
Abstract
Derivation of pluripotent stem cells (iPSCs) induced from somatic cell types and the subsequent genetic modifications of disease-specific or patient-specific iPSCs are crucial steps in their applications for disease modeling as well as future cell and gene therapies. Conventional procedures of these processes require co-culture with primary mouse embryonic fibroblasts (MEFs) to support self-renewal and clonal growth of human iPSCs as well as embryonic stem cells (ESCs). However, the variability of MEF quality affects the efficiencies of all these steps. Furthermore, animal sourced feeders may hinder the clinical applications of human stem cells. In order to overcome these hurdles, we established immortalized human feeder cell lines by stably expressing human telomerase reverse transcriptase, Wnt3a, and drug resistance genes in adult mesenchymal stem cells. Here, we show that these immortalized human feeders support efficient derivation of virus-free, integration-free human iPSCs and long-term expansion of human iPSCs and ESCs. Moreover, the drug-resistance feature of these feeders also supports nonviral gene transfer and expression at a high efficiency, mediated by piggyBac DNA transposition. Importantly, these human feeders exhibit superior ability over MEFs in supporting homologous recombination-mediated gene targeting in human iPSCs, allowing us to efficiently target a transgene into the AAVS1 safe harbor locus in recently derived integration-free iPSCs. Our results have great implications in disease modeling and translational applications of human iPSCs, as these engineered human cell lines provide a more efficient tool for genetic modifications and a safer alternative for supporting self-renewal of human iPSCs and ESCs.
Introduction
H
The second challenge in utilizing iPSCs for disease modeling and for treating genetic diseases is the relative inefficiency of genetic modifications in these cells. It is essential to genetically modify disease-specific iPSCs in order to understand molecular pathogenesis using iPSC-based disease modeling. Applications of iPSCs in cell replacement therapy may also require corrections of genetic lesions before transplantation. There have been significant advances in genetic engineering in hESCs and iPSCs, including lentivirus- or retrovirus-mediated gene transfer, DNA transposon-mediated gene transfer, and, more recently, homologous recombination (HR)-mediated gene targeting. The current efficiencies of these technologies are still being improved and often require special culture systems that support drug selection in order to enrich the cell populations that have undergone the desired genetic engineering. An efficient selection system that is supportive of the clonal expansion of iPSCs is especially crucial in HR-mediated gene targeting, because HR at a specific site is not an efficient process even with the recent development of zinc finger nuclease (ZFN) technology [8 –15]. There have been several successful reports of HR-mediated gene targeting in hESCs and iPSCs, but all of the previously reported experiments have been done with the use of MEFs that carry drug-resistance genes. This underlies the need to develop systems that can sufficiently support human iPSC clonal growth and have the potential to be compatible with clinical production under current Good Manufacturing Practice (cGMP) standards. We have previously demonstrated that human adult marrow stromal cells [also called mesenchymal stem cells (MSCs)] could efficiently support undifferentiated growth of hESCs [16] and that self-renewal of hESCs can be promoted by modulating the Wnt signaling pathway [17,18]. It has also been reported that MSCs facilitated the derivation of hESCs from cryopreserved poor-quality embryos [19]. Here, we report our successful approach to genetically engineer the human adult MSCs to develop an immortalized human feeder system, and investigate its utility in supporting derivation of safer human iPSCs and in promoting genetic modifications in human iPSCs.
Materials and Methods
Immortalization and engineering of human MSC
Bone marrow-derived MSCs were established from CD34-depleted mononuclear cells (MNCs) (AllCells) as previously described [16], from adult donors (such as a male donor #BM2426). Primary MSCs and immortalized MSCs were cultured in Dulbecco's modified Eagle's medium (DMEM) (low glucose) supplemented with 10% fetal bovine serum and 1 ng/mL basic fibroblast growth factor (bFGF). For immortalization, hMSCs (p2) were plated at a density of 105 cells per 10 cm2 in cell culture dishes, and cultured for 2 days before they were transduced by a retroviral vector co-expressing the human telomerase reverse transcriptase (hTERT) and the hygromycin resistance (HygroR ) gene. Two days later, the cells were then selected with hygromycin B (50 μg/mL) for 2 weeks during proliferation. Three passages later, the resulting hMSC-TERT cells were transduced by a retroviral vector co-expressing Wnt3a and the neomycin resistance (NeoR ) gene. The transduced cells were selected by both hygromycin B and G418 (a synthetic form of neomycin, 500 μg/mL) in every other passage until continuously proliferating TW2R cells were obtained (the parental MSCs without any drug selection ceased active proliferation by this time). The TW3R cell line was created by transducing TW2R with a lentiviral vector expressing puromycin resistance (PuroR ) gene. The retroviral and lentiviral vectors were previously described [18].
Undifferentiated culture of hESCs and iPSCs
The human H9 ESC line was obtained from WiCell Research Institute at passage 23. Human ESC and iPSCs were maintained on MEFs with a human ESC culture medium that consisted of KNOCKOUT DMEM (Invitrogen) supplemented with 20% KNOCKOUT Serum Replacement (KSR) (Invitrogen), 2 mM L-glutamine (Invitrogen), 2 mM nonessential amino acids (Invitrogen), 1× antibiotic/antimycotic mix (Invitrogen), 0.1 mM β-mercaptoethanol (Sigma), and 10 ng/mL bFGF (Peprotech). Unless otherwise noted, hESCs and iPSCs were passaged via Collagenase IV (Sigma) digestion. MEF (p3), hMSC (p4 to p7), hMSC-TERT (p7), W3R (>p30) TW2R (>p30), and TW3R (>p30) were used as feeder cells after irradiation at 50 Gy using a gamma-irradiator. To determine the clonal efficiency of hESCs and iPSCs on feeders, H9 ESCs and BC1 iPSCs were cultured on Matrigel for 2 passages before being dissociated by Accutase (Invitrogen). The dissociated cells were then plated onto various feeder plates at a density of 10,000 (H9) or 20,000 (BC1) cells/well in 24-well plates coated with feeders. After 5 days of culture using standard human ESC culture medium, the undifferentiated colonies were counted after alkaline phosphatase staining as previously described [17].
To determine the long-term effects of feeders on pluripotency, the human H9 ESC line and the BC1 iPSC line labeled with green fluorescent protein (GFP) reporter were cultured on human feeders or MEFs in standard human ESC medium. The undifferentiated state of cells was analyzed by flow cytometry after staining with TRA-1-60 antibody (Millipore) and a secondary Alexa 647 conjugated antibody (Invitrogen). The percentage of undifferentiated cells was determined as # of TRA-1-60 positive cells divided by # of GFP positive cells.
Generation and characterization of iPSCs derived on immortalized MSCs
Cord blood MNCs were expanded in an erythroblast expansion condition for 8 days, then reprogrammed by the pEB-C5 plasmid (10 μg for 2×106 cells) as previously described [20]. Briefly, frozen MNCs from a cord blood donor GB/CB were thawed and cultured in the serum-free medium supplemented with the following: SCF (50 ng/mL), IL-3 (10 ng/mL), EPO (2 U/mL; R&D Systems), IGF-1 (40 ng/mL), and dexamethasone (1 μM; Sigma). Eight days later, expanded MNC were nucleofected by 10 μg pEB-C5 plasmid via the CD34 nucleofection kit and the T-016 program (Lonza Walkersville). The transfected MNCs were cultured in the expansion medium for 2 days before being plated onto CF-1 MEF or TW3R feeder cells with standard human ESC medium in the presence of 0.25 mM sodium butyrate [21]. Fourteen days postnucleofection, colonies stained positive for TRA-1-60 were counted, subsequently picked, and expanded on TW3R feeder cells with standard human ESC culture conditions. After 6 passages, pluripotency markers analysis, embryoid body (EB) differentiation, and teratoma formation were performed as previously described [22 –25]. Detection of vector DNA integration by polymerase chain reaction (PCR) was also performed as previously described [20].
Stable transfection of human iPSCs by piggyBac transposition
BC1 iPSCs (passage 58) were dissociated with accutase, and 2×106 cells were nucleofected with the piggyBac-GFP transposon and an improved piggyBac transposase plasmid [26] at 1:1 ratio (5 μg each) using mouse ES nucleofection buffer and program A-023 (Lonza Walkersville). The nucleofected cells were plated immediately onto 4 wells of TW3R feeders in a 6-well plate with 10 μM Rho-associated kinase (ROCK) inhibitor Y27632 (Stemgent). Four days after plating, puromycin (1 μg/mL) was added to the medium for 1 week to select stable transfected cells. The selected cells were then expanded and maintained without puromycin on TW3R. At day 11 and day 29 post transfection, the cells were harvested by Accutase for fluorescence-activated cell sorting analysis of TRA-1-60 staining and GFP expression.
Gene targeting into the AAVS1 locus in human iPSCs
BC1 iPSCs (passage 67) were dissociated into a single-cell suspension by accutase digestion. After washing with phosphate-buffered saline, 6×106 cells were resuspended in 100 μL mouse ES nucleofection solution (Lonza Walkersville) with 20 μg of the AAVS1-CAGG-GFP vector and the 2 AAVS1 ZFN mRNAs (6 μg; Sigma). The cells were nucleofected with Nucleofector II device using program A-023. After nucleofection, the cells were plated immediately onto puromycin-resistant TW3R or DR4 MEF feeders (GlobalStem) with 10 μM ROCK inhibitor Y27632 (Stemgent). Puromycin (1 μg/mL) selection started 4 days after nucleofection and continued for 7 days. Individual iPSC clones were picked and then expanded and maintained on TW3R or CF1 MEF feeder without puromycin.
Genomic DNA was isolated from the expanded iPSC clones at passage 8 for PCR screening and southern analysis. To screen for potential clones with HR, PCR was carried out using the following primers (as shown in the diagram in Fig. 6A): (Forward) 5′-CTGCCGTCTCTCTCCTGAGT-3′; (Reverse) 5′-GTGGGCTTGTACTCGGTCAT-3′. The iPSC clones that generated a ∼1 kb band in the PCR reaction were used for southern blot to confirm HR-mediated gene targeting. For southern blot analysis, 10 μg of genomic DNA from each cell line was digested with Sph I (shown in Fig. 6A) and separated on 0.8% agarose gel before transfer. The southern blot using a 705-bp 5′ probe (shown in Fig. 6A) was carried out as previously described [11]. As shown in Fig. 6A, hybridization to the wild-type (WT) genomic DNA would generate a 6.5 kb band in the blot, and a targeted insert (TI) at the AAVS1 locus would generate a 3.8 kb band.
Results
Engineered human MSC cell lines support long-term self-renewal of human pluripotent stem cells
We have previously reported that human MSCs derived from adult bone marrow support human ESC self-renewal [16]. However, the proliferation rate of primary MSCs and their ability to support undifferentiated growth of hESCs diminish significantly after 5–6 passages. Subsequently, cultured MSCs started to display a senescent phenotype featuring increased cell size, decreased nucleus/plasma ratio, and irregular cell morphology. To generate a renewable or ideally immortalized human cell line that can better support human pluripotent stem cell expansion, we sequentially transduced adult bone marrow-derived MSCs with retroviral vectors expressing hTERT (co-expressing the HygroR
gene) and Wnt3a (co-expressing the NeoR gene) (Fig. 1A). After drug selections by hygromycin B and G418, a stable cell line that expresses hTERT, Wnt3a, and 2 drug-resistance genes (thus named TW2R) was established. After an extended period of cell proliferation for more than 140 days (Fig. 1A), it still maintained a fibroblast-like morphology homogeneously, similar to that of primary MSCs at early passages. The TW2R cell line had been maintained for more than 40 passages over 200 days and had a constant doubling time at about 9 h (Fig. 1B). The TW3R cell line was derived by introducing a lentiviral vector expressing only the PuroR gene into TW2R cells, in order to enhance the cell line's ability to support additional applications that require puromycin drug selection (Fig. 1A). Both immortalized cell lines can be cultured over a long term while maintaining a morphology and growth rate similar to primary and early passage (p1-p4) MSCs (Fig. 1A,B). They also maintain a normal karyotype (Supplementary Fig. S1; Supplementary Data are available online at

Establishment of genetically engineered human cell lines from primary MSCs.
To examine the ability of the engineered human cell lines to support self-renewal of human pluripotent stem cells, first we plated a single-cell suspension of H9 hESCs (p58-p65) at the same density on various feeders. After a 5-day culture, we assessed undifferentiated ESC clones that stained positive with alkaline phosphatase (AP). Results showed that there were comparable numbers of ESC colonies on MEFs to those on Wnt3a-producing human cell lines TW2R, TW3R, and W3R, a previously established human feeder cell line from adult dermal fibroblasts (Fig. 2A). The high passage MSCs (p5 or higher; data not shown) and MSCs transduced with only hTERT did not support the clonal growth of hESCs under the condition (Fig. 2A). We also used BC1, a human iPSC line which is free of reprogramming vector sequences [20], demonstrating that all 3 Wnt3a-expressing human cell lines (TW2R, TW3R, and W3R) support human iPSC clonal growth better than MEFs (Fig. 2B). Three consecutive passages of GFP-labeled BC1 iPSCs on TW3R feeders yielded a higher percentage of undifferentiated iPSCs than on W3R or MEF feeders by flow cytometric analysis after TRA-1-60 staining (Fig. 2C). The TW3R feeder cells performed the best for both human iPSCs and ESCs, and, therefore, we focused on TW3R for long-term cultures and other applications.
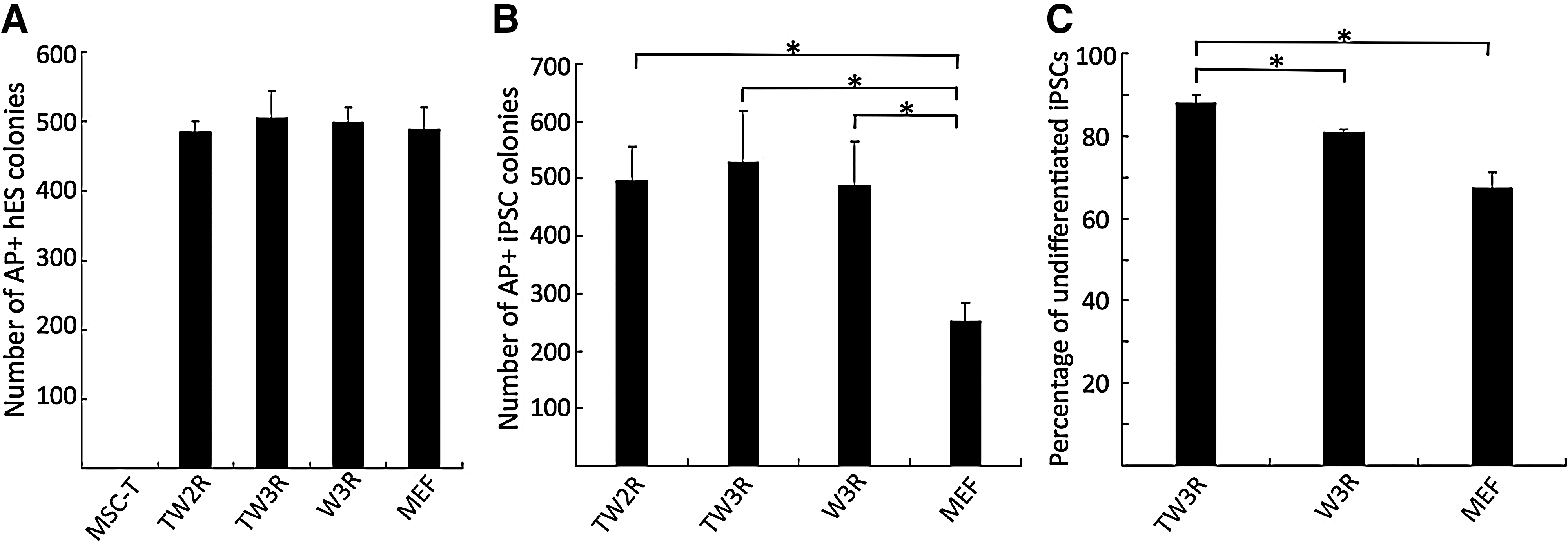
Engineered human feeders support clonal expansion of human ESCs and iPSCs.
To assess the ability of TW3R human feeders in supporting long-term self-renewal of human pluripotent stem cells, GFP-labeled H9 ESCs were cultured on TW3R or MEFs for 12 consecutive passages before their pluripotency was analyzed (Fig. 3). Flow cytometric analysis showed that hESCs at different passages on TW3R maintained high percentages of undifferentiated cells comparable to those on MEFs (Fig. 3A). After 12 passages on TW3R feeders, the hESCs displayed a typical undifferentiated morphology with expression of pluripotency markers TRA-1-60, SSEA4, OCT4, and NANOG (Fig. 3B). These iPSCs also formed EBs with cell types expressing markers of all 3 embryonic germ layers (Fig. 3C). To more definitively assess their pluripotency, the H9 ESCs after 12 passages on TW3R were injected into immuno-deficient mice for teratoma formation. Analysis of the teratoma showed tissue types of the 3 germ layers, demonstrating their pluripotency (Fig. 3D). Importantly, a normal karyotype was maintained in the hESCs over the long-term culture on TW3R feeders (Fig. 3E).

TW3R human feeders support long-term culture of undifferentiated human ESCs. H9 hESCs were cultured on TW3R or MEF for 12 consecutive passages.
TW3R human feeders support efficient derivation of integration-free human iPSCs from un-fractionated blood cells by an episomal vector
The majority of the reported generation of human iPSCs have been conducted on animal sourced feeders or matrix with a few exceptions where fibroblast reprogramming was supported by autologous fibroblasts [6,7]. One important question we sought to answer is whether the renewable TW3R feeders were capable of supporting the derivation of human iPSCs, as this would potentially provide a universal system for generating safer human iPSCs from various types of tissues. Therefore, we investigated whether the TW3R feeder cells could be used to support the generation of iPSCs from blood cells. Un-fractionated cord blood MNCs were first expanded using a previously established protocol before being transfected by a single EBNA1/OriP based episomal vector (pEB-C5) expressing 5 reprogramming factors Oct4, Sox2, Klf4, c-Myc, and Lin28 [20]. The transfected cells were cultured for 2 days before being transferred to either MEFs or TW3R feeders under the hESC culture condition (Fig. 4A). Starting from day 6–8 post-transfection, iPSC-like colonies emerged on both types of feeders; the majority of the sizable colonies on TW3R emerged 1 day earlier than those on MEFs. Reprogrammed cells committed as candidate iPSC clones were identified by live staining with the TRA-1-60 antibody recognizing a surface-antigen on pluripotent cells [28,29]. Colony counting on day 14 showed that the reprogramming efficiency on TW3R was ∼1,100 colonies from 2×106 transfected MNCs, comparable to or slightly lower than that observed on MEF feeders (Fig. 4B). Since there were abundant TRA-1-60+ candidate iPSC colonies formed on TW3R feeders, it is not critical whether TW3R human feeder cells are indeed weaker than MEFs under the current condition. To characterize these TRA-1-60+ candidate iPSC colonies formed on TW3R feeders, we picked individual colonies and expanded them on the same TW3R feeders for establishing iPSC lines. Complete characterization of a representative iPSC line, CN2, after 6 passages of expansion is shown (Fig. 4C–G). It displayed an undifferentiated morphology and expression of typical pluripotency markers TRA-1-60, SSEA4, OCT4, and NANOG (Fig. 4C). The pluripotency of CN2 was demonstrated by in vitro EB differentiation (Fig. 4D) and in vivo teratoma formation after an injection into immuno-deficient mice (Fig. 4E). This cell line also maintained a normal karyotype (Fig. 4F). Furthermore, PCR was performed as previously described on 2 iPS clones, CN2 and CNP5, to detect possible vector integration in their genome [20]. Our results showed that no vector DNA was detected in either iPSC line after 8 passages (Fig. 4G). These data demonstrated that integration-free human iPSCs can be also generated efficiently on TW3R feeders by episomal-vector mediated reprogramming.

Efficient derivation of integration-free human iPSCs on TW3R human feeders.
TW3R human feeders support stable nonviral gene transfer and expression in human iPSCs delivered by a piggyBac transposon vector
In addition to commonly used retroviral- or lentiviral-mediated gene delivery, nonviral methods such as piggyBac DNA transposon/transposase system have been recently improved and been reported to stably deliver genetic elements into human hematopoietic stem/progenitor cells and hESCs [30 –32]. The advantages of the piggyBac transposon-based vector system over viral systems include the ability to deliver larger DNA fragments and the potential for removing mutagenic insertions or other types of footprints in the host genome [26,31,32]. Efficient delivery of the transposon vectors to human ESC and iPSCs requires complete dissociation of hESC or iPSC colonies before transfection; however, the dissociated single cells after treatment typically grow poorly when plated at a low density even with a ROCK inhibitor. Therefore, it is important to improve survival and clonal expansion of undifferentiated pluripotent stem cells after plasmid delivery in order to achieve efficient and stable transfection.
To test whether the TW3R human feeder system can effectively support the selection and expansion of human iPSCs stably transfected with a piggyBac transposon, we used a piggyBac vector PB-CAG-GFP that co-expresses the GFP gene (under the CAG promoter) and PuroR-HSV-Δtk dual selection gene [33] (under the PGK promoter) to transfect BC1 human iPSCs (Fig. 5A). After co-transfection with a second vector expressing an improved transposase gene [21,26], the BC1 iPSCs were plated onto TW3R feeder cells for recovery followed by puromycin selection. Although GFP+ iPSC colonies were also observed without puromycin selection within 7 days after transfection, a majority of the colonies remained GFP− under this condition. In contrast, all colonies in the puromycin-selected condition showed GFP expression (Fig. 5B). The puromycin-selected cells were further expanded and analyzed by flow cytometry after staining with TRA-1-60 antibody to eliminate feeder cells from the analysis. Our results of the day 11 culture showed that nearly 90% of the TRA-1-60+ cells were also GFP+ after puromycin selection, increased from 16.3% in the condition without puromycin (Fig. 5C). Importantly, the transgene expression can be maintained over a long-term culture on TW3R feeders, which enables puromycin selection, as flow cytometric analysis of the day-29 culture showed that 95.6% of iPSCs (TRA-1-60+) maintained GFP expression (Fig. 5C). In the absence of continuous puromycin selection, <5% of iPSCs expressed GFP due to spontaneous gene silencing (Fig. 5C). Moreover, the expanded BC1-piggyBac-GFP cell line maintained typical undifferentiated morphology and expression of pluripotency markers such as TRA-1-60, SSEA-4, OCT4, and NANOG, in addition to the GFP transgene (Fig. 5D).
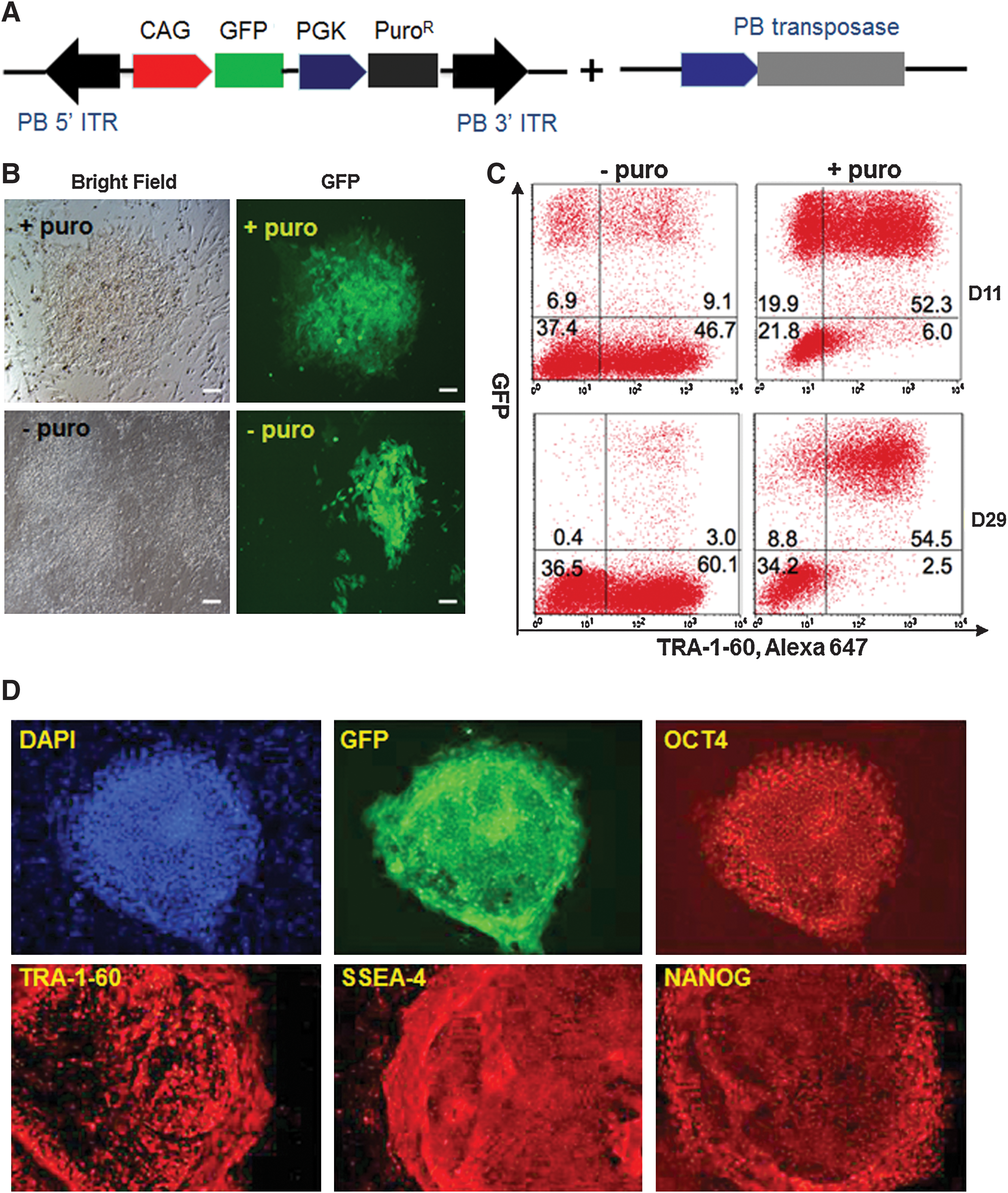
TW3R human feeders support efficient and stable gene transfer to human iPSCs by a piggyBac vector.
TW3R support enhanced gene targeting through HR in human iPSCs
Viral and nonviral mediated gene transfer, whether for transgene overexpression or shRNA-induced gene knockdown, has been invaluable to biomedical research. More precisely, controlled gene targeting or editing through HR would eliminate some of the clinical concerns associated with these established technologies, such as un-controlled transgene dosages and potential insertional mutagenesis. The possibility of precisely repairing disease-causing mutations in patient-specific iPSCs also created great hopes for personalized cell replacement therapy. Studies based on gene targeting in mouse ESCs have revolutionized our understanding of gene functions [34]. With our ability to derive iPSCs from patient somatic cells, it was anticipated that gene targeting technology in human iPSCs would have a great impact on understanding molecular pathogenesis of human diseases as well. Applying the same HR-mediated gene targeting technology to human stem cells, however, has been met with numerous technical difficulties. One of the major rate-limiting factors of this technology is the poor clonal growth of hESCs and iPSCs as compared with that of mouse ESCs. As a result, the overall efficiencies of reported HR in hESCs and iPSCs have been extremely low [8,9,11,12,14], and drug-mediated selection for rare targeted clones is critical. A recently developed ZFN technology has been shown to vastly enhance gene-targeting efficiencies in hESCs and iPSCs, although the absolute efficiencies are still far from satisfactory [9,10,14]. Therefore, efficient gene targeting in human iPSCs would require optimal culture conditions that support clonal expansion of targeted iPSCs, in addition to the improvement of targeting vectors and ZFNs. We hypothesized that the TW3R human feeder cells may improve gene targeting in human iPSCs by providing a consistent and reliable feeder that is resistant to multiple drug treatments and is supportive of the clonal growth of human iPSCs as well as ESCs.
To test this hypothesis, we conducted experiments in BC1 iPSCs to target the AAVS1 site within the first intron of the PPP1R12C gene located on chromosome 19. The AAVS1 locus has recently gained significant interest for gene therapy purposes, as it provides a safe harbor for stable integration and expression of transgenes. It has been shown to have an open chromatin structure with insulator elements that favors stable transgene expression [10,11,35,36]. In addition, disruption of 1 copy of the PPP1R12C gene has no reported adverse effect on cellular function; therefore, it is considered a safe harbor for transgene delivery [37]. We recently showed gene targeting to the AAVS1 locus in X-CGD patient-derived iPSCs that are cultured on a special type of MEF feeders (DR4), illustrating the potential of this approach in gene therapy [11]. This approach of site-specific targeting of the gp91phox therapeutic minigene into the AAVS1 locus enabled sustained expression of the transgene and restored neutrophile ROS production in re-differentiated iPSCs [11]. To target this strategy with TW3R feeder cells, we co-nucleofected BC1 iPSCs with (1) two mRNAs that encode a pair of ZFNs specifically recognizing sequence within the AAVS1 locus [10,11]; and (2) a targeting construct with 2 homologous arms flanking the ZFN recognition sequence (within the intron 1) as we and others have previouslydone [10,11]. The donor construct also carries a GFP gene under the CAG promoter, and the PuroR gene that would be expressed by the endogenous promoter if HR occurs (Fig. 6A). After nucleofection, iPSCs were plated either onto DR4 mouse feeders or onto TW3R feeders, for recovery and subsequent drug selection. After 7 days of puromycin selection, GFP-expressing iPSC colonies were observed in both culture conditions (Fig. 6B). However, the GFP+ colonies were significantly more abundant on TW3R than on DR4 MEFs (Fig. 6C). To confirm that HR-mediated gene targeting indeed occurred in the iPSCs, we first screened the puromycin-resistant iPSC clones from TW3R co-cultures with PCR primers designed to specifically amplify the genomic region after a correct HR event (Fig. 6A). Six out of 24 picked clones showed the correct size (1 kb) of the predicted PCR product (Fig. 6D), and were selected for further confirmation by Southern analysis. The Southern blot analysis utilized a 5’ probe (shown in Fig. 6A) to hybridize genomic DNAs after Sph I restriction digestion, and showed that an HR event at this AAVS1 site (targeted insertion) had occurred in at least 1 allele in all 6 examined clones, as compared with the parental BC1 iPSCs (Fig. 6E). Clones with both alleles targeted were identified, such as clones 5 and 17, while clones 10 and 16 showed one WT allele and a targeted allele without other alleles. On further culture of the clones with a single targeted allele at the AAVS1 safe harbor site, clones such as #16 displayed typical undifferentiated morphology, showed expression of markers characteristic of undifferentiated hESCs (Fig. 6F), and formed teratomas that consist of tissue types of all 3 embryonic germ layers (Fig. 6G).

TW3R human feeders support gene targeting to the AAVS1 locus in human iPSCs.
Discussion
In this study, we established a human feeder system with the aim of overcoming several major hurdles associated with the commonly used MEF feeders in translational applications of human iPSCs. Although culture systems with complete elimination of feeders have been utilized in laboratories, these feeder-free culture systems either still contained animal components or are currently inefficient to support clonal expansion of undifferentiated pluripotent stem cells. Efficient genetic modifications in human iPSCs are crucial for stem cell-based disease modeling, and the ability to achieve high efficiencies without animal sourced feeders is essential for translating stem cell research into therapies. As an alternative to the current feeder-free culture conditions, several types of human cells have been reported to be supportive of hESC growth [16,19,38 –45]. Human feeders have the potential to be cGMP-compliant and have been used to derive multiple clinical-grade human ESC lines [46]. Based on these considerations, we attempted to immortalize and engineer human adult marrow MSCs, a cell type that at early passages exhibits multi-lineage differentiation potential [27] and has been previously shown to be supportive to hESC growth [16,19,44,45]. As a result, we generated a renewable cell source that can support the derivation, expansion, and genetic modifications of human iPSCs.
It has been previously reported that the expression of hTERT in human somatic cells can result in immortalization without inducing transformation. We, therefore, chose hTERT as the initial reagent to immortalize primary human MSCs to preserve their fibroblastic phenotype and ability to survive (despite cycle arrest) after DNA cross-linking treatment, 2 characteristics that are important for functioning as effective feeder cells. In contrast to our previous study with human skin fibroblast immortalization [17], various batches of hTERT-transduced MSCs not only gained the ability to proliferate over a long term but also acquired a transformed-like morphology and sensitivity to irradiation-induced cell death, or behaved as primary MSCs but did not acquire the ability for extended growth (Fig. 2A). Addition of the Wnt3a transgene to the hTERT-expressing MSCs significantly enhanced the immortalization process and the resultant feeder lines TW2R and TW3R; both retained a fibroblastic MSC-like morphology, demonstrated the ability to support long-term undifferentiated expansion of hESCs and iPSCs (Figs. 2, 3). Recent data suggest that the Wnt canonical pathway activated by Wnt3a and other ligands suppresses MSC differentiation to osteogenic or adipogenic lineages. We have previously demonstrated that modulating Wnt canonical signaling promoted human ESC self-renewal on human feeders that also provide other soluble secreted proteins to support the long-term growth of human ESCs [18]. However, the mechanism of Wnt3a promoting immortalization of MSCs is currently not fully understood. Similarly, it is important to determine the influence on differentiation by the Wnt3a-producing TW3R feeder cells, even though they provide a growth advantage for undifferentiated human iPSC and ESCs. However, the influence on the differentiation propensity of co-cultured human iPSC and ESCs by TW3R feeder cells is likely to be minor or nondeterministic, because the standard in vitro and in vivo pluripotency assays did not detect overt bias toward 1 of the 3 germ layers. Previous reports have demonstrated the feasibility and advantages of maintaining hESCs in media conditioned by hMSCs [44], and it has been recently shown that maintenance of hESCs in MSC-conditioned medium augments hematopoietic specification, while direct co-culture of hESCs on MSC feeders did not predispose hESCs toward hematopoietic specification [47]. If this phenomenon were also true for TW3R feeder cells, it may provide a potential advantage of using TW3R for the objective to differentiate co-cultured hESCs and iPSCs more efficiently to the hematopoietic lineage. The influence of TW3R human feeders on differentiation of hESCs and iPSCs to other lineages, especially those that are difficult to achieve, needs to be determined for various differentiation projects with different objectives.
To demonstrate the ability of TW3R human feeders to support derivation of human iPSCs, unfractionated cord blood cells were used as source for somatic cell type. Advantages of using neonatal cord blood cells for generating iPSCs include (1) cord blood may contain a fewer number of acquired somatic mutations compared with other adult cell types from the same individuals; (2) a large number of cord blood samples have already been preserved in cord blood banks. Cord blood samples cryopreserved for more than 20 years have been successfully reprogrammed to iPSCs [48]. In addition, we have previously shown that an episomal vector-based system efficiently reprogrammed unfractionated cord blood cells without the need for enrichment of progenitor cells [20]. The combination of cord blood cells and the virus-free episomal vectors are more likely to produce safer and more clinically relevant iPSCs for future regenerative medicine. Moreover, autologous fibroblastic feeders that have been used in some other reported MEF-free reprogramming studies cannot be generated from cord blood cells. Our results showed that comparable reprogramming efficiencies to what achieved using MEFs were observed with TW3R as supporting feeders (Fig. 4). These episomal vector-generated iPSCs were also free of exogenous vector sequence as determined by a sensitive PCR assay, suggesting that the engineered human feeder cell lines are capable of generating safer iPSCs that have great potentials in therapy.
One of the major advantages of using immortalized TW3R human feeder cells that were engineered to carry 3 drug-resistance genes against hygromycin, neomycin, and puromycin selections is for gene targeting and other forms of genetic modifications in human iPSCs and ESCs.
All the successful human stem cell gene-targeting studies reported so far have used mouse feeders, the most established culture condition, for selection of targeted clones. The need for MEFs, however, would significantly hinder the translation of this technology to clinical applications. Even for basic research purposes, the variability of different preparations of MEFs, especially for specialized DR4 MEFs that are derived from a different strain of transgenic mice, has frequently resulted in inconsistent experimental results. In order to make gene-targeting technology more robust and practically applicable to clinical applications, we developed the TW3R feeder system to eliminate the need for mouse feeders. Our results showed that targeting of the AAVS1 “safe harbor” locus in human iPSCs under this condition is fairly efficient, as we have obtained multiple clones of iPSCs that have targeted insertion either in a single allele or in both endogenous alleles after double hits (Fig. 6E). The observed higher gene targeting efficiency in TW3Rs was likely due to the consistency in TW3R feeder quality and the higher capability for supporting clonal expansion of iPSCs.
In summary, we have demonstrated that the engineered human feeder cell lines can serve as renewable sources for supporting efficient generation and long-term maintenance of virus-free integration-free iPSCs. Combined with their ability to support genetic modifications in human iPSCs, they provide a foundation for developing cGMP-compliant conditions for human iPSC-based gene and cell therapies.
Footnotes
Acknowledgments
The authors thank Dr. Jizhong Zou for his expert advice and assistance of gene targeting in human iPSCs used in this article. Research conducted in the Johns Hopkins University was supported in part by NIH grants (R01 HL073781 and RC2 HL101582 to L.C.; T32 HL007525 to Z.Y.) and Maryland Stem Cell Research Fund (2011-MSCRFE-0087). C.Z. and Y.A.Z. were supported in part by the National Basic Research Program of China (Project Code: 2007CB947704) and High Level Talent Fund of the Beijing Healthcare System (Project Code: 2009-2–14).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.