy and apoptosis. Here we showed that autophagy was induced in MSCs under H/SD and it served as a protective mechanism for MSC apoptosis under such a condition. ATV could enhance H/SD-induced autophagy via AMPK/mTOR pathway, which mediated, at least partly, the protective effects of ATV on MSC apoptosis under H/SD. To our knowledge, this is the first report showing the interaction of autophagy with apoptosis in MSCs under H/SD condition and autophagy activation as a protective mechanism of ATV in MSCs.
Autophagy is essential under both physiological and pathological conditions. The basal autophagy exerts “quality control” function, that is, eliminating old organelles or turning over long-lived proteins, to maintain cellular homeostasis [27,28]. Meanwhile, autophagy could be dramatically induced by multiple stresses used for cell survival or death, which depends on the context [7,8]. The relationship between autophagy and death has been characterized in many contexts and has been discussed comprehensively; however, it has not been extensively studied in MSCs. Hypoxia and serum deprivation are both energy-limiting stresses, and further they could impair the function of mitochondria and stimulate the production of radicals. Under such a condition, one well-established view is that adaptive autophagy is essential for cell survival by energy supply and damaged organelle or radical elimination [29 –31]. Accordingly, the protective role of autophagy has been previously reported in models of ischemic injury in heart and brain [9,10] and under conditions of H/SD in cell culture [32,33]. More recently, Sanchez et al. reported that autophagy could be utilized to provide needed energy and secret anti-apoptotic factors for survival in MSCs during long period of serum deprivation [30]. In agreement with these results, we found that autophagic pathway was activated effectively by H/SD as shown by increased autophagolysosomes (AVO-positive MSCs), autophagosomes (double-membraned structure), and LC3-II/LC3-I ratio (autophagic marker). Moreover, suppressing autophagy by its inhibitor 3-MA resulted in increased apoptotic levels in H/SD-treated MSCs; in contrast, rapamycin, an autophagy inducer, could decrease the degree of apoptosis. These data suggested that autophagy was a pro-survival mechanism rather than a pro-death pathway in MSCs under H/SD. Thus, modulating autophagy may be a new target to reduce apoptosis of MSCs under H/SD in vitro or, more importantly, to solve the “low survival” problem of transplanted MSCs in vivo.
In the cardiovascular field, much hope has been laid on stem cells to repair and regenerate the damaged myocardium after AMI. However, the postinfarct microenvironment is deleterious to implanted stem cells and may lead cell death, which impairs the therapeutic benefits of stem cell transplantation. ATV is one of the most prescribed statins in clinic with multiple biological activities independent of cholesterol-lowering action, and could promote MSC survival through various mechanisms both in vivo and in vitro [3,13]. Recent investigations, mainly conducted in cancer cells, demonstrated that the activation of autophagy by statins was cytoprotective and could delay apoptosis in response to various stresses, including serum deprivation [17 –20]. In the present study, conducted in MSCs, we showed that ATV increased AVO-positive MSCs, LC3-II/LC3-I ratio, and autophagosomes, and autophagy inhibitor 3-MA abrogated these changes observed in the 1 μM ATV group, suggesting that autophagy does indeed be induced by ATV. Further, ATV-mediated anti-apoptosis effects on MSCs under H/SD were significantly diminished by 3-MA, confirming our hypothesis that autophagy activation may serve as one important mechanism of ATV to promote MSC survival during H/SD.
Of note, the level of beclin1, a well-known autophagy-related protein, was not related with the autophagic activity observed in our study. However, previous studies have shown that when autophagy is induced by serum deprivation or other stresses, the expression of beclin1 is augmented significantly [10,30,32,34]. This discrepancy may be due to the different cell types, stimuli, and duration of treatment applied in the studies. A study conducted by Carloni et al. showed that, even exposed to the same hypoxia-ischemia stimulus for the same duration of treatment, the expression of beclin1 was only induced in neurons but not in other cells [10]. In addition, the time point when autophagic markers increased also differed widely from cell to cell [10,35,36]. Thus, although the increased expression of beclin1 was observed in MSCs exposed to serum deprivation for 28 days, the duration of 6 h in our study may be too short to cause obvious elevation of beclin1 expression.
To date, many signaling pathways have been suggested in autophagy regulation and AMPK is thought to be an important positive regulator of autophagy via its indirect inhibition to mTOR under serum deprivation and other starvation stresses [21]; meanwhile, we found that rapamycin treatment did not further enhance the autophagic activity of MSCs under H/SD compared with ATV alone, indicating that ATV may utilize the same way as rapamycin does, that is, through mTOR inhibition, to promote autophagy in MSCs under H/SD. Therefore, we monitored the expression and activation levels of AMPK/mTOR pathway to further explicit the mechanisms underlying ATV-induced autophagy. Interestingly, we found that 1 μM ATV treatment increased the AMPK activation and correspondingly reduced the mTOR activation significantly. Moreover, AMPK inhibition not only counteracted the mTOR suppression, but also eliminated the autophagy induction by 1 μM ATV, suggesting that AMPK/mTOR pathway serves as a potential mechanism of ATV to promote autophagy in MSCs under H/SD. There may be many other mediators responsible for the autophagy promotion of ATV. For example, mevalonate metabolic pathway and nuclear factor-kappa B have been reported to participate in statin-induced autophagy [18 –20]. In addition, PI3K/Akt, ERK1/2 and p53 signaling pathways, which are reported to mediate the protection of statins against apoptosis in many published studies, are found to be able to signal autophagy as well [21,37,38]. Thus, further investigation is needed to identify other signaling pathways by which ATV utilizes autophagy to defend MSC apoptosis in response to H/SD.
As we know, autophagy could use for both cell survival and death. Previous studies suggested that prolonged autophagy may digest essential components for cell survival and leads cell death [7,12]. Thus, we also examined the role of autophagy in MSC apoptosis followed by longer H/SD exposure (48 h). Intriguingly, we found that during longer exposure to H/SD, the population of necrotic or apoptotic MSCs in terminal stages was enhanced by autophagy inhibition but reduced by autophagy activation, although the early apoptotic rate of MSCs did not differ among groups, suggesting that, just as Carloni et al. reported in their study [10], autophagy may slow down the progression from early apoptosis to necrotic or apoptotic cell death. Moreover, ATV promoted the autophagy activation and the inhibiting autophagy eliminated the protective effects of ATV on necrotic or apoptotic death in MSCs, confirming that autophagy activation was still a potential mechanism of ATV to promote MSC survival even under a longer H/SD exposure. However, ATV did not activate AMPK effectively in MSCs treated with H/SD for 48 h (data not shown), indicating that there may be some other molecules, other than AMPK, accounting for the autophagy activation by ATV under such a condition. Further studies will be needed.
We should acknowledge that there are some limitations of this study. First, we did not employ Baf-A1, another autophagy inhibitor to block the fusion of autophagosomes with lysosomes, so we could not exclude the possibility that the increased LC3-II/LC3-I and autophagosomes were due to the autophagic flux impairment induced by ATV. In addition, autophagy and apoptosis could interact with each other. But in the present study, we had not detected the role of apoptosis in autophagy, and further studies are required in the future.
In conclusion, for the first time, we found that autophagy plays a protective role in H/SD-induced apoptosis of MSCs. ATV could effectively promote autophagy via AMPK/mTOR pathway, which may be a novel mechanism of ATV to enhance MSC survival during H/SD. Regulating autophagic activity may provide an attractive strategy to defense MSC death, promoting their applications for cellular therapies in regenerative medicine.
Footnotes
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (81070169 and 81170129 to Y.J. Yang), China Health & Medical Development Foundation (2008-zhfj2 and 2011-H25 to Y.J. Yang), and China Postdoctoral Science Foundation 20100470010 (to H. Wang).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.