
Abstract
Epigenetic changes are regarded as emerging major players for hematopoietic stem cell (HSC) biology. Although some histone deacetylase (HDAC) inhibitors, such as valproic acid (VA), induce differentiation and apoptosis in a variety of leukemic cells in vitro, they produce a favorable effect on the expansion of normal HSCs. In this study, we have identified the VA target HDAC3 as a negative regulator of umbilical cord blood HSC expansion. We demonstrate that knockdown of the transcript dramatically improves CD34+ cell expansion, which correlates with a higher potential to generate colony-forming units in functional assays. We show that this effect is mediated at the level of primitive hematopoietic cells and that it is not due to negative effects on specific cell commitment or alterations in the cell cycle. HDAC3 inhibition does not block commitment to the monocytic lineage and the maturation of monocyte precursors, which are the main inhibited pathways in the presence of VA. Therefore, our results identify HDAC3 as a promising target for therapies aiming to expand HSCs.
Introduction
T
In vivo, HSCs initiate hematopoiesis through an organized hierarchy in which the differentiation potential becomes more restricted as the precursors evolve into different hematopoietic lineages. Human HSCs can be found within a highly heterogeneous population of CD34+ Lin− cells and, in fact, HSCs capable of long-term hematopoietic activity in vitro and long-term engraftment in humans have been identified within the CD34+CD90+ population [4 –6]. However, the massive in vitro proliferation showed by HSCs is coupled with a significant loss of stem cell properties mainly due to differentiation into more committed precursors. Thus, to successfully produce competent HSCs ex vivo, it is essential to provide conditions that inhibit at the same time cell death, differentiation, and loss of in vivo repopulation ability, and to induce self-renewal divisions. Several culture conditions and cytokine combinations have already been tested in an attempt to expand HSC cultures while avoiding differentiation. Notably, it has been observed that valproic acid (VA) maintains increased CD34 expression during in vitro HSC culture [7,8]. VA is a short-chain fatty acid used in humans for decades as an anticonvulsant and as a mood stabilizer [9]. Although it is now clear that VA may act through more than one target [10], the efficiency as a strong inhibitor of histone deacetylase (HDAC) activity is well documented. In addition to selectively inhibiting the catalytic activity of class I HDACs (HDAC1, 2, 3, and 8), VA induces proteasomal degradation of HDAC2 [11,12]. In fact, molecules with HDAC inhibitory activity are known to be potent inducers of growth arrest and differentiation potential of tumor cells.
Reversible acetylation of histones and sequence-specific transcription factors represents a posttranslational modification with significant impact on many biological processes. Histone acetylation and deacetylation promote the activation and repression of the transcriptional machinery, respectively [13,14]. Moreover, deacetylation of specific transcription factors, for example, p53, Sp1 or E2F, reduces their DNA binding capacity and subsequent transcriptional activity [15 –17]. Two opposing classes of enzymes, histone acetyltransferases and the aforementioned HDAC, regulate this process and are now emerging as critical regulators of cell proliferation, differentiation, and cell death. Mechanisms that control the activity of these enzymes represent a class of anticancer strategies that hold particular potential. Surprisingly, while the clinical use of VA for the treatment of both solid and hematological tumors is increasing, the specific effect on the molecular basis of hematopoiesis and on the self-renewal maintenance of normal HSCs [7,8], and more precisely, the contribution of each HDAC family member in HSC differentiation, remain unclear. In this study, we demonstrate that the inhibition of HDAC3 is sufficient to induce a quantitative expansion of cord blood CD34+ HSCs independently of cell proliferation and differentiation.
Materials and Methods
Isolation and culture of human HSCs from umbilical cord
Human cord blood was obtained from Inbiobank Stem Cell Bank (
CD34+ cells were cultured at a density of 1.2×104 cells/cm2 in 0.5 mL/cm2 of Stemline II culture medium (Sigma) supplemented with 50 ng/mL stem cell factor, 20 ng/mL interleukin-3, 10 ng/mL thrombopoietin, 10 ng/mL Flt-3/Flk-2 ligand (R&D), and 20 ng/mL interleukin-6 (Sigma). VA (Sigma) was added when needed at a final concentration of 1 mM.
Colony formation assays were performed using Human Methylcellulose Enriched Media (R&D), following manufacturer's instructions. Briefly, methylcellulose-based media were thawed and mixed with the cytokines (as detailed previously) and cells, and the mixture was plated in 35-mm cultured dishes. Each dish contained 1.1 mL of mixture and 500 cells. Colonies were counted and classified according to morphology on day 15.
Giemsa staining
Cytospin slides of sorted CD14+ and CD15+ cells (254 g, 5 min; Shandon Cytospin 4) were fixed in methanol and air-dried before incubating them in May-Grünwald solution for 1 min. Slides were then washed for 1 min with deionized water, incubated in 1:2 diluted Giemsa solution for 5 min, rinsed in distilled water, and dried before analysis.
shRNA cloning and lentiviral transduction
Specific shRNAs were cloned in the lentiviral plasmid pLVTHM (Addgene plasmid 12247,
Lentiviral particles were produced as previously described [19]. Briefly, HEK293T cells were seeded in high-glucose Dulbecco's modified Eagle medium containing 10% fetal bovine serum (FBS). pRRE, pREV, pVSV-G, and the lentiviral vector pLVTHM or pLVTHM-ΔCD7 were transfected in the packaging cell line. Titration of lentiviral supernatants was performed in fresh HEK293T by our Viral Vector Unit. To test the efficiency of the shRNA sequences, HEK293T cells were transduced with lentiviral vectors containing the shRNA sequences and the expression of the target genes was measured by quantitative polymerase chain reaction (qPCR). Lentiviral transduction of HSCs was performed by incubating cells for 24 h with lentiviral particles at a multiplicity of infection of 50 in HSC culture medium.
Quantitative PCR
Total RNA was extracted using Trizol reagent (Invitrogen) followed by RNA purification using RNAqueous Micro Kit (Ambion). cDNA was obtained using the GeneAmp reverse transcriptase kit, following manufacturer's instructions. Quantitative PCRs were performed on these cDNAs using the following oligonucleotide pairs: HDAC1: forward GCAAGCAGATGCAGAGATTCAAC, reverse AGCCCAATTCACAGCGATGT; HDAC2: forward CTCAAAGGTGATGGAGATGTATCAA, reverse CAACCCAGTCTATCACCAGATAATGA; HDAC3: forward GTGGCCGCTACTACTGTCTGAA, reverse GGTGCTTGTAACTCTGGTCATCAA; HDAC8: forward GGAGATGGTGTAGAAGACGCATT, reverse CGTCACCTGTTCCTGGGAAA; and CD34: forward TTTGCTTGCTGAGTTTGCTG, reverse ATTTGAAAATGTTCCCTGGGT.
Flow cytometry and cell division tracking
For cell surface marker expression analysis, cells were suspended in phosphate-buffered saline (PBS)/bovine serum albumin (BSA) 1%–FBS 1%–sodium azide 0.1%, supplemented with normal mouse serum (10%) and incubated for 10 min. Appropriate conjugated antibodies (BD Biosciences) were added and cells were incubated for 10 min at room temperature and washed in PBS/BSA 1%. Carboxyfluorescein acetate succinimidyl ester (CFSE) staining was done following manufacturer's protocol (CellTrace CFSE Cell Proliferation Kit; Invitrogen). Briefly, cells were suspended at a concentration of 106 cells in PBS/0.1% BSA (Invitrogen), CFSE was added at a final concentration of 2 μM, and cells were incubated at 37°C for 10 min. Reaction was stopped adding cold medium and cells were washed in medium 3 times. Cell surface marker expression and CFSE fluorescence were analyzed in an FACSCanto cytometer (BD Biosciences). Analysis of flow cytometry data, including CFSE tracking assays, was performed using FlowJo software (Treestar).
Statistical analysis
When indicated, mean values were compared using Student's t-test for independent samples. P-values below 0.05 were considered statistically significant.
Results
VA inhibits differentiation of CD34+ cord blood cells toward the monocyte lineage
Within freshly isolated cord blood CD34+ cells, 2 populations can be distinguished by virtue of CD45RA expression (Fig. 1A, left panel). The CD34+CD45RA+ subpopulation has been described as more mature than the CD34+CD45RA− cluster [5,21 –23], but it does not yet express any of the lineage markers (CD14, CD15, CD19, CD56, or CD235a). To determine which lineages can be generated from CD45RA+ cells in our culture conditions, cord blood cells were sorted for CD34 and CD45RA phenotype (Fig. 1A, right panel), cultured for 5 or 7 days, and analyzed by flow cytometry using various late-lineage markers. As shown in Fig. 1B, sorted CD34+CD45RA+ cells were able to generate mainly monocyte precursors showing typical morphology and CD14 marker expression. CD14+ cells were detected only at a later stage within the most immature CD34+CD45RA− population cultures, after the acquisition of CD45RA expression (Fig. 1B, upper panels). To further investigate the colony-forming potential and lineage commitment of these freshly isolated CD34+ progenitors, we utilized a semisolid culture system. CD34+CD45RA− cells formed all types of myeloid colonies at a high plating efficiency (41%), while CD34+CD45RA+ cells showed less efficiency in colony formation (16%) and a differentiation potential restricted to monocyte/granulocyte colonies (granulocyte-macrophage colony-stimulating factor [GM-CSF], monocytes colony-stimulating factor [M-CSF], and granulocytes colony-stimulating factor [G-CSF]) (Fig. 1C). These results agree with the observation that CD34+CD45RA+ cells represent a more mature subpopulation of CD34+ cells and suggest that they constitute an intermediate step between HSCs and more mature lineages.

Sorting of human cord blood progenitor cells based on CD45RA expression and differentiation capacity in culture.
It has been previously reported that VA stimulates expansion of HSCs and a concomitant decrease of CD14+ monocyte/macrophage lineages [7,8]. As expected, VA treatment was accompanied by the maintenance of CD34 expression in CD34+CD45RA− cells cultured for 7 days. Remarkably, VA seriously impaired the differentiation of CD34+CD45RA− cells into CD34+CD45RA+ population (Fig. 2A). This inhibition was concomitant to an increase in the percentages of the most primitive CD34+CD90+ population (Fig. 2A). CD90 expression could not be observed in the CD34+CD45RA+ cells, reinforcing the idea that this population represents a more mature population (Fig. 2B). As expected, emergence of CD14+ cells from sorted CD34+CD45RA+ cells was totally abolished (Fig. 2B). Additionally, differentiation into other lineages, such as CD235a+ erythrocytes from the most immature CD34+CD45RA− population or from CD34+CD45RA+ progenitors, was not affected by the presence of VA (data not shown). Thus, this data suggests that VA acts independently at 3 levels: maintenance of the most immature population CD34+CD45RA−, inhibition of early generation of CD34+CD45RA+ precursors, and the subsequent differentiation toward CD14+ mature cells.
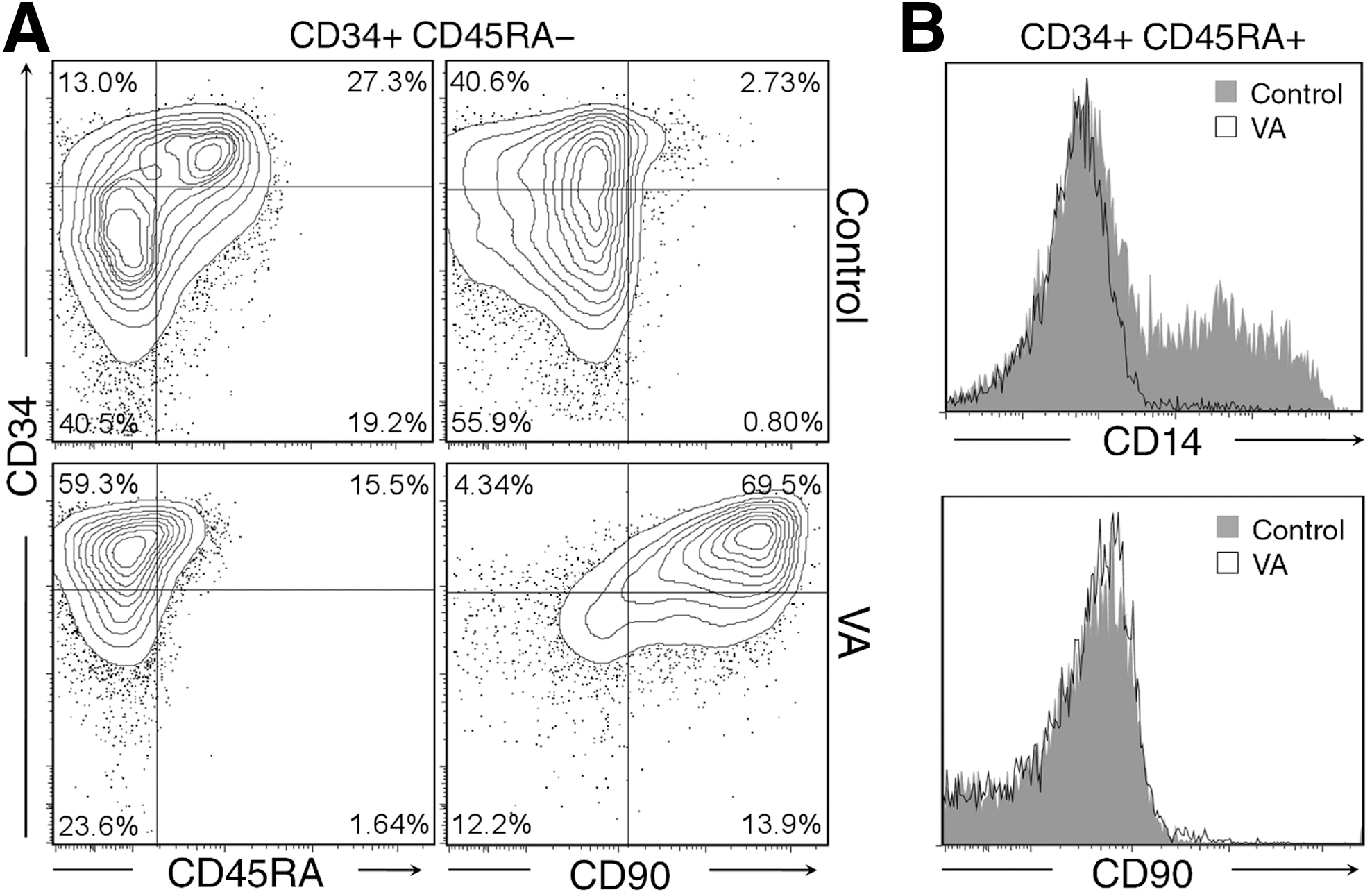
Valproic acid inhibits differentiation of CD34+CD45RA− cord blood cells toward the monocyte lineage.
VA favors expansion of CD34+CD45RA− population
Next, we focused on the effect displayed by VA on the expansion of CD34+ cells from each sorted population (CD34+CD45RA+ and CD34+CD45RA−). Overall, CD34+CD45RA− cells showed a higher proliferation status regarding the more mature CD34+CD45RA+ population. VA treatment slightly inhibited the growth of CD34+CD45RA− population, although this effect was more robust in the CD34+CD45RA+ cell culture (Fig. 3A). However, a more detailed analysis showed that the expansion of CD34-expressing cells from sorted CD34+CD45RA− population was significantly increased in the presence of VA (35-fold expansion in the presence of VA versus 16-fold expansion in the control culture), which was practically negligible in the CD34+CD45RA+ population (Fig. 3B). More importantly, the observed CD34+CD45RA− expansion was highly enriched in the early stem cell fraction of CD34+CD45RA− CD90+ population (Fig. 3C). We further examined the effect of VA treatment on CD34 expression throughout cell division history using CFSE staining. CD34+CD45RA− cells divided in a highly synchronous manner. As shown in Fig. 3D, CD34+CD45RA− cells undergo 3 to 9 divisions, regardless of the presence of VA. Nevertheless, VA was able to maintain a subpopulation with high level of CD34 expression after 5 days of culture (Fig. 3C, 68% vs. 38% observed in untreated cells). The frequency of cells in each generation in the presence of VA indicated that over 80% of CD34+CD45RA− population that had undergone 3 to 7 divisions still maintained high level of CD34 expression, which drastically went down in the untreated population (Fig. 3E). Thus, the lower rate of proliferation induced by VA seems to correlate with the immaturity of the cells generated in culture.

Valproic acid favors self-renewal of CD34+CD45RA− population.
To further track the effect of VA on the emergence of a more mature population, CD34+CD45RA− as well as CD34+CD45RA+ cells were stained with CFSE. As shown in Fig. 4A, in untreated cells, commitment from the CD34+CD45RA− to the CD34+CD45RA+ population was mainly observed in the early round of cell division (rounds 4 to 7), whereas generation of CD14+ cells was only seen when CD34+CD45+ precursors had already undergone 6 to 9 cell divisions (Fig. 4B). More importantly, VA inhibited the differentiation from CD34+CD45RA− to CD34+CD45RA+ cells (Fig. 4A) and from CD34+CD45RA+ to CD14+ mature cells independently of the number of cell divisions (Fig. 4B). In addition, VA treatment was not accompanied by an increase in cell death (data not shown). Taken together, these data demonstrate that VA inhibition of the generation of CD34+CD45RA+ cells from the more immature CD34+CD45RA− cell population and the subsequent generation of its CD14+ progeny are independent of the slowing down of proliferation.

VA treatment results in division-independent lineage inhibition.
HDAC3 knockdown promotes expansion of CD34+ cells without affecting monocyte lineage commitment
Next, we wanted to understand the mechanisms underlying the effect of VA on hematopoietic progenitor cells. To identify the specific HDAC class I family member (HDAC1, 2, 3, and 8) responsible for the observed effects of HDAC inhibitors in HSCs, we performed a gene silencing approach using a GFP-expressing lentiviral vector (pLVTHM) to transduce cells with specific short hairpin RNAs. At first, we checked the RNAi silencing efficiency using RT-qPCR in HEK293-transduced cells as they present high expression levels of HDAC1, 2, 3, and 8. Specific shRNAs reduced HDAC RNA expression levels to <20% of control in all of them (Supplementary Fig. S1; Supplementary Data are available online at

Histone deacetylase 3 (HDAC3) silencing promotes expansion of CD34+ CD90+ cells.
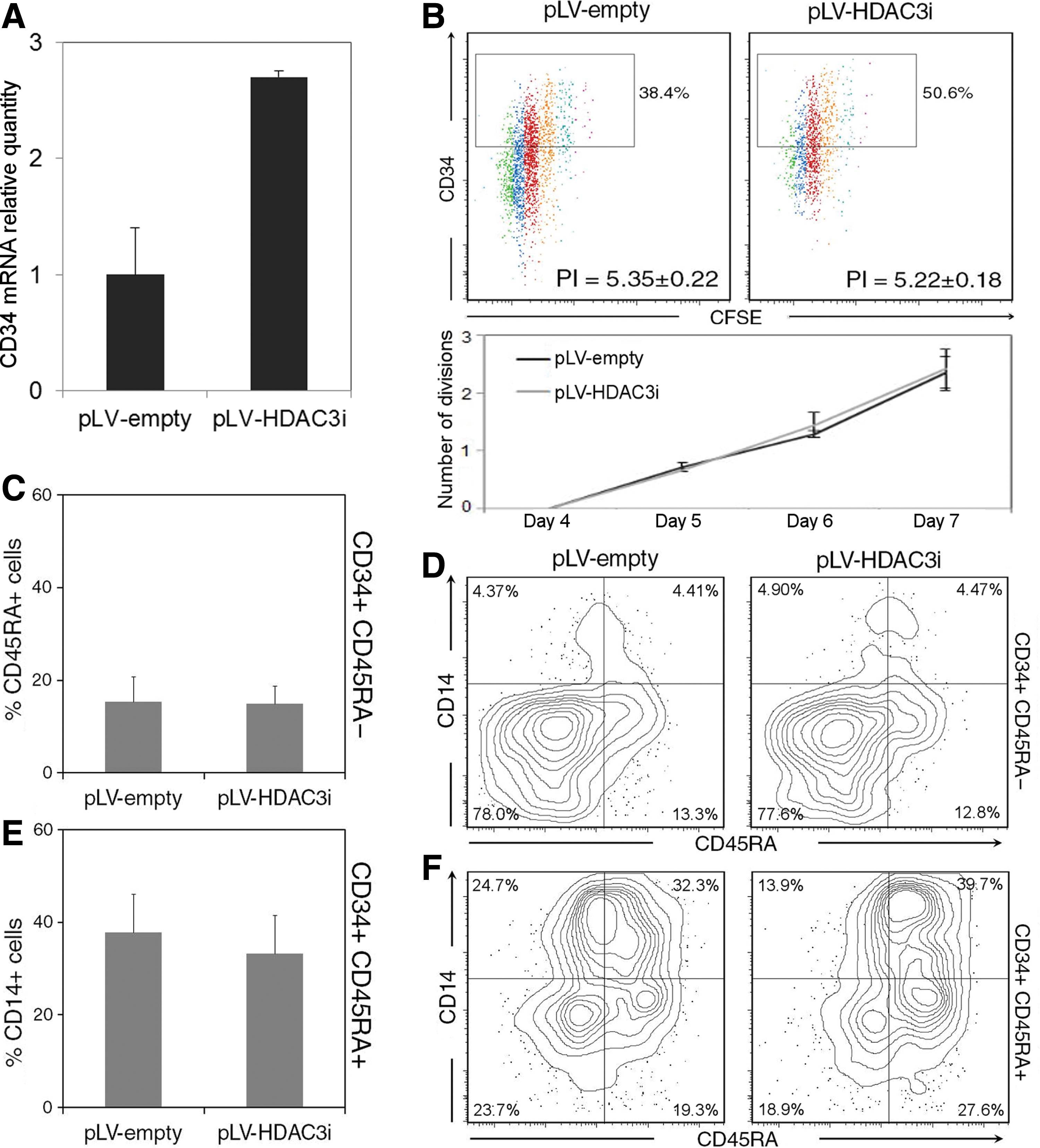
HDAC3 silencing does not affect proliferation nor lineage development.
Next, we wanted to evaluate the effects of HDAC3 inhibition on lineage commitment in liquid culture. To assess the influence of HDAC3 inhibition in differentiation, we analyzed the ability of shRNA-transduced CD34+CD45RA− and CD34+CD45RA+ subpopulations to generate CD34+CD45RA+ progeny and CD14+ mature cells, respectively (Fig. 6C–F). First of all, sorted and transduced CD34+CD45RA− cells were cultured for 7 days and analyzed by flow cytometry. Differentiation of CD34+CD45RA− cells toward CD45RA+ or CD14+ lineage is not impaired upon inhibition of HDAC3 (Fig. 6C, D). Similarly, differentiation of CD34+CD45RA+ cultures to CD14+ cells is not affected in the absence of HDAC3 activity (Fig. 6E, F). To functionally assess the effect of HDAC3 knockdown, specific colony-forming assays in methylcellulose were performed with transduced cells. As shown in Fig. 7A, knockdown of HDAC3 not only maintains the differentiation potential to all lineages but also increases the colony-forming potential of CD34+ cultures compared with the empty vector. The test showed that the higher number of colonies in HDAC3-transduced cells was mainly obtained from the granulocyte–macrophage, granulocyte, and macrophage colony-forming units (Fig. 7B). Additionally, all the generated colonies were morphologically normal (Fig. 7C).

HDAC3i-transduced cells retain a higher number of progenitors and the multilineage developmental potential. Colony-forming unit (CFU) assay in methylcellulose media. Seven-day liquid-cultured cells were sorted upon GFP expression and assayed for CFU.
VA treatment and HDAC3 inhibition effects are additive. When HDAC3-knockdown cells were treated with VA, the expansion of the CD34+ population increased even further, reaching 60% of the cells at day 7 of culture (Supplementary Fig. S3). This result suggests that downregulation of HDAC3 is indeed essential to expand HSCs, and that this effect may be dose dependent. To rule out any possible CD34− to CD34+ reprogramming effect caused by the inhibition of HDAC3, emerging CD34− cells from shRNA-treated cultures were sorted out at day 7 and further cultured for 3 days with or without VA. Flow cytometry analysis showed that CD34+ cells could not be found in these cultures, indicating that CD34+ subpopulation comes entirely from the direct expansion of initial CD34+ cells (Supplementary Fig. S4).
Discussion
HSCs are multipotent cells that have the capacity for self-renewal and for differentiation into the major cell types of the hematopoietic system, for example, erythrocytes, lymphocytes, and granulocytes. The molecular mechanisms regulating gene transcription that results in HSC differentiation and cell lineage specification are slowly being unraveled. This knowledge will facilitate the development of therapies to amplify HSCs in vitro or in vivo for clinical purposes. In this study, we have characterized the contribution of HDACs, which are involved in the proliferation and differentiation of many cell types by modifying gene expression and chromatin structures, to human HSC differentiation and self-renewal. Indeed, several studies have shown that HDAC inhibitors (VA, trichostatin A, or sodium butyrate) stimulate the expansion of HSCs in vitro. The current study is the first to identify HDAC3 as the specific HDAC involved in this process. We have shown that silencing of HDAC3 mirrors the positive effect of VA in expansion of human CD34+ HSCs.
Although the precise biological role of CD34 is still unresolved and little is known about the effect of cultivation on the relationship between phenotype and function in human CD34 in vitro, CD34 is the most important marker of early hematopoiesis in experimental and clinical studies. CD34 expression defines most immature progenitors as well as repopulating-capable stem cells in freshly isolated samples [24]. Current CD34+ cell culture techniques result in an increase in the total number of cells and progenitors cells, but the frequencies of cells positive for this marker decrease to very low levels after a few days of culture. It has been shown as well that CD34 expression decreases progressively with proliferation, which correlates with the loss of self-renewal property. On the contrary, the maintenance of surface CD34 expression in vitro has been associated with the appearance of hematopoietic functions, such as cloning capacity, the ability of sustaining long-term culture [25], and perhaps more interestingly, long-term reconstitution ability in animal model (SCID-Hu) [26,27]. In this regard, VA treatment has shown to significantly increase the CD34+ cell expansion in vitro and to enhance surface CXCR4 expression, thereby promoting chemotactic responses [7,8,28]. In fact, it has been previously reported that VA-treated cells reconstitute hematopoiesis in immunodeficient mice with higher efficiency than control cells [29], which correlates with an improvement in the replating efficiency of HSCs in vivo, as well as the long-term potential of HSCs [7]. In the present work, we have shown that the inhibition of HDAC3 expression, but not of any other examined class I HDACs family members (HDAC1, 2, and 8), is sufficient to upregulate the transcript and to maintain high-level protein expression of CD34 marker even after several rounds of cell division. Additionally, inhibition of HDAC3 expression and VA treatment allow the expansion of a significant CD90+ population within the sorted CD34+CD45RA− subset culture, which is completely absent in cultures from sorted CD34+CD45RA+ cells. Most primitive HSCs have been identified within CD34+ CD90+CD38− population. Unfortunately, serum-free condition gives rise to CD38 downregulation from the cell membrane of committed progenitors in culture [30,31]. All new synthetic media do not include serum in their composition (e.g., Stemline II used in this study). Thus, CD38 is not a reliable stemness marker in cultured cells. The expression of CD34 and CD90 after in vitro culture is considered the most reliable predictor for nonobese diabetic/severe combined immunodeficiency (SCID) repopulation potential [26]. To our knowledge, this is the first report of upmodulation of both antigen markers by specific HDAC3 inhibition in normal hematopoietic early progenitors; these data also indicate that the HDAC3-induced gene set may be dependent on the maturation stage of the HSCs.
As stable shRNA expression after lentiviral transgene integration into the whole-cell genome is established first after 2 to 3 days, we cannot rule out whether the upregulation mediated by the absence of HDAC3 is due to an increase in the transcription of CD34 and CD90, or alternatively, to an amplification of a cell population expressing these markers. However, both transcripts have been found to be upregulated in VA-treated CD34+ cells in <24 h with no sign of proliferation (data not shown), suggesting that HDAC3 activity may work in a similar fashion. Another open question is which pathway modulates transcription and expression of CD34 and CD90 in HSCs. Previous studies have reported that VA-treatment induces the upregulation of HOXB4, a key transcription factor in the regulation of the self-renewal of HSCs [8]. However, we could not observe a similar effect after HDAC3 knockdown (data not shown), indicating that the expansion signaling pathways through HDAC3 may be promoted by other triggers.
We have further investigated the hierarchical relationships between the CD45RA± subpopulations of CD34+ cord blood and observed that CD34+CD45RA− population gives rise to the CD34+CD45RA+ subpopulation in vitro, which in turn is a candidate for monocyte progenitors in our culture condition. In agreement with Majeti et al. the CD34+CD45RA− population is capable of expansion and gives rise to the CD34+CD45RA+ subpopulation [5]. Additionally, we have found that CD34+CD45RA+ cells represent a precursor of a more mature subpopulation of monocytes as evident by morphology, phenotype, and colony-forming potential. The major difference between both reports is that our population of CD34+CD45RA+ cells is more heterogeneous and probably contains more committed cells, which allowed us to identify its immediate progeny. The fact that CD34 is highly expressed in knockdown cells and that HDAC3 inhibition does not seem to affect monocyte lineage commitment or maturation of monocyte precursors (neither CD34+CD45RA+ cell generation nor CD14+ monocyte maturation, respectively) suggests that the HDAC3 controls the HSC expansion without affecting the differentiation potential. Knockdown of HDAC1, 2, nor 8 in CD34+ cultures is able to mimic the effect of VA in monocyte differentiation (Supplementary Fig. S5), suggesting that a combination of multiple HDAC activities may be responsible for the differentiation process. However, functional assays based on the ability of progenitor cells to differentiate in response to the cytokine cocktail demonstrate that inhibition of HDAC3 expression correlates with a higher potential to generate colony-forming units. The quality of the expanded CD34+ cells is not impaired in HDAC3i culture, as demonstrated by their ability to fully form all lineages. The GM/G/M-CFU increase could be due to that HDAC3 inhibition provides a specific restriction of these differentiation pathways, which in turn gives rise to the expansion of CD34+ cells. In this scenario, the development of culture conditions that restrain the differentiation to other hematopoietic lineages will be essential for HSC expansion in vitro. Alternatively, the virus-mediated constitutive expression of shRNA HDAC3 may cause a bias toward specific lineages in methylcellulose cultures. Conditional viral-vector-based strategies are needed to clarify this issue [32].
The absence of HDAC3 does not affect cell cycle entry as all the CD34+CD45RA− cells that are present in the expansion culture have undergone multiple rounds of cell division as shown by CFSE membrane labeling (proliferation index) and no subpopulation of cells refractory to cycle induction was evident. However, HDAC3 knockdown gives rise to a higher transient accumulation of CD34bright cells in each generation with a similar proliferation index as control cells. Thus, HDAC3 silencing is able to maintain a CD34+ population despite a similar proliferation rate comparing to control cells. In contrast, treatment of CD34+CD45RA+ with VA revealed a significant reduction in fold expansion after culture, with no adverse effects on cell viability. Thus, VA treatment decreases the proliferative capacity of CD34+CD45RA+ progenitors and leads to a defect in monocyte development.
The CD34+ cell expansion by VA treatment is in contrast to the effect observed on growth arrest, differentiation, and apoptotic cell death of leukemic precursors. Inhibition of an alternative class I HDAC by VA, other than HDAC3, might explain this contradictory result. Additionally, VA-containing cultures have shown the greatest fold expansion of CD34+ cells compared with cultures containing other chromatin-modifying agents (e.g., SAHA, trichostatin A, etc.) [33,34]. A higher specificity for one or another HDAC by these agents based on their structures and functionalities may be responsible for the observed effect. In conclusion, this study represents the first evidence for a relevant role of HDAC3 in human HSCs. Our findings identify HDAC3 activity as a negative regulator of in vitro HSC expansion. We show that simultaneous VA treatment and HDAC3 silencing further enhances maintenance of the CD34+ population. This suggests that HDAC3 downregulation is crucial in the HSC expansion process, and that this occurs in a dose-dependent manner. The fact that the specific inhibition of HDAC3 holds expansion-promoting ability, together with the fact that silencing has no adverse effects on cell differentiation, makes it a promising target for HSC expansion and might represent a new tool for therapeutic use. It will be interesting to determine in future studies which elements are, in fact, involved in regulating a subset of HDAC3 target genes that encode proteins involved in HSC self-renewal. Our findings add new insight into the epigenetic regulation of normal hematopoiesis and ultimately contribute to develop a stem cell expansion protocol for human HSCs using developmental fate determinants.
Footnotes
Acknowledgments
The authors would like to thank Pilar Olaizola of Hospital Donostia for obtaining CB samples; we thank Cell Cytometry and Microscopy Unit for cell sorting analysis. This work was supported by the “Obra Social KUTXA,” the Spanish Ministry of Innovation and Science, the Basque Country Government and the Diputación Foral de Guipúzcoa. C. Elizalde was supported by a contract from ADEGI “Asociación de Empresarios Guipuzcoanos.”
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.