
Abstract
Human mesenchymal stem cells (hMSCs) are attractive candidates for cell-based tissue repair approaches and have been used as vectors for delivering therapeutic genes to sites of injury. It is believed that hMSCs are able to detect and respond to shear stress due to blood and interstitial fluid flow through mechanotransduction pathways after transplantation. However, information regarding hMSC migration under shear stress and its mechanism is still limited. In this study, we examined the effect of shear stress on hMSC migration and the role of mitogen-activated protein kinases (MAPKs) in their migration. Shear stress between 0.2 and 10 Pa, which was produced by the flow medium, was exerted on fluorescently labeled hMSCs. Cell migration was evaluated using the scratch wound assay, and images were captured using a microscope equipped with a digital 3CCD camera. The results showed that hMSCs subjected to a shear stress of 0.2 Pa caused notably faster wound closure than statically cultured hMSCs, while migration in the 0.5- and 1-Pa shear stress group did not differ significantly from that in the control group. Shear stress >2 Pa markedly inhibited hMSC migration. hMSCs subjected to a shear stress of 0.2 Pa displayed an increase in extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinases (JNK), and p38 MAPK activation for up to 60 min, while a shear stress of 2 Pa abrogated the activation. JNK and p38 MAPK inhibitors completely abolished the effect of shear stress on hMSC migration, while significant differences were observed between the ERK1/2 inhibitor-treated static control and shear stress groups. Taken together, these results demonstrate that low shear stress effectively induces hMSC migration and that JNK and p38 MAPK play more prominent roles in shear stress-induced migration than ERK1/2.
Introduction
M
Several reports highlight the capacity of MSCs to migrate home and long-term engraftment into the appropriate target tissues. For example, Devine et al. studied the distribution of MSCs in baboons using intravenous infusion and found that both allogeneic and autologous MSCs appeared to distribute into the kidney, lung, liver, thymus, and skin after lethal total body irradiation, thereby having a high level of engraftment, while the nonconditioned group appeared to have a low level of engraftment [7]. Morigi et al. reported that MSCs are able to sense renal injury, which causes them to migrate to the site of damage, differentiate into tubular epithelial cells, and restore renal structure and function [8]. The engraftment level of donor-derived MSCs was increased 23-fold in injured mice than in noninjured mice, and these MSCs also reduced the degree of bleomycin-induced inflammation and collagen deposition [9]. Furthermore, MSC migration is known to contribute to several important pathological processes, including vascular disease, chronic inflammatory disease, cancer, and osteoporosis [10 –13]. Thus, understanding the basic mechanisms underlying MSC migration can help improve the development of methods for therapeutic treatment and tissue regeneration.
Extensive research on stem cells focuses on the chemical microenvironment and biochemical signals that are believed to guide the physiological processes of stem cells. However, mechanical stimulation is also known to play an important role in the biological activity of these cells [14 –16]. As an important mechanical factor, shear stress has been demonstrated to induce the promotion of migratory responses in many types of cells. More specifically, in the physiological shear stress model, Albuquerque et al. found that a laminar shear stress of 0.3 Pa significantly enhanced the migration of human umbilical vein endothelial cells (ECs) compared with a shear stress of 1.2 or 2 Pa [17]. Garanich et al. quantified the migratory activity of rat aortic smooth muscle cells (SMCs) and suggested that shear stress suppressed SMC migration by up-regulating the cellular production of nitric oxide (NO) [18]. In a recent study of the migration of ECs using the scratch wound assay under 3 conditions of laminar shear stress (0.56, 1.00, and 1.53 Pa), the percentage of wound closure by ECs increased in a magnitude-dependent manner compared with that of wound closure by the static control cells. More specifically, cells on the upstream side of the wound migrated farther than those on the downstream side, which indicates that laminar shear flow provides a traction force for asymmetric cell migration along the direction of flow [19]. Studies have shown that shear stress can regulate MSC proliferation and differentiation into osteoblasts, ECs, or cardiomyocytes [20 –22], suggesting that MSCs are also sensitive to mechanical stress produced by fluid flow. However, the mechanism of the effect of shear stress on human MSC (hMSC) migration still remains unknown.
As a classical signaling pathway, the mitogen-activated protein kinase (MAPK) pathway, which includes ERK1/2, JNK, and p38 MAPK, has been demonstrated to control major cell functions, such as proliferation, differentiation, and migration [23]. In breast cancer cells, ERK1/2 activation leads to expression of the activator protein-1 (AP-1) components Fra-1 and c-Jun, both of which are necessary for cell migration [24]. In contrast, ERK1/2 is not associated with osteopontin-stimulated vascular SMC migration, whereas JNK and p38 MAPK play a crucial role during this process [25]. Ryu et al. demonstrated that with the stimulation of stromal cell-derived factor-1 (SDF-1), phosphoinositide 3-kinase (PI3K)/Akt, ERK, and p38 MAPK signal transduction pathways are involved in the regulation of umbilical cord blood-derived MSC (hUCB-MSC) migration, and knockdown of Akt, ERK, or p38 MAPK completely inhibits the migratory ability [26].
This evidence indicates that shear stress probably utilizes the MAPK pathway to regulate hMSC migration. To prove this hypothesis, we investigated the effect of shear stress on hMSC migration during wound healing and the role of signal transduction involving ERK1/2, JNK, and p38 MAPK of the MAPK family in hMSC migration.
Materials and Methods
Cell culture
hMSCs (Lonza) were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (JRH Bioscience), 100 U/mL penicillin, 100 μg/mL streptomycin, and 10 ng/mL human basic fibroblast growth factor (bFGF; Austral Biologicals) at 37°C in a humidified incubator (Thermo Scientific) with 5% CO2. The culture medium was replenished 2 to 3 times a week, and subculture was performed by digestion with 0.05% trypsin–0.02% ethylenediaminetetraacetic acid when the cells were nearly confluent. For experiments, hMSCs at passages 4–8 were plated onto glass-based dishes (φ35, Asahi Techno Glass) coated with 0.1% gelatin.
Flow-exposure experiment
hMSCs were exposed to steady laminar shear stress using a parallel flow chamber system [27]. In brief, the flow chamber was connected to a recirculation flow loop, and a peristaltic pump (Masterflex) was used to propel the culture medium from a reservoir, which dampened pulsatility before the fluid entered the flow chamber. The culture medium was then allowed to pass into another reservoir, which opened to the atmosphere (Fig. 1). The culture medium was maintained at 37°C and equilibrated with 5% CO2 throughout the experiment. Because serum can influence cell migration to a great extent and to specifically study the effect of shear stress on hMSC migration, the culture medium used for flow–exposure experiments was serum-free but contained Insulin–Transferrin–Selenium-X Supplement (Invitrogen). The magnitude of shear stress (0.2–10 Pa) was controlled by changing the flow rate.

Schematic diagram of the device used in the flow–exposure experiment. The device consisted of a flow chamber, a roller pump, and 2 reservoirs. One of the reservoirs (the pulse damper) was used to dampen the pulsatility of fluid flow caused by the roller pump, and the other reservoir was open to the atmosphere. The culture medium in the flow circuit was equilibrated with 95% air and 5% CO2. The system was maintained at 37°C.
Scratch wound migration assay
hMSC migration was evaluated using the monolayer scratch wound assay. The cells were seeded onto glass-based dishes and cultured until confluence. Mitochondria were fluorescently labeled with Mitotracker (Invitrogen). Next, the confluent cells were wounded with a linear scratch ∼200 μm in width by a plastic cell scraper (Corning), and laminar shear stress was applied in a direction perpendicular to the wound edges. Images of the wounds were acquired 0, 1, 3, 6, and 10 h after the onset of flow by a microscope (IX-81; Olympus) with a 3CCD camera (ORCA-3CCD; Hamamatsu Photonics). Using ImageJ, the levels of wound closure could be assessed by calculating the ratio of the closure area to the initial wound area as follows:
Inhibition of MAPK activation
The ERK1/2 inhibitor U0126, p38 MAPK inhibitor SB203580 (Cell Signaling Technology), or JNK inhibitor SP600125 (Sigma) was dissolved in dimethyl sulfoxide, aliquoted, and stored at −20°C. These inhibitors were then diluted immediately before use in the cell culture medium and applied to hMSCs at a final concentration of 10 μM 1 h before shear stress.
Fluorescence staining
After flow exposure, hMSCs were fixed with 4% paraformaldehyde for 15 min at room temperature and washed 3 times with phosphate-buffered saline (PBS). hMSC membranes were then permeabilized with 0.1% Triton X-100 for 10 min. The membranes were then blocked with Block Ace (Dainippon Pharmaceutical, Japan) for 1 h at room temperature and incubated overnight at 4°C with a rabbit monoclonal antibody against phospho-ERK1/2 (p-ERK1/2), ERK1/2, JNK or p38 MAPK, mouse monoclonal antibody against p-JNK (Cell Signaling Technology), or rabbit polyclonal antibody against p-p38 MAPK (Abcam). Thereafter, the cells were incubated with an Alexa Fluor 488–conjugated or Alexa Fluor 555–conjugated antibody (Invitrogen) for 1 h at room temperature. To stain the nuclei, hMSCs were treated with 1 μg/mL 4′,6-diamidino-2-phenylindole (Invitrogen) for 3 min. Fluorescence images of cells were obtained by confocal laser scanning microscopy (Olympus).
Bromodeoxyuridine incorporation assay
Cell proliferation was evaluated using the bromodeoxyuridine (BrdU; Sigma) incorporation assay. Cells were treated with 10 μM BrdU during the flow–exposure experiment. After removal of the culture medium, the cells were fixed with acetone for 10 min at 4°C and washed 3 times with PBS. After incubation with 2 M HCl for 30 min, hMSCs were treated with Tris-HCl (pH 7.5) for 5 min and then blocked with Block Ace for 1 h at room temperature. Subsequently, the cells were incubated with an anti-BrdU antibody (Sigma) for 1 h, followed by a second antibody (Alexa Fluor 555–conjugated antibody) for 1 h. Nuclei staining and observation were performed as described above for fluorescence staining.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot analysis
Cells were washed with ice-cold PBS and scraped immediately after exposure to laminar shear stress for 10, 30, 60, or 120 min. Proteins were extracted using cell lysis buffer (62 mM Tris-HCl (pH 6.8), 10% glycerol, 2% sodium dodecyl sulfate (SDS), and 2% β-mercaptoethanol) supplemented with phenylmethylsulfonyl fluoride and phosphatase inhibitor cocktails I and II (Sigma-Aldrich). Protein concentration was determined using a bicinchoninic acid protein assay (BCA method; Bio-Rad). After electrophoretic separation by 8% SDS–polyacrylamide gel electrophoresis, proteins were electroblotted onto polyvinylidene fluoride membranes (GE healthcare). The membranes were blocked with Tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% bovine serum albumin for 1 h at room temperature. ERK1/2, p-ERK1/2, JNK, p-JNK, p38 MAPK, and p-p38 MAPK antibodies and a rabbit monoclonal antibody against β-actin (Cell Signaling Technology) were used according to the manufacturer's protocols and were incubated overnight at 4°C with slight shaking. Thereafter, the membranes were washed in TBST buffer and further incubated with a horseradish peroxidase-conjugated antibody (goat anti-rabbit IgG; Sigma-Aldrich) for 1 h at room temperature. To observe the protein bands, an enhanced electrogenerated chemiluminescence system (Amersham) was used. The bands were semiquantitatively evaluated by densitometry. The levels of ERK1/2, p-ERK1/2, JNK, p-JNK, p38 MAPK, and p-p38 MAPK proteins were determined by normalizing to those of the housekeeping protein β-actin.
Statistical analysis
Results were analyzed by Student's t-test and analysis of variance (ANOVA). Bonferroni post hoc tests were used when the p-value (by ANOVA) indicated a statistical difference among groups. Data are expressed as mean±SD. P<0.05 was deemed statistically significant.
Results
Shear stress regulates hMSC migration
Fluorescence images of hMSCs exposed to a shear stress of 0.2, 0.5, 1, and 2 Pa are presented in Fig. 2A. In the absence of flow (control), cells reached confluence 10 h after wound formation. Under a shear stress of 0.2 Pa, hMSCs closed ∼98% of the original wound at 6 h, whereas under the static condition (control), hMSCs closed only 62% of the original wound (Fig. 2B). Wound closure in the 0.5- and 1-Pa shear stress groups was not significantly different from that in the control group. In stark contrast, a shear stress of 2 Pa markedly inhibited hMSC migration. We also studied the effects of shear stress >2 Pa (i.e., up to 10 Pa) on hMSC migration and confirmed that greater shear stress inhibited cell migration to an extent similar to that observed under a shear stress of 2 Pa, and almost no detachment of cells was observed with the stimulation of higher shear stress (data not shown). Because proliferation itself can promote wound closure, it is necessary to exclude the effect of proliferation when evaluating shear stress-induced cell migration. Using the BrdU incorporation assay, we observed no proliferative cells in the control group or in the 0.2-Pa shear stress group at 6 h (data not shown). These results thus indicate that low shear stress, rather than high shear stress, accelerated hMSC migration and that this effect was independent of the change in cell number.
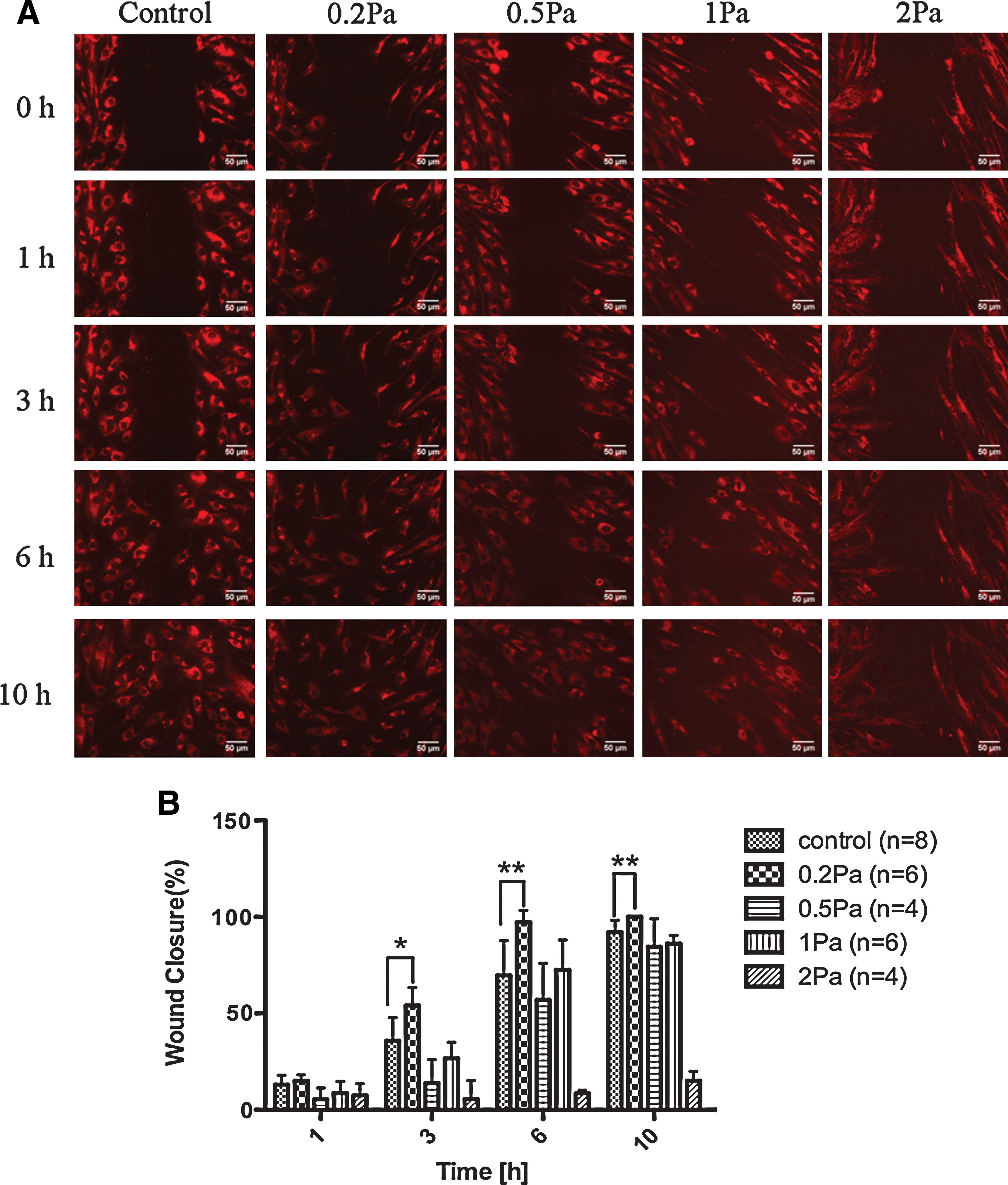
Effects of shear stress on hMSC migration.
Shear stress activates MAPK expression in hMSCs
We examined ERK1/2, JNK, and p38 MAPK phosphorylation by Western blot analysis and immunostaining. We found that ERK1/2 activation was increased at 60 min followed by a slight decrease in the 0.2-Pa shear stress group, but a significant difference was observed in the cells at 2 h compared with the control group. Expression of total ERK1/2 (t-ERK1/2) was relatively stable under shear stress (Fig. 3A). In the shear stress groups, JNK phosphorylation proceeded faster than ERK1/2 phosphorylation and reached a peak at 30 min, which was more than 3-fold (P<0.01) higher than that in the control group. No significant difference was observed in t-JNK expression between all detected time points (Fig. 3B). In the shear stress groups, p-p38 MAPK activation also increased rapidly and peaked at 30 min, which was 2.7-fold higher than that in the control group. A significant difference was observed at 2 h (2.1-fold higher) compared with the control group, while p38 MAPK expression remained the same (Fig. 3C). Figure 4 shows typical immunofluorescence staining results for MSCs cultured with or without shear stress. Higher p-ERK1/2, p-JNK, and p-p38 MAPK expression was observed in the shear stress groups, which further shows that MAPK can be activated by shear stress. To confirm the effect of magnitude of shear stress on MAPK activation, we studied ERK1/2, JNK, and p38 MAPK phosphorylation in hMSCs exposed to 2-Pa shear stress. The results showed that the activities of ERK1/2, JNK, and p38 MAPK in hMSCs subjected to shear stress were not markedly different from those in the control group (Fig. 5A–C). This strongly indicates that lower shear stress promoted hMSC migration via MAPK signal molecules.
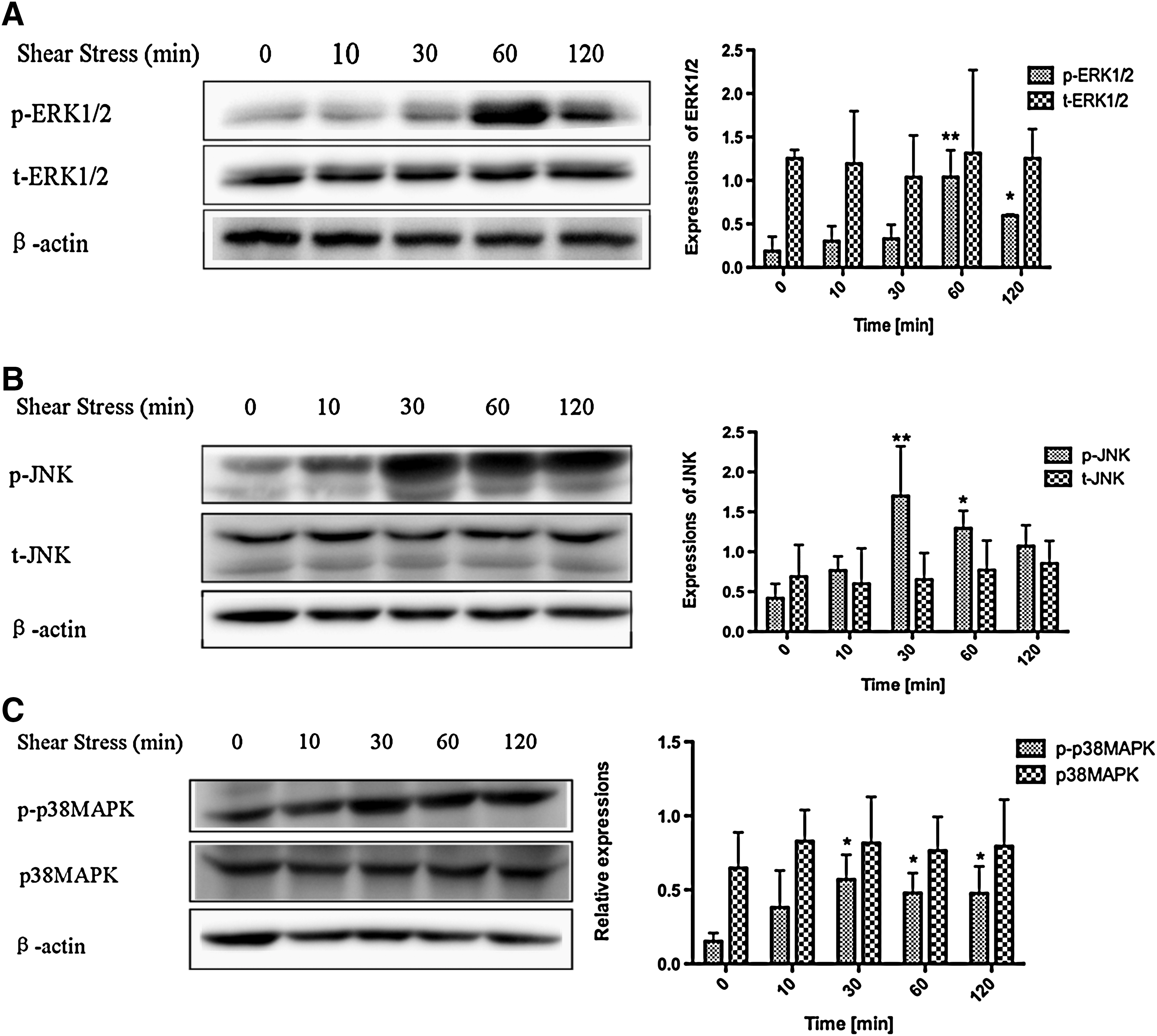
Effects of shear stress on the activation of MAPK proteins in hMSCs exposed to a shear stress of 0.2 Pa for 10, 30, 60, and 120 min.

Representative fluorescence images of total and phosphorylated ERK1/2, JNK, and p38 MAPK in hMSCs exposed to a shear stress of 0.2 Pa for 1 h. Blue, nucleus; red, phosphorylated ERK1/2, JNK, or p-p38 MAPK; green, total ERK1/2, JNK, and p38 MAPK. Bars=50 μm. Color images available online at

Effects of shear stress on the activation of MAPK proteins in hMSCs exposed to a shear stress of 2 Pa for 10, 30, 60, and 120 min.
MAPK regulates shear stress-induced hMSC migration
To examine whether ERK1/2, JNK, and p38 MAPK phosphorylation was essential for shear stress-induced hMSC migration, we used specific inhibitors of ERK1/2 (U0126), JNK (SP600125), and p38 MAPK (SB203580) to inhibit their activation. At the inhibitor concentration of 10 μM, shear stress-induced ERK1/2, JNK, and p38 MAPK phosphorylation was completely abolished by each inhibitor (Fig. 6A–C). Based on the usage of inhibitor-containing media for flow–exposure experiments, we found that U0126 significantly decreased the wound closure rate, and cell confluence was delayed until 10 h in the 0.2-Pa shear stress group. Interestingly, hMSC migration was observed in the 0.2-Pa U0126-treated shear stress group compared with the U0126-treated control group, and differences in the wound closure rate were apparent 6 h after wound formation (Fig. 7A, B). Treatment with the JNK inhibitor completely abolished the migratory ability of hMSCs, and shear stress did not compensate for the effect of inhibition (Fig. 7C). The same phenomenon was observed when p38 MAPK activation was inhibited (Fig. 7D). These results indicate that JNK and p38 MAPK play a more prominent role in shear stress-induced hMSC migration than ERK1/2.
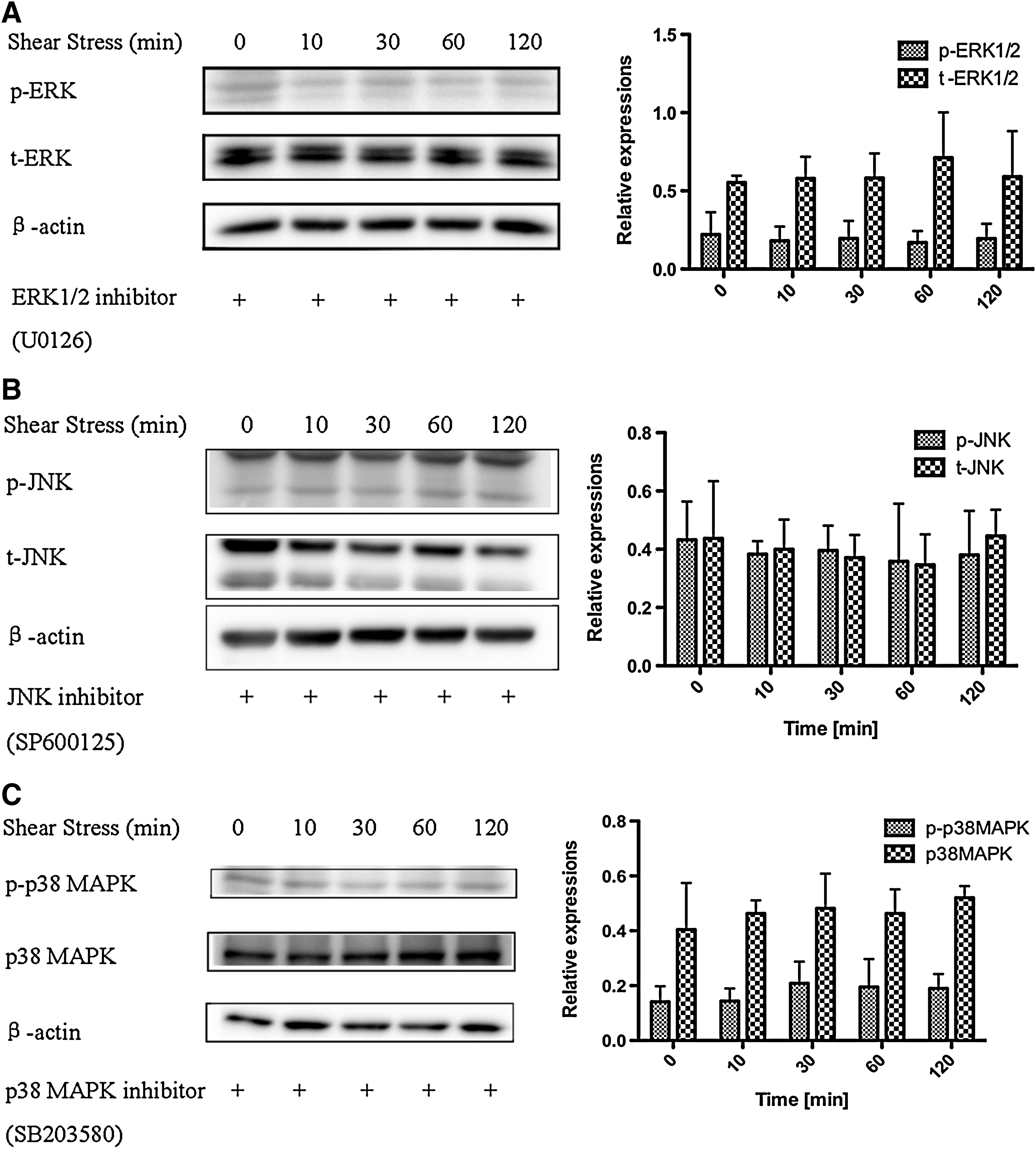
Effects of shear stress on the activation of MAPK family proteins in hMSCs exposed to a shear stress of 0.2 Pa for 10, 30, 60, and 120 min with the inhibitors of ERK1/2 (U0126), JNK (SP600125), and p38 MAPK (SB203580) at the concentration of 10 μM.
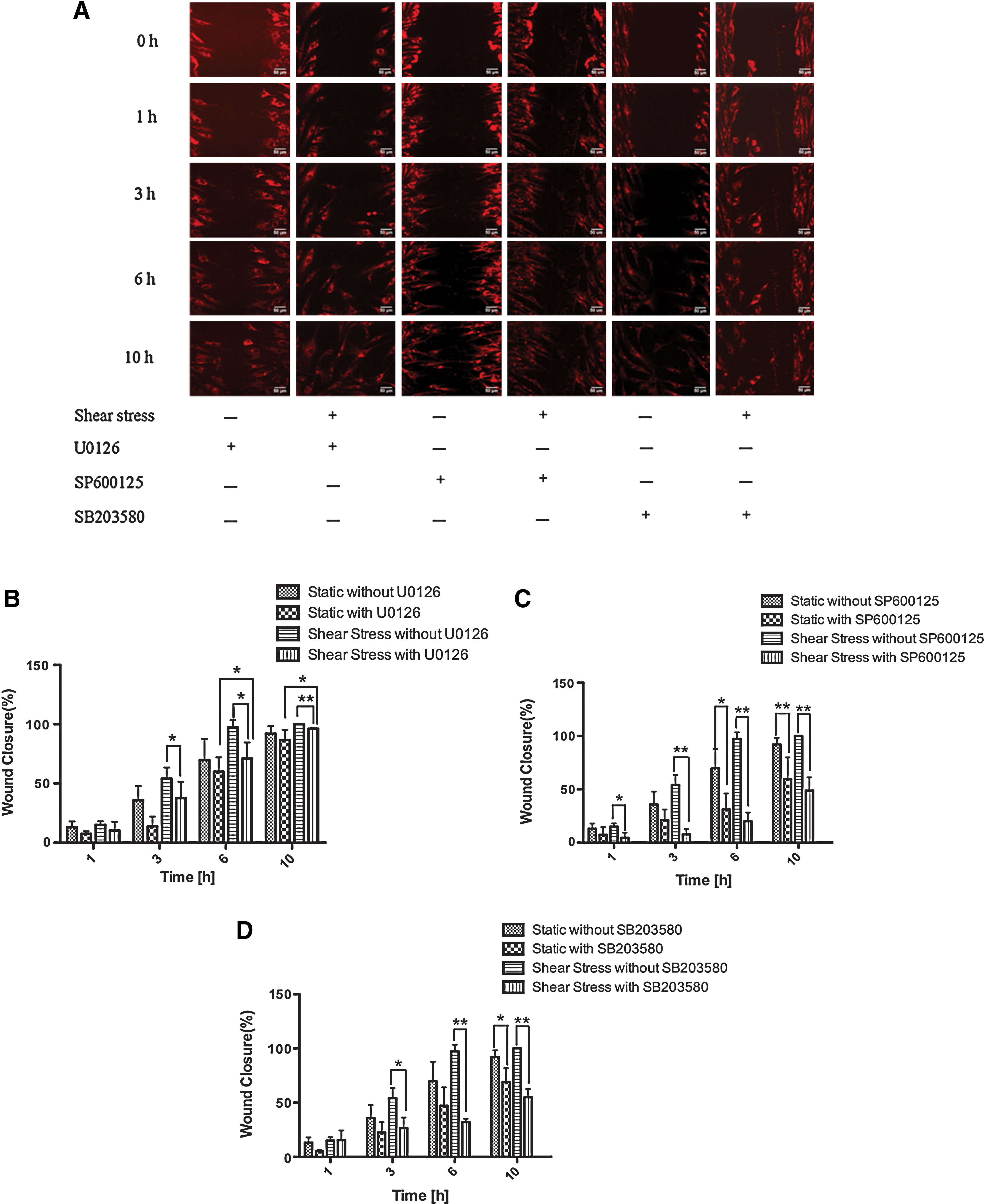
Effects of MAPK inhibitors on hMSC migration.
Discussion
Among stem cells that are currently being tested in clinical trials for cardiovascular repair, lung fibrosis, spinal cord injury, and bone and cartilage repair, MSCs are the most widely studied [28]. These cells are released from the bone marrow, circulate in the blood, and contribute to tissue repair under homeostatic conditions, whereas inflammatory stimulations can lead to their mobilization and activation, thereby causing a change from promoting tissue repair to tissue remodeling and fibrosis [29]. As the drag force created by flow through blood vessels, shear stress is also believed to play critical roles in normal physiological responses of MSCs as well as in disease pathology. Thus, if hMSCs are to be used in regenerative applications, it is highly important to consider the effect of shear stress on hMSC migration, the ability of these cells to migrate to damaged tissues, and the related signal molecules [30]. In the present study, we investigated hMSC migration under various magnitudes of shear stress using a parallel plate flow chamber system and found that a low shear stress of 0.2 Pa promoted hMSC migration, while shear stress >2 Pa markedly inhibited cell migration. We also examined MAPK activation under shear stress and used selective inhibitors of ERK1/2, JNK, and p38 MAPK to determine the effect of these MAPKs on shear stress-induced hMSC migration. Our results showed that shear stress could lead to activation of MAPKs, especially JNK and p38 MAPK, which were critical for shear stress-induced hMSC migration. To our knowledge, this is the first study of shear stress-induced hMSC migration and its related mechanisms.
In vitro, the effects of shear stress on various cell types have been widely studied. Liu et al. demonstrated that the migration speed of human trophoblast cells (TCs) increased under a shear stress of 3 Pa, and this magnitude of shear stress is considered physiologically relevant for TCs [31]. A laminar shear stress of 1.2 Pa, which is within the physiological range occurring in arteries, significantly increased the migration speed of bovine aortic ECs [32], and with the same degree of shear stress, SMCs displayed reduced migratory activity [33]. Interestingly, interstitial shear stress, which is much lower than 1.2 Pa in arteries, can promote rat aortic vascular SMC motility in collagen gels [34]. Recent evidence indicates that MSCs are present not only in bone marrow but also in the medial layer of normal healthy vessels as a subpopulation of SMCs [35]. Thus, MSCs are normally not directly exposed to a shear stress of flowing blood in the vascular system because the EC layer, which lines all blood vessels, provides a contact surface for blood flow, thereby shielding the underlying MSCs. However, like SMCs, MSCs are believed to be exposed to shear stress caused by interstitial flow under physiological conditions, and when endothelial injury and denudation occur, these cells may be exposed directly to an amount of shear stress similar to that experienced by ECs in intact blood vessels. We found that greater shear stress inhibited hMSC migration, while lower shear stress (0.2 Pa) enhanced migratory ability. Together with the findings of Tarbell's group, which showed that interstitial shear stress on SMCs is ∼0.1 Pa [36 –38], these results reflect the idea that conditions promoting cells migration are naturally present in the human body. Unlike a previous report showing that ECs on the upstream side of the wound migrated farther than those on the downstream side with the stimulation of shear stress [19], we found the downstream hMSCs migrated rapidly than the upstream cells subjected to a shear stress of 0.2 Pa (data not shown). Such directed movement of cell motility simply not caused by drag force which created by fluid flow, but induced as a cell response to fluid shear stress, called as mechanotaxis, which has been reported by several previous studies [39,40].
It remains unclear as to how the mechanical signal of shear stress was detected by hMSCs and which signal molecules were involved in this process. Although MAPKs have been well characterized as important mediators of multiple processes and have been shown to play a critical role in proliferation, differentiation, and apoptosis in various cell types [41], their effect on MSC migration remains controversial. Zha et al. demonstrated that migration of Snail-mediated human bone marrow MSCs was not affected by pretreatment with the ERK1/2 inhibitor PD98059 and the p38 MAPK inhibitor SB203580 [42]. Similarly, treatment with the p38 MAPK inhibitor SB203580 or the ERK1/2 inhibitor U0126 had no effect on platelet-derived growth factor-induced human adipose tissue-derived MSC (hASC) migration, while pretreatment with SP600125, an inhibitor of JNK, completely blocked the migratory ability of hASCs [43]. As revealed by immunofluorescence staining and western blot analysis in the present study, ERK1/2, JNK, and p38 MAPK activation was apparent within 60 min of exposure to lower shear stress (0.2 Pa). To further determine whether hMSC migration was mediated by shear stress-induced ERK1/2, JNK, and p38 MAPK activation, we examined the migratory ability of hMSCs in the presence of MAPK inhibitors. Interestingly, compared with the U0126-treated control group, cell migration increased in the 0.2-Pa shear stress group. In contrast, previous studies demonstrated that ERK1/2 played an important role during SDF-1-stimulated hUCB-MSC migration and that ERK knockdown completely inhibited the migratory ability [26]. Tamama et al. also found that epidermal growth factor (EGF) caused robust ERK phosphorylation in rats and immortalized human bone marrow MSCs as well as stimulated the motility of these cells [44]. These differences probably depend on differences in stimulation and the sources of the cells used in the experiments. Because many studies showed that cytokines such as tumor necrosis factor-α and SDF-1α enhanced MSC migration through MAPK signaling pathways [45,46], our findings raise the question whether the signaling pathways governing migration due to applied mechanical forces are the same as those involved in responses to chemical cues. In the present study, we also added conditioned media (CM) collected from sheared hMSCs (2 Pa for 24 h) to static hMSCs in the scratch wound migration assay and found that the CM significantly inhibited the migration of static hMSCs (data not shown). This suggests that shear stress-stimulated hMSCs secrete soluble factors that regulate their migration. Thus, our results comply with those of previous studies, which state that MAPKs act as hub or node molecules that mediate chemical, physical, and various types of stimuli, thereby forming a molecular crosstalking network to induce cell biological processes. Moreover, the application of a shear stress of optimum magnitude may induce the production of cytokines or growth factors that trigger MAPK activation and the regulation of the migratory ability of hMSCs. Migration is an exquisitely coordinated process and signaling pathways are not linear but form a complicated net structure; therefore, it is not surprising that other signaling pathway, such as the phosphoinositide 3-kinase (PI-3K)/Akt signaling pathway and the wingless/int-1 (Wnt) signal transduction pathway, also have been shown to be related to MSC migration [47,48]. However, whether these signaling pathways mediate shear stress-induced hMSC migration requires further study.
Conclusion
This study confirms that lower shear stress induces hMSC migration, while greater shear stress hinders hMSC migration. It also demonstrates that ERK1/2, JNK, and p38 MAPK are involved in the mechanotransduction pathway. These findings serve as a starting point to understand the effect of shear stress on MSC migration. We still have much to learn with respect to the specific mechanisms of shear stress-induced MSC migration and further studies on the same will undoubtedly provide more insight into the effects of the physical microenvironment on cell biological reactions and offer a rational base for developing new stem cell-based therapeutic strategies in tissue engineering and tissue repair.
Footnotes
Acknowledgments
This study was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (no. 20001007), the Natural National Science Foundation (nos. 30770530 and 11032012), and the Fundamental Research Funds for the Central Universities (CDJXS11232243) of China.
Author Disclosure Statement
The authors have nothing to disclose and no competing financial interests exist.