
Abstract
Although many techniques can be used to generate multitype-induced pluripotent stem (iPS) cells from multitype seed cells, improving the efficiency and shortening the period of cell reprogramming remain troublesome issues. In this study, to generate iPS cells, CD34+ cells, isolated from human amniotic fluid cells (HuAFCs) by flow cytometry, were infected with retroviruses carrying only one reprogramming factor (Oct4) and cultured on human amniotic epithelial cell (HuAEC) feeder layers. Approximately 4 to 5 days after viral infection, some embryonic stem cell (ESC)-like colonies appeared among the feeder cells. These colonies were positive for alkaline phosphatase and expressed high levels of ESC pluripotent markers (Nanog, Sox2, Oct4, CD133, and Rex1). Moreover, these iPS cells exhibited high levels of telomerase activity and had normal karyotypes. Additionally, these cells could differentiate into cell types from all 3 germ layers in vivo and in teratomas. In summary, we report a novel way of iPS generation that uses CD34+ HuAFCs as seed cells. Using this method, we can generate human iPS cells with greater efficiency and safety (the oncogenic factors, c-Myc and Klf4, were not used), and using the minimum number of reprogramming factors (only one factor, Oct4). Besides, HuAECs were used as feeder layers to culture human iPS cells, which could not only avoid contamination with heterogeneous proteins, but also maintain iPS cells in a self-renewing and undifferentiated state for a long time.
Introduction
B
Since the use of iPS cells can overcome the ethical problems associated with the use of ESCs and reduce the possibility of immune rejection, if solutions become available for the technical limitations (such as genetic modification by viral infection, long-term safety, and the production of the right types of cells), then iPS cells may become an attractive source for cell therapy [20,21]. To this aim, the first integration-free iPS cells were generated from adult mouse hepatocytes using nonintegrating adenoviral vectors, and from mouse embryonic fibroblasts (MEFs) transfected with plasmids [11,12,22 –25]. Subsequently, Zhou et al. used recombinant proteins to successfully reprogram mouse and human fibroblasts into iPS cells without any viral or plasmid vectors [12,26,27]. Furthermore, iPS cells have also been generated by the integration of microRNAs (miRNAs) or suicide genes [17,28,29]. Although integration-free iPS cells have been successfully generated from plasmids, proteins, miRNAs, or suicide genes, reprogramming efficiencies (∼0.001%) with currently used nonintegrating methods are several orders of magnitude lower than with approaches involving viral integration [12,30]. To solve this problem, several small molecular drugs, such as histone methyltransferase inhibitors [31], an L-channel calcium agonist [31,32], Wnt inhibitors [33], zinc finger nucleases [14], rapamycin [34], lithium [35], and vitamin C [36], have been used to increase the efficiency of reprogramming during the generation of iPS cells [37]. In addition, Woltjen and Hoon et al. used either the Bmi1 protein, the piggyback transposon, or the Glis1 transcription factor but not Yamanaka factors to reprogram fibroblasts into iPS cells [24,38 –40]. To date, iPS cells, which share the key properties of unlimited self-renewal and pluripotency with ESCs, have been generated in different species by various methods [7,9,24,28,41 –45]. iPS cells can be generated from patient- or disease-specific sources, which makes them attractive for personalized medicine, drug screening, and tissue engineering [2,46 –54], as well as for gene and cellular therapy in a wide range of human diseases, including Parkinson's and other neurodegenerative diseases, cardiac and vascular disorders, and diabetes [28,55].
Kim et al. were the first to report that exogenous expression of just one Yamanaka factor, the germline-specific transcription factor Oct4 (official symbol Pou5f1, also known as Oct3/4), was sufficient to generate iPS cells from adult mouse neural stem cells [51,56]. In addition, Jin and coworkers. reported for the first time the rapid and efficient generation of iPS cells from human amniotic fluid-derived cells via the ectopic expression of Yamanaka factors; typical single iPS cell colonies could be picked 6 days after viral infection with high efficiency. Thus, the results indicated that the cell type could significantly influence reprogramming efficiency and kinetics [57]. Moreover, our previous research revealed that human amniotic fluid contains multiple cell types (CD44+, CD34+, and CD105+) derived from the developing fetus, and these cells can give rise to various cell types, including adipose, muscle, bone, and neuronal cells [58,59]. In view of previous results, in this study, we isolated the CD34+ subpopulation cells from the human amniotic fluid-derived cells (HuAFCs), and used these CD34+ HuAFCs as seed cells to generate iPS cell lines through retroviral delivery of only one Yamanaka factor (Oct4). Strikingly, 4 days after infection, typical human ESC-like colonies could be picked for expansion and further characterization. Furthermore, assays showed that the HuAFD-CD34-iPS cells expressed pluripotency markers and demonstrated differentiation potential both in vitro and in vivo. Therefore, the shorter time required for and the higher efficiency of induction of pluripotency make CD34+ HuAFCs an attractive cell type for producing human iPS cells.
Materials and Methods
Isolation of CD34 positive cells from human amniotic fluids
Human amniotic fluid was obtained by ultrasound-guided amniocentesis performed on pregnant women for routine prenatal diagnosis purposes at gestational ages ranging from 18th–22th weeks. All of the human samples were obtained after approval from the Ethical Review Board of the Shanghai First Maternity and Infant Hospital (Shanghai, China) and after obtaining written informed consent from subjects [57]. CD34+ subpopulation cells were isolated from that human amniotic fluid using 4 μL of the primary monoclonal antibodies (rabbit anti-human CD34-fluorescein isothiocyanate [FITC], eBioscience) stored at 4°C in phosphate-buffered saline (PBS) for 30 min in a volume of 1 mL as previously described [57,59]. After the reaction, the cells were washed twice in PBS, placed as the secondary monoclonal antibodies (Goat anti-mouse or Goat anti-rabbit coupled to magnetic microbeads, Miltenyi Biotec), incubated at 10°C in PBS for 15 min, and then washed twice in PBS. Single cells were plated at 1,000 cells/mL in Dulbecco's modified Eagle's medium (DMEM) (HyClone), supplemented with 10 ng/mL basic fibroblast growth factor (bFGF), 10 ng/mL epidermal growth factor (EGF) (all from Sigma-Aldrich), 10% fetal bovine serum, and 2 mM L-glutamine (all from HyClone). All CD34+ cells were cultured in a humidified incubator, at 37°C with 5% CO2, until they were 80% confluent.
Preparation of human amnion epithelial cells
Human placentas were obtained with written and informed consent from pregnant woman in the International Peace Maternity and Child Health Hospital, Shanghai Jiaotong University, and Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine. They were negative for HIV-I, hepatitis B, and hepatitis C. Appropriate use of human amnion was approved by the institutional ethics committee. Amnion membranes were mechanically peeled from chorines of placentas obtained from women with an uncomplicated Cesarean section. The epithelial layers with basement membrane attached were obtained and used to harvest human amniotic epithelial cell (HuAECs) as previously described with some modification [60,61]. Briefly, the membrane was placed in a 250-mL flask containing DMEM medium and cut with a razor to yield 0.5−1.0 cm2 segments. The segments were digested with 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) at 37°C for 45 min. The resulting cell suspensions were seeded in a 6-well plate in DMEM medium supplemented with 10% fetal bovine serum (PAA), 100 μg/mL streptomycin, 100 U/mL penicillin, and 0.3 mg/mL glutamine, and incubated in a humidified tissue culture incubator containing 5% CO2 at 37°C. The HuAECs grown to a density of 80%–90% were used as feeder layers for human iPS cells culture.
Generation of human iPS cells using Oct4 expression retrovirus
Reprogramming was conducted as described [3,29,57]. Briefly, 5×106 CD34+ HuAFCs were transduced with pMX retrovirus to overexpress Oct4 [The Oct4 retrovirus was kindly provided by Dr Tianjin Liu (Laboratory of Molecular Cell Biology, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences)]. One days later, transduced Oct4-CD34-HuAFCs were fed with ES medium and cultured on HuAECs [DMEM:F12(1:1) medium supplemented with 15% KnockOut™ Serum Replacement, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 0.1 mM beta-mercaptoethanol, Penicillin (25 U/mL)-streptomycin (925 mg/mL), 10 ng/mL, and bFGF and mixtured, but without leukemia inhibitory factor (LIF)], and the media were changed every other day.
Induction of haematopoietic differentiation
The method was conducted as described [41,46,52]. Human iPS cells were replaced with haematopoietic differentiation medium with cytokines (SCF, G-CSF, FLT3LG, IL-3, IL-6, and BMP-4; R & D Systems) and for erythroid/megakaryocytic differentiation, the medium was supplemented with haematopoietic cytokines plus 3 U/mL EPO and changed every 6 days. This was followed by collection for molecular and functional analysis.
Alkaline phosphatase staining
Alkaline phosphatase (AP) activity of human iPS cells, which were cultured on HuAECs, was determined using the AP substrate kit (Beyotime Institute of Biotechnology) according to the manufacturer's instructions [62].
Immunofluorescence staining
The cultured cells were washed thrice with fetal calf serum and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 30 min. After blocking, the cells were incubated first with rabbit anti-human Oct3/4 polyclonal antibody (1:200; Chemicon), rabbit anti-human Sox2 polyclonal antibody (1:200; Chemicon), rabbit anti-human Nanog polyclonal antibody (1:200; Chemicon), and rabbit anti-human Rex1 polyclonal antibody (1:200; Chemicon) overnight at 4°C, and then with Cy3-conjugated goat anti-rabbit immunoglobulin (IgG) antibody (1:200; Abcam) and 5 μg/mL 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) at room temperature for 30 min. Then, the cells were thoroughly washed with TBST (Tris buffered saline and 0.05% Tween 20) and viewed through a fluorescence microscope (DMI3000; Leica).
RNA extraction and analysis by quantitative real-time polymerase chain reaction
Total RNA from each cell was isolated using Trizol Reagent (Invitrogen) according to the manufacturer's protocol. The RNA samples were treated with Dnase I (Sigma-Aldrich), quantified, and reverse transcribed into cDNA using the ReverTra Ace-α First Strand cDNA Synthesis Kit (TOYOBO). Quantitative real-time polymerase chain reaction (qRT-PCR) was conducted using a RealPlex4 real-time PCR detection system from Eppendorf Co. LTD (Germany), with SyBR Green RealTime PCR Master MIX (TOYOBO) used as the detection dye. qRT-PCR amplification was performed over 40 cycles with denaturation at 95°C for 15 s and annealing at 58°C for 45 s. Target cDNA was quantified using the relative quantification method. A comparative threshold cycle (Ct) was used to determine gene expression relative to a control (calibrator), and steady-state mRNA levels are reported as an n-fold difference relative to the calibrator. For each sample, the marker genes Ct values were normalized using the formula ΔCt=Ct_genes–Ct_18sRNA. To determine relative expression levels, the following formula was used ΔΔCt=ΔCt_iPS–ΔCt_HuAFCs. The values used to plot the relative expressions of markers were calculated using the expression 2−ΔΔCt. The mRNA levels were calibrated based on the levels of 18s rRNA. The cDNA of each gene was amplified using primers as described [63].
qRT-PCR validation miRNA expression
In accordance with manufacturer's instructions and previous description [64], qRT-PCR was conducted in a RealPlex4 real-time PCR detection system from Eppendorf Co.LTD, using as SyBR Green RealTime PCR Master MIX (TOYOBO) as detection dye. The qRT-PCR amplification for 40 cycles was performed by denaturation as 95°C for 10 s and annealing at 57°C for 20 s. Target cDNA was quantified using the relative quantification method. A comparative threshold cycle (Ct) was used to determine gene expression relative to the control (calibrator). Thereby, steady-state mRNA levels were expressed as an n-fold difference relative to the calibrator. For each sample, the miRNAs Ct values were normalized using the formula ΔCt=Ct_miRNA–Ct_18srRNA. To determine relative expression levels, the following formula was used: ΔΔCt=ΔCt_iPS–ΔCt_HuAFCs and the values to plot relative miRNAs expression were calculated using the expression 2−ΔΔCt. The miRNAs levels were calibrated by 18S rRNA. The primers of miRNAs cDNA were amplified as described [64].
Bisulfite conversion of genomic DNA and methylation-specific PCR
Each of the cells was lysed in DNA lysis buffer (0.5% sodium dodecyl sulfate, 0.1M EDTA, 10 mM Tris-HCl pH8.0, and 100 ng/mL Proteinase K, all from Sigma-Aldrich) and incubated at 55°C for 2 h. The treatment of genomic DNA and methylation-specific PCR assay was done as previously described [3,6,10,60]. The specific primers for Nanog and Oct-4 were designed as previously described [3,6,10,60]. The PCR products were separated by 12g/L ethidium bromide containing agarose gel electrophoresis with 1×Tris Acetate EDTA (TAE) buffer and visualized under UV illumination.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) experiments were carried out by using anti-acetylated histone H3 antibody (Upstate Biotechnology), anti-trimethylated H3-K4 antibody (Abcam), anti-trimethylated H3-K27 antibody (Abcam), and normal rabbit IgG (Upstate Biotechnology) as a negative control. Briefly, all steps were conducted as previously described [3,6,10,60]. The cells were fixed by 1% formaldehyde for 30 min at 37°C and then quenched by 125 mM glycine for 10 min at room temperature to form DNA-protein cross-links. Samples were sonicated on ice until chromatin fragments became 200–1,000bp in size and incubated with antibodies at 4°C overnight. The PCR amplification was performed under the following conditions: 33 cycles run by denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s.
Teratoma formation
All animal procedures were carried out at the Shanghai Tongji University with the Institutional Animal Care and Use Committee approval in accordance with institutional guidelines. The 1×106 human iPS cells were inoculated into the hind leg of severe combined immunodeficient (SCID) mice. Teratomas were embedded in paraffin and histologically examined after hematoxylin and eosin staining. The procedure of the teratoma formation experiment was performed as described [60].
Flow cytometry analysis for mouse SSCs markers expression
All cells were suspended (1×104 cells/mL) and stained with the blood cells' markers primary antibody (CD71; FITC conjugate, BD Biosciences; CD14, CD15, CD45; APC conjugate, BD Biosciences) on ice in Dulbecco's phosphate-buffered saline containing 10% bovine serum albumin. Staining was compared with an isotype control antibody (mouse IgG1-FITC, mouse IgG1-APC BD Biosciences) to correct for nonspecific binding. Evaluation of antibody staining by flow cytometry and cell sorting by flow cytometry were performed using a fluorescence activated cell sorting vantage SE (BD).
Statistical analysis
Each experiment was performed as least thrice, and data are shown as the mean±SE where applicable, and differences were evaluated using Student's t-tests. The probability of < 0.05 was considered statistically significant.
Results
Isolation and enrichment of CD34+ cells from HuAFCs
We used a magnetic activated cell sorting system to isolate and enrich a specific subpopulation of cells, namely the CD34+ cells, from HuAFCs. The flow cytometry results showed that CD34+ cells represented 96.875%±1.253% of HuAFCs after sorting, compared with 0.183%±0.032% of HuAFCs before sorting (Fig. 1). These findings indicated that the CD34+ cells could be successfully enriched from human amniotic fluid using magnetic activated cell sorting.

Generation of iPS cells from CD34+ HuAFCs via overexpression of the reprogramming factor Oct4.
Generation and characterization of iPS cells derived from CD34+ HuAFCs
iPS cells were generated from the CD34+ HuAFCs after infection with lentiviral constructs encoding only Oct4. Since we utilized Oct4-GFP expression as a reprogramming marker in accordance with previous studies, after infection, the CD34+ HuAFC cells gave rise to GFP-positive cells on day 4 and GFP-positive colonies on day 5. In contrast to CD34+ HuAFCs, the GFP-positive cells are first observed in human amniotic epithelial cells (HuAECs; used as feeder layers to culture iPS cells in this study) on day 4 after infection with the Oct4-encoding lentivirus (Figs. 1, 2). On day 7, the morphology of iPS cell colonies derived from CD34+ HuAFCs and growing on HuAECs could be observed clearly. Under the microscope, we found that these colonies appeared more isolated and rounded, consistent with more undifferentiated cells (Fig. 1).
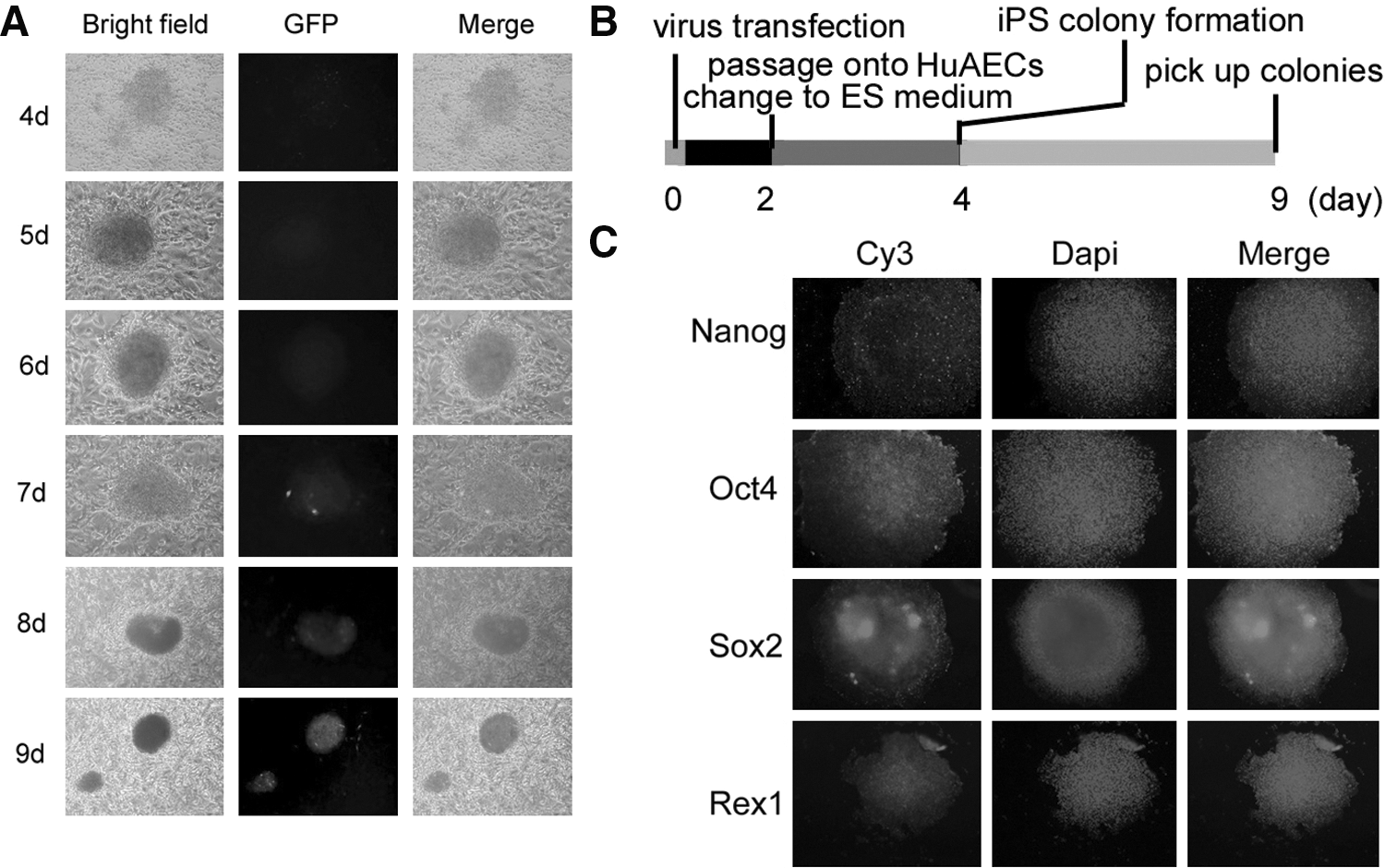
Characterization of reprogrammed human iPS cells.
Furthermore, since AP levels increase as stem cells acquire pluripotency, we measured the AP activity of these human iPS cells. The Deep Blue staining on the surface of the colonies indicates that the levels of AP in these cells was very high (Fig. 1). In addition, the cells were karyotyped after passage 10, and the human iPS colonies were found to have a normal karyotype of 46XY (Fig. 1). We also analyzed the expression of several stem cell markers using qRT-PCR and immunofluorescence (IF) staining to evaluate the degree of differentiation and stemness of human iPS colonies grown on the HuAECs. IF staining revealed that the expression levels of Nanog, Oct4, Sox2, and Rex1 were increased in iPS colonies (Fig. 2). The qRT-PCR results showed that the expression of these stem cells markers was ∼5- to 120-fold higher in human iPS cells than in HuAFCs. Relative mRNA expression is shown after normalization to 18S rRNA, which served as an internal control. However, the oncogenes, c-Myc and Klf4, were not expressed in either iPS cells or HuAFCs (Fig. 3). In addition, the high level of telomerase activity in human iPS cells suggests that their replicative lifespan will exceed that of somatic cells. These results demonstrate that human iPS cells cultured on HuAECs had more stemness and pluripotency.
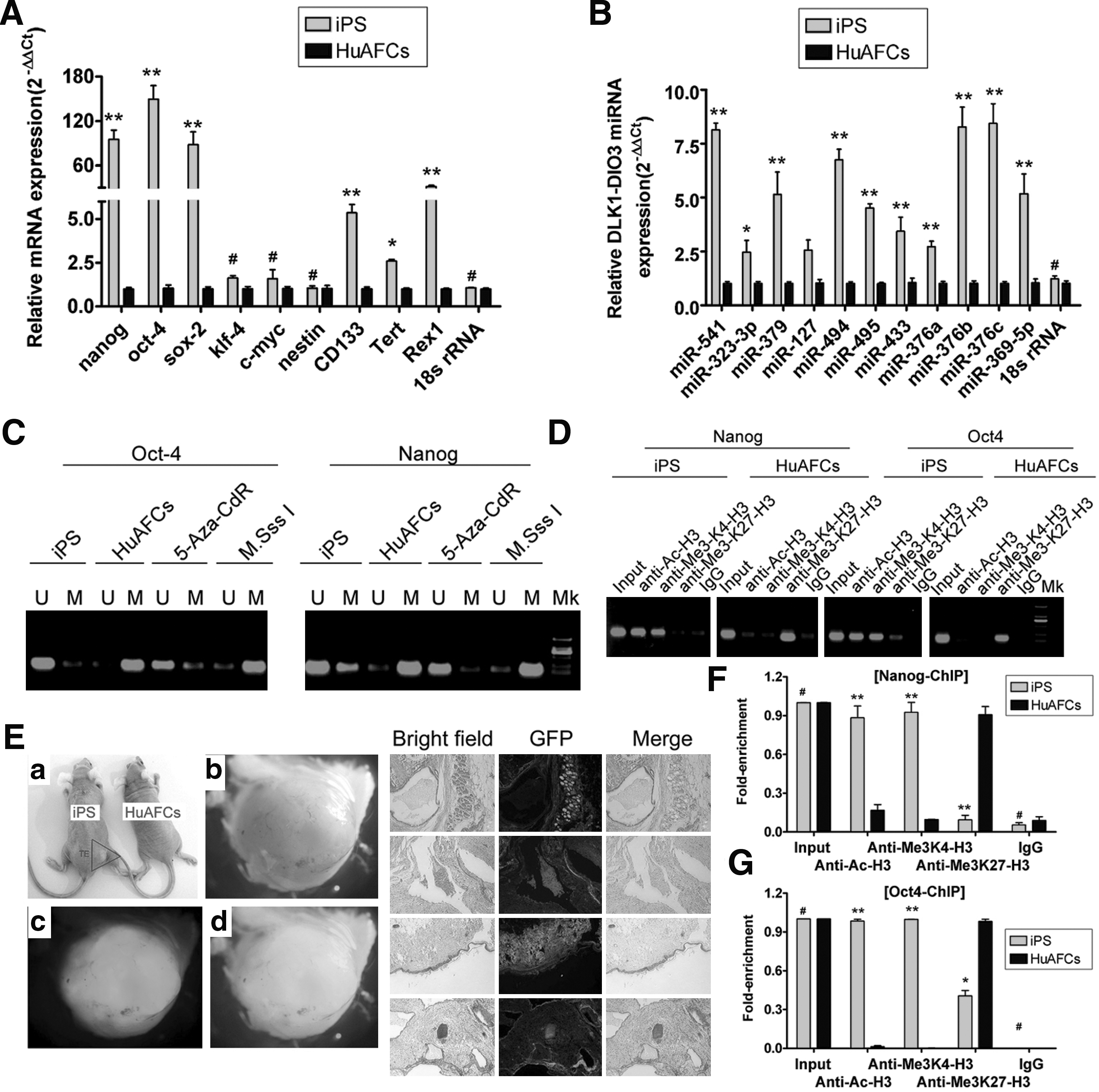
Epigenetic modification and in vivo pluripotency differentiation abilities of human iPS cells.
To further evaluate the pluripotent potential of human iPS cells derived from CD34+ HuAFCs, 3×107 of these cells were injected into the hind leg of mice with SCID. After 7 weeks, teratomas could be observed on the legs of SCID mice injected with human iPS cells (Fig. 3). The iPS-derived teratomas contained cellular representatives of all 3 germ layers; hematoxylin and eosin staining demonstrated the presence of immature tissues, such as neurocoele and glands, muscle, lipocytes, and bone cells (Fig. 3). Therefore, human iPS cells derived from CD34+ HuAFCs that were cultured on HuAEC feeder layers maintain pluripotency.
Change in the epigenetic status of iPS cells derived from CD34+ HuAFCs during reprogramming
The methylation state of CpG (the CpG sites were regions of DNA where a cytosine nucleotide occurs next to a guanine nucleotide in the linear sequence of bases along its length) dinucleotides in the Oct4 and Nanog promoters of human iPS cells and CD34+ HuAFCs was analyzed by sodium bisulfite treatment and sequencing of genomic DNA. In HuAFCs, the Nanog and Oct4 promoter regions had highly methylated CpG islands, whereas in human iPS cells cultured on HuAECs, the promoters of Nanog and Oct4 were moderately hypomethylated (Fig. 3). The negative control, human iPS cells treated with 5-aza-2′-deoxycytidine for 48 h, and the positive control, human iPS cells treated with CpG methyltransferase, showed hypomethylation and hypermethylation, respectively, of the CpG islands in the Nanog and Oct4 promoters (Fig. 3). In addition, ChIP assays were performed to evaluate histone H3 acetylation levels of the Nanog and Oct4 promoters of human iPS cells cultured on HuAECs and HuAFCs (Fig. 3). The assay showed that histone H3 appeared relatively hyperacetylated and methylated at the K4 locus of the Nanog and Oct4 promoters in human iPS cells cultured on HuAECs compared with the same region in HuAFCs. However, in human iPS cells, H3-K27 trimethylation at the Nanog and Oct4 promoters was remarkably low compared with the same region in HuAFCs. Consistent with the epigenetic remodeling that occurs during reprogramming, the CpG islands of promoters of endogenous Oct4 and Nanog were demethylated, and Oct4 mRNA was transcribed from the endogenous locus, which can be distinguished from the virally expressed human Oct4. Genomic integration of the Oct4 transgenes was confirmed, and the expression of both transgenes was efficiently silenced in these iPS cells. The expression of the genomic imprinted Dlk1-Dio3 miRNA cluster serves as a useful indicator for the quality of iPS cells clones [12,64,65]. The results of an miRNA-specific qRT-PCR indicated that the expression of Dlk1-Dio3 miRNA in human iPS cells cultured on HuAECs was ∼2- to 8-fold higher than that in HuAFCs. Relative mRNA expression is shown after normalization to 18S rRNA, which served as an internal control. Thus, the epigenetic status of these human iPS cells was more frequently and faithfully reprogrammed.
In vitro differentiation of iPS cells from CD34+ HuAFCs into multilineage blood cells
The successful differentiation of iPS cells into somatic cells in vitro effectively demonstrates the pluripotent nature of these cells. Therefore, in this study, we induced the differentiation of iPS cells derived from CD34+ HuAFCs into multilineage blood cells in vitro. The iPS cells were cultured on specific induction medium and on day 20, some colony-forming units, including colony-forming unit-megakaryocyte and erythrocytic burst-forming units, were observed under the microscope (Fig. 4). Fluorescence-activated cell sorting analysis of blood cell markers suggested that the expression of these markers (CD14, a monocyte marker; CD15, a granulocyte marker; CD45, a pan-hematopoietic marker; and CD71, an erythroblast marker) on human iPS cells after induction was about 10- to 40-fold higher than that of markers on human iPS cells which were not induced (Fig. 4). In addition to the multilineage, blood cell markers expressed in different degrees on the hemocyte-like cells derived from human iPS cells, Giemsa–Wright staining demonstrated the presence of multilineage blood-like cells, such as granulocyte-like cells, erythroblast-like cells, lymphocyte-like cells, monocyte-like cells, and so on, under the microscope (Fig. 4). Thus, these results indicated that human iPS cells derived from the CD34+ HuAFCs could differentiate into multilineage blood-like cells by in vitro induction.

Differentiation of human iPS cells into multilineage blood-like cells in vitro.
Discussion
Since the generation of the first iPS cells generated by Yamanaka and Takahashi [3,4,25] in 2006, this technique has developed quickly. Today, iPS cells have been reported for many species used in preclinical modeling [1,25]. However, if iPS cells are to have a clinical application, then the techniques for their generation need to be highly efficient, safe, and not involve heterogeneous proteins [21,66]. In light of these conditions, in this study, we generated iPS cells from the CD34+ subpopulation of cells isolated from human amniotic fluid. The cells were infected with retroviruses carrying only one reprogramming factor (Oct4) and cultured on HuAEC feeder layers. The implications of these modus operandi are discussed next.
First, the use of only one Yamanaka reprogramming factor (Oct4) that generates the iPS cells is safer. In previous reports, 4 Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc) have been used to induce the reprogramming of somatic cells into iPS cells [3,4]. However, c-Myc and Klf4 are oncogenic factors, and previous studies indicate that c-Myc might cause tumor development in vivo, and Klf4 could induce tumor formation in offspring [51,67,68]. On the other hand, the transcription factor Oct4 functions as a major regulator in maintaining the pluripotency and self-renewal capacities of ESCs [67]. Therefore, Oct4 is the most important factor in the generation of iPS cells. In our study, ∼4 days after CD34+ HuAFCs had been infected with retrovirus carrying only the Oct4 gene, some ESC-like colonies appeared among the feeder cells. These colonies were positive for both AP and other pluripotent markers specific to ESCs, which were expressed at a high level. Moreover, these cells could differentiate into cells from all 3 germ layers in vivo and in teratomas. Thus, our results showed that human iPS cells can be generated using the Oct4 factor alone.
Second, the CD34+ HuAFCs were chosen as seed cells in order to improve the efficiency and reduce the time taken to generate iPS cells. To reference the previous studies, Takahashi and Yamanaka et al. reported that the pluripotent cells were provided by the direct reprogramming of murine somatic fibroblast cells into iPS cells using the 4 factors (Oct3/4, Sox2, c-Myc, and Klf4); the process would need more than 30 days [3,4,6,10]. In addition, although Jin and coworkers. reported for the first time the rapid and efficient generation of iPS cells (the reprogramming process only needed 6 days) from human amniotic fluid-derived cells, they also used 4 Yamanaka factors [57]. On the other hand, although there are multifarious ways to generate iPS cells, the induction efficiency is as low as 0.001%, and the induction process takes from 1 to 5 weeks (with 1 week being the minimum) [32]. HuAFCs are presently used for the routine prenatal genetic diagnosis of a wide range of fetal abnormalities [58,59]. However, amniotic fluid contains multiple cell types derived from the developing fetus, such as CD34+, CD44+, and CD105+ cells, which can give rise to various types of cells (including adipose, muscle, bone, and neuronal cells) [58,59] that have some stemness. In our study, after infection of CD34+ HuAFCs with Oct4, the cells gave rise to GFP-positive cells on day 4 and GFP-positive colonies on day 5. On day 7, the morphology of colonies of iPS cells derived from CD34+ HuAFCs growing on HuAECs feeders could be clearly observed. However, according to other reports, the time taken for the reprogramming of other seed cells was longer than that for CD34+ HuAFCs. Therefore, our method of iPS generating has a remarkable advantage. Moreover, we considered the possibility that CD34+ HuAFCs might represent a more advanced stage in the reprogramming process and that terminally differentiated cells have to go through a stage such as this in order to acquire pluripotency.
Third, the use of HuAECs as feeder layers can maintain the pluripotency of human iPS cells for a long time and avoid contamination from heterogeneous proteins. To date, many studies have indicated that the in vivo proliferation and differentiation of human iPS cells is dependent on a specific microenvironment, including various cytokines, LIF, and other unknown factors. In order to maintain the self-renewal and proliferation and inhibit the differentiation of iPS cells in vitro, we should provide a similar microenvironment with the essential ingredients for growth. In many experiments, mouse ESCs have been typically cultured on a feeder layer of MEFs, with the addition of LIF and growth factors, such as EGF and bFGF, to maintain the ESCs in an undifferentiated, self-renewing state. These techniques require many animals for the time-consuming preparation of MEFs and costly growth factors and LIF. Addition of growth factors can result in extraneous contamination, which may induce iPS differentiation. In order to avoid the disadvantages associated with the use of MEFs, we chose HuAECs as the feeder layer. HuAECs are temporary specialized fetal cells, derived from the placenta, that can maintain the pluripotency of early epiblast cells. Previous studies have indicated that HuAECs express many growth factors such as LIF, EGF, bFGF, transforming growth factor-α/β, and BMP-4 as well as stem cell markers, including Nanog, Oct4, and Nestin [61,69,70]. Grueterich et al. reported that amniotic membrane culture conditions could promote limbal stem cell expansion [71]. Chen et al. reported that HuAECs can be used as a human feeder layer equivalent for effective ex vivo expansion of adult human limbal epithelial stem cells [72]. Lekhanont et al. developed a serum- and feeder-free technique of culturing human corneal epithelial stem cells on amniotic membranes [73]. The procedures for culturing mouse ESCs would be simplified if the human placental amnion, routinely discarded as medical waste, could be used as a feasible abundant source of feeder cells [61]. The findings of our previous studies suggested that the expression of LIF by HuAECs could maintain mouse and human ESCs and mouse spermatogonial stem cells in an undifferentiated, proliferative state capable of self-renewal [60,61,74]. This study indicates that the human placental amnion could be used as an abundant source of feeder cells for human iPS culture procedures. The use of the human placental amnion is free from ethical constraints and has low toxicity and high safety due to the presence of few exogenous foreign proteins.
At last, we should emphasize that although our iPS cells could generate teratoma in SCID mice, the benign teratoma did not explain that these iPS cells have a safety problem. Since the iPS cells could generate teratoma, these cells could differentiate into cell types from all 3 germ layers in vivo and in teratomas, which indicated that the iPS cells had strong pluripotency. Moreover, there was no evidence that the heterogeneity or/and malignant cells in these teratoma (maybe, we could also comprehend that the teratoma generated with our iPS cells was not an immature teratoma). Therefore, in this regard, we may as well say that the iPS cells produced with this method are safe.
In conclusion, we used CD34+ HuAFCs as seed cells and HuAECs as feeder layers to successfully generate human iPS cells via the administration of a single Yamanaka factor (Oct4), which was a highly efficient and safe method.
Footnotes
Acknowledgments
This work was supported by a grant from the Shanghai Committee Medical Science Foundation of China (No.10411967100) to Te Liu.
Author Disclosure Statement
No competing financial interests exist.