
Abstract
Hedgehog (Hh)-glioma-associated oncogene homolog (Gli) signaling is implicated in a large number of human cancers such as leukemia. In this study, we investigated the effects of the potent Hh antagonist GDC-0449 on the BCR-ABL-positive cell line OM9;22 and primary samples when leukemia cells were protected by a feeder cell line (S9 cells). The numbers of OM9;22 cells significantly increased with S9 cells. Treatment of OM9;22 cells with GDC-0449 caused cell growth inhibition and induced apoptosis. Moreover, GDC-0449 inhibited the colony growth of Philadelphia chromosome (Ph)-positive primary samples. We next investigated the effects of a combination of GDC-0449 and dasatinib on these cell lines. The growth inhibition typically promoted by dasatinib was significantly reduced in the presence of S9 cells. Treatment of Ph-positive leukemia cells with GDC-0449 and dasatinib in the presence of S9 caused significantly more cytotoxicity than that caused by each drug alone. Inhibition of Gli1 or Gli2 by siRNA transfection reduced the growth of the Ph-positive cell line K562 and increased cytotoxicity of dasatinib. Moreover, colony formations of Gli1 or Gli2 knockdown cells were also reduced. Data from this study suggest that administration of the Hh inhibitor GDC-0449 inhibits BCR-ABL-positive cell growth and enhances the cytotoxic effects of dasatinib in the presence of feeder cells.
Introduction
T
The introduction of imatinib, the most successful molecular targeted therapy against CML, has revolutionized the treatment of patients with CML. Imatinib binds to the inactive conformation of the BCR-ABL protein and prevents its conformational switch to the active form. Imatinib inhibits the BCR-ABL pathway and induces apoptosis in CML cells. The new tyrosine kinase inhibitors (TKIs) dasatinib and nilotinib are effective in most patients with CML [4]. Despite such therapeutic interventions, CML is often not curable, largely because of the innate sensitivity of CML stem cells, particularly in their quiescent state [5]. Residual CML cells are present in niches of the bone marrow and are considered quiescent [6]. These leukemia stem cells are contained within a niche in the bone marrow and are often impervious to current treatments; thus, they maintain their proliferative activity when treatment ceases [7]. Therefore, new approaches against leukemia stem cells are required to treat Ph-positive leukemia patients.
The hedgehog (Hh)-signaling pathway identified in mammalian cells is a highly conserved signaling system that regulates the progenitor cells of proliferation and maintenance [8]. Three Hh ligands, namely Sonic Hh (Shh), Indian Hh, and Desert Hh, have been identified in humans. These Hh proteins are expressed in stromal cells of the bone marrow and are secreted as soluble ligands [9]. When Hh ligands bind to the PATCHED protein, which is a 12-pass transmembrane-spanning receptor, this complex represses the activity of smoothened (Smo) on target cells; therefore, if the inhibition of Smo is relieved, the Hh pathway will be activated. The downstream molecules of the glioma-associated oncogene homolog (Gli) protein play key roles in mediating the Hh-signaling pathway [9]. Although the Hh-signaling pathway plays an important role in tissue growth and repair, it is almost silent in adult tissues.
Recent studies have implicated the Hh pathway in the maintenance of stem cells in multiple myeloma and CML [10,11]. Therefore, Hh signaling has been recently shown to be required for cancer stem cell self-renewal, including leukemia. A novel Smo inhibitor, GDC-0449, is now in phase II clinical trials for several cancers, such as basal cell carcinoma [12]. We hypothesize that Hh pathway inhibitors are good therapeutic candidates for reducing leukemia cells and tumor burden, because Hh signaling is shown to regulate the self-renewal and survival of leukemia cells. Moreover, it remains unclear that how the tumor microenvironment contributes to the maintenance of Ph-positive leukemia cells. In this study, we investigated the effects of the ABL TKI dasatinib in combination with the Hh inhibitor GCD-0449 on Ph-positive leukemia cells. We found that co-treatment of GCD-0449 and dasatinib significantly induced cell growth inhibition of Ph-positive cells in the presence of feeder cells (S9 cells).
Materials and Methods
Drugs, reagents, and cells
The Hh inhibitors GDC-0449 (Selleck chemicals LLC, Houston, TX) and [N-[3-(1H-Benzimidazol-2-yl)-4-chlorophenyl]-3,4,5-triethoxybenzamide] SANT-2 (Enzo Life Sciences, Plymouth Meeting, PA) and the BCR-ABL TKI dasatinib (Selleck Chemicals LLC) were diluted in dimethyl sulfoxide. The Hh ligand Shh was obtained from StemRD (Burlingame, CA) and dissolved in deionized water. The human Ph-positive cell line OM9;22 was described previously [13]. OM9;22 and another Ph-positive cell line, K562 cells, were grown on a Roswell Park Memorial Institute (RPMI) 1640 medium, including 10% fetal bovine serum supplemented with 1% penicillin/streptomycin. The human feeder cell line S9 was obtained from ATCC and grown in an LHC-8 medium (Invitrogen, Carlsbad, CA). Antiphospho-v-crk sarcoma virus CT10 oncogene homolog (avian)-like (Crk-L) (No. 3181), Akt (No. 9271), mitogen-activated protein kinase (MAPK, No. 9101), and anticleaved poly(ADP-ribose) polymerase (PARP, No. 9541) antibodies were purchased from Cell Signaling Technologies (Beverly, MA). Shh (sc-9204, 1:500), Gli1 (sc-20687, 1:500), Gli2 (sc-28674, 1:500), c-myc (sc-788 1:500), cyclin D (sc-246 1:500), β-tubulin (sc-9104, 1:500), and β-actin (sc-47778 1:500) antibodies, and siRNA or shRNA of Gli1 (sc-270268, sc-270268-V) and Gli2 (sc-37913, sc-37913-V) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Western blotting
Western blotting analysis was performed according to previously described methods [14]. In brief, protein contents of the lysates were determined using a protein assay kit (Bio-Rad Laboratories, Hercules, CA). Proteins were loaded onto polyacrylamide gels and then transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were probed with the primary antibodies of interest at the appropriate dilution for 2 h at room temperature. Blots were then probed with secondary antibodies conjugated to horseradish peroxidase and developed using an enhanced chemiluminescence system (Amersham Pharmacia Biotech, Bucks, UK).
Primary samples
This study was approved by the institutional review board of the Tokyo Medical University, and informed consent was provided by all patients in accordance with the Declaration of Helsinki. In this study, de novo CML samples were used. Primary samples were obtained from the peripheral blood or bone marrow of patients with CML. Mononuclear cells were isolated from the blood samples and separated using a Lymphosepar (Immuno-Biological Laboratories Co., Gunma, Japan). The cells were then cultured in an RPMI 1640 medium containing 10% fetal calf serum. In the experiments, 2×105 mL−1 mononuclear cells was used.
Cell proliferation assay
The cells were seeded in 24- or 96-well plates at a density of 2×105 mL−1 in the presence or absence of S9 feeder cells. The cells were treated with the inhibitor drugs at the indicated concentrations: dasatinib, 1–100 nM; and GDC-0449, 100 nM to 10 μM. Viable cells were counted by trypan blue exclusion or fluorescence-activated cell sorting.
Colony formation assay
The cells were seeded in the MethoCult GFH4434 medium (Stem Cell Technology, Vancouver, Canada) containing appropriate concentrations of GDC-0449 in 6-well culture plates. Cells were incubated for more than 14 days in a humidified atmosphere with 5% CO2 at 37°C, and the colonies were counted on a reverse microscope. Cell aggregates containing ≥40 cells were considered colonies. In some experiments, colonies were picked up and replated. Each experiment was carried out in duplicate.
Apoptosis analysis
The cells were treated with 20 μM GDC-0449 for 24 h. An annexin V/propidium iodide apoptosis assay was performed according to the manufacturer's instructions (Becton, Dickinson and Company, Franklin Lakes, NJ). The cells were gently mixed and immediately analyzed by flow cytometry.
siRNA and shRNA transfection
siRNA and shRNA of Gli1 or Gli2 were purchased from Santa Cruz Biotechnology. Although the Gli1 or Gli2 siRNA design is important, the siRNA sequence was not available in the manufacturer's protocol. The K562 cells were transfected with siRNA of Gli1 and Gli2 by electroporation. In brief, cells were electroporated in 400 μL of culture medium at a density of 2×107 per mL in electroporation cuvettes and mixed with 1 nmol of siRNA. The cells were electroporated using a Bio-Rad Gene Pulser II System at 0.2 kV and 950-μF capacitance. Fifteen minutes after electroporation, the cells were diluted in the culture medium and incubated at 37°C, 5% CO2, and 92% humidity. After 48 h, the cells were treated by dasatinib and immunoblot. Transfection of shRNA was described by the manufacturer's protocols. The transfected cells were analyzed for protein expression of the targeted protein using immunoblotting with a specific antibody.
Statistical analysis
Student's t test was used to determine if the effects of the drug treatments among the different groups were statistically significant compared to those in the control. Values of P<0.05 were considered statistically significant.
Results
Enhanced OM9;22 cell proliferation in the presence of S9 cells and Shh
The bone marrow stromal cells create a permissive environment in which myelopoiesis and lymphopoiesis occur by the production of cytokines and growth factors. Therefore, in the first series of experiments, we analyzed the proliferation of the Ph-positive cell line OM9;22 and Ph-positive primary samples in the presence of feeder cells (S9). We found that the proliferation of OM9;22 and primary cells significantly increased in the presence of S9 cells compared to that in the absence of the S9 feeder cells (Fig. 1A, B). We next examined whether Shh was involved in OM9;22 cell proliferation. We found that compared to control cells, OM9;22 cell proliferation significantly increased in cells treated with 10 ng mL−1 Shh for 24 h (Fig. 1C). We next investigated the efficacy of 100 nM dasatinib in the presence of Shh for 24 h. We found that the efficacy of dasatinib was reduced in the presence of Shh (Fig. 1C). In primary samples, we found that Shh was not involved in cell proliferation, but dasatinib activity was reduced in the presence of Shh (Fig. 1D).
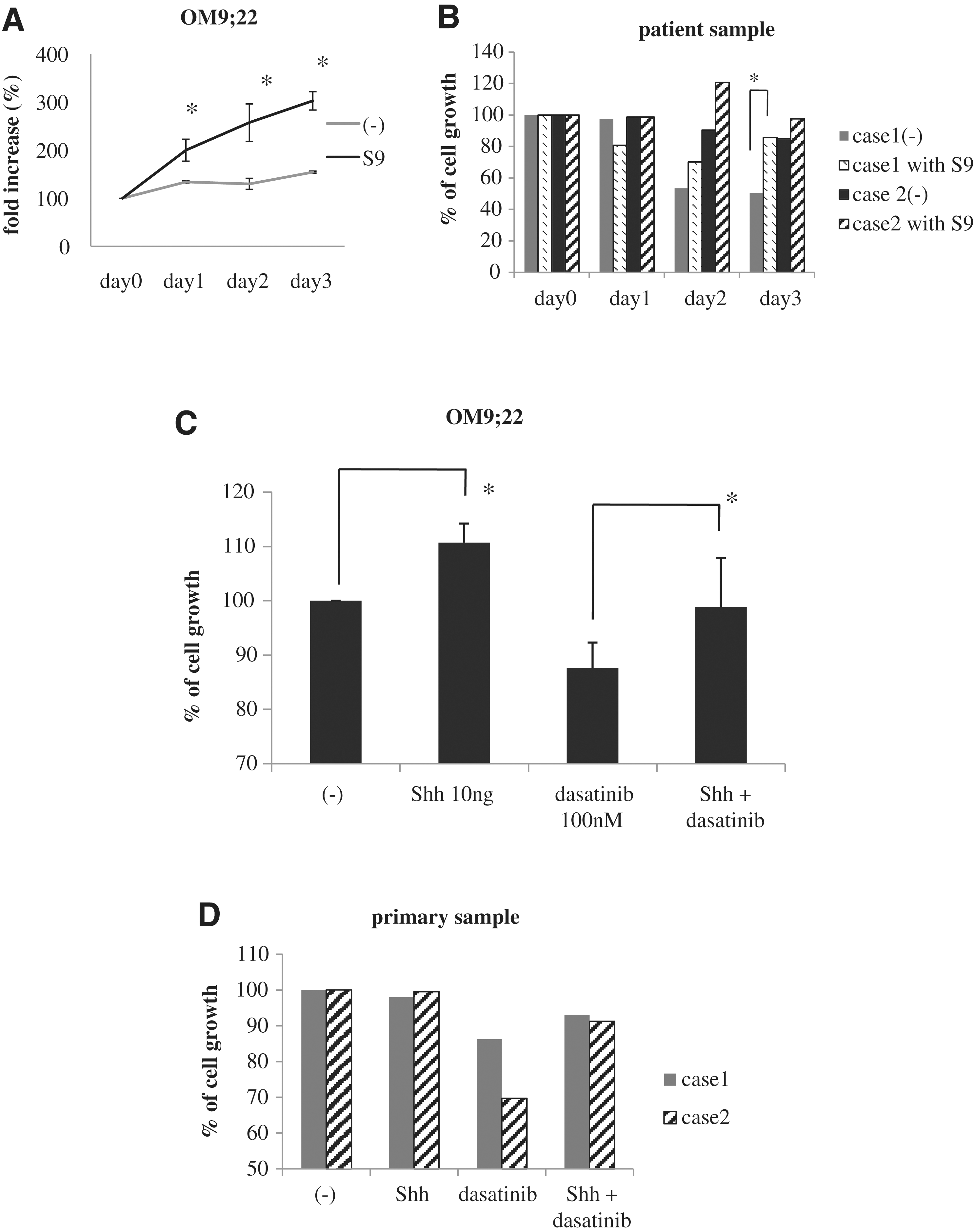
Ph-positive cell proliferation is enhanced in the presence of S9 cells.
Efficacy of dasatinib on cell proliferation in the presence of feeder cells
We next examined whether dasatinib activity was reduced in the presence of S9 cells. The number of OM9;22 cells decreased in a dose-dependent manner after treatment with dasatinib for 72 h. Cell growth inhibition by dasatinib was significantly reduced in the presence of S9 cells (Fig. 2A). We next examined the levels of intracellular signaling molecules of the Hh pathway in S9 cells to determine which molecular pathways might be affected and produce this effect. Because dasatinib is a dual src/Abl kinase inhibitor, we expected dasatinib to inhibit the src kinase activity of the S9 cells. In this experiment, S9 cells were treated with dasatinib at a concentration of 100 nM for 24 h. In western blot analysis, src phosphorylation was decreased in response to dasatinib treatment in a dose-dependent manner. However, Shh levels were unaltered by dasatinib treatment. The Hh-Gli inhibitor GDC-0449 is reported to have a median maximal plasma level of 23.0 μM in a phase I study [12]. Thus, a 10–20 μM concentration of GDC-0449 was used in this study to examine the effects of GDC-0449 on S9 cells. The Shh levels in the S9 cells were decreased after GDC-0449 treatment in a dose-dependent manner (Fig. 2B).
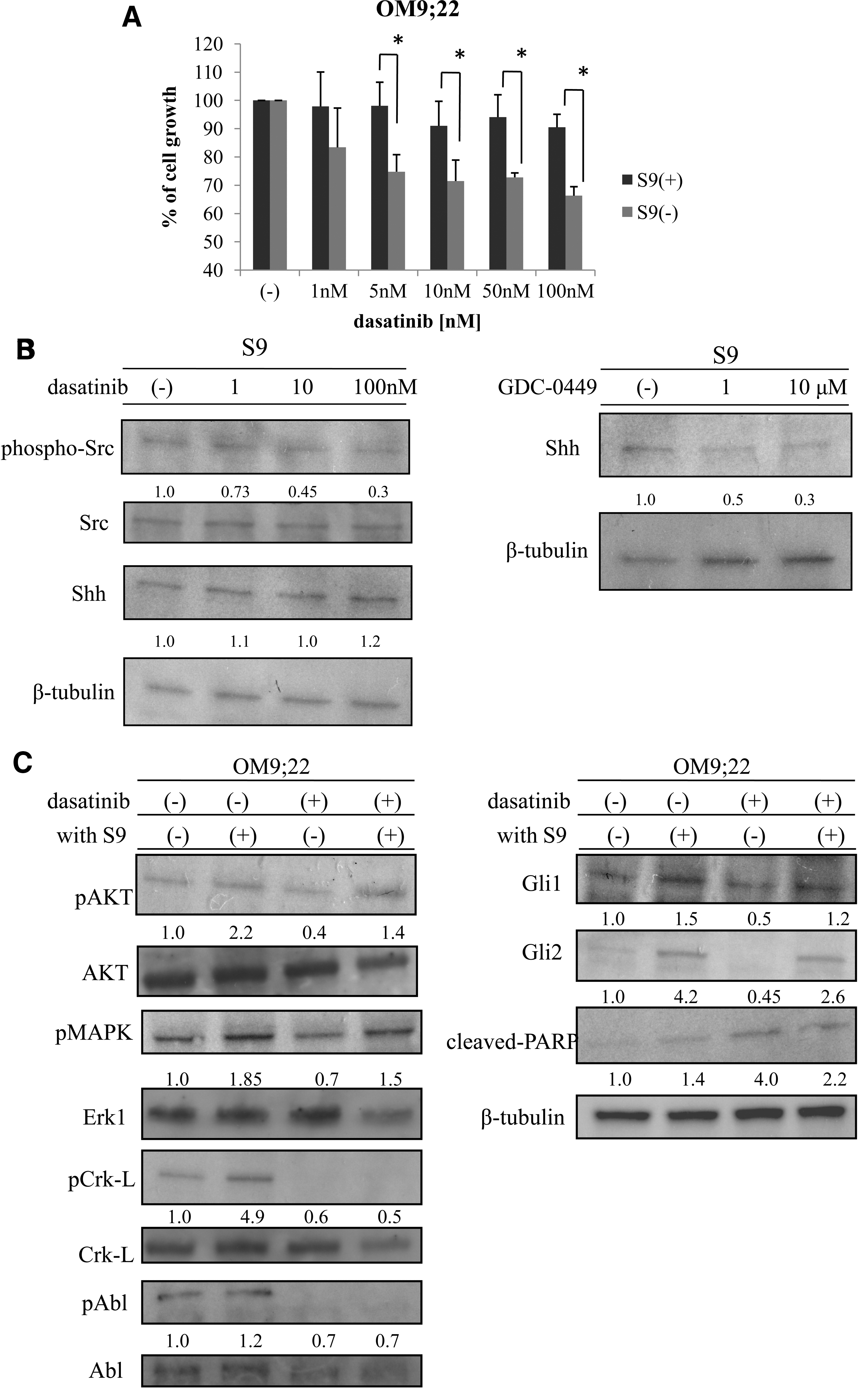
Cell proliferation inhibition of OM9;22 by dasatinib is reduced in the presence of S9 cells.
Intracellular signaling of OM9;22 cells in the presence of S9 cells
Since OM9;22 cell proliferation was increased in the presence of feeder cells, we next examined the intracellular signaling of OM9;22 cells in the presence of the S9 feeder cells with or without dasatinib treatment. Compared to the controls, phosphorylation of Akt, MAPK, Crk-L, and Abl was increased in the presence of S9 cells. The downstream targets of Hh signaling, Gli1 and Gli2, were also increased. The activated form of PARP was found after dasatinib treatment; however, PARP activity was reduced in the presence of S9 cells (Fig. 2C).
Efficacy of GDC-0449 on Ph-positive cell proliferation
We next investigated the efficacy of GDC-0449 on Ph-positive cell proliferation. OM9;22 cells were treated with GDC-0449 at concentrations of 100 nM to 10 μM, and a cell proliferation analysis was performed. After 48-h treatment with GDC-0449, OM9;22 cell proliferation was inhibited in a dose-dependent manner (Fig. 3A). Another Hh inhibitor, SANT-2, was also effective against OM9;22 cell proliferation, and GDC-0449 was also found to inhibit OM9;22 cell proliferation in the presence of S9 cells (Fig. 3A). In western blot analysis, Crk-L, which is the downstream target of BCR-ABL, was not inhibited by the presence of GDC-0449. However, cleaved PARP was detected, as was apoptosis of OM9;22 cells after GDC-0449 treatment (Fig. 3B, C). We also evaluated the GDC-0449 efficacy using Ph-positive primary samples. After exposure to 10 μM GDC-0449, the percentage of colony formation was reduced in the treated cells compared to the control cells (Fig. 3D). The inhibition rate of colony numbers of primary cells was 40%–60%, suggesting that the activity of the Hh pathway is modulated in Ph-positive leukemia cells. We also found that expression of Gli1, which is the downstream molecule of the Hh pathway, was reduced after GDC-0449 treatment in OM9;22 and primary samples (Fig. 3E).

GDC-0449 inhibits Ph-positive cell proliferation.
Effects of co-treatment with GDC-0449 and dasatinib on Ph-positive cells
We examined the efficacy of dasatinib and GDC-0449 combination treatment against OM9;22 cell proliferation. We found that the combination of GDC-0449 and dasatinib significantly enhanced antileukemic activity (Fig. 4A). We next examined this using primary samples. The clinical details of patients who provided samples are described in Fig. 4B. Mononuclear cells (2×105 mL−1) were treated with dasatinib and/or GDC-0449 at the indicated concentrations. We found that GDC-0449 and dasatinib combination treatment inhibited primary sample's cell growth (Fig. 4C). These results suggest that compared to each treatment alone, co-treatment with GDC-0449 and dasatinib has superior antileukemic activity against BCR-ABL-positive leukemia cells.
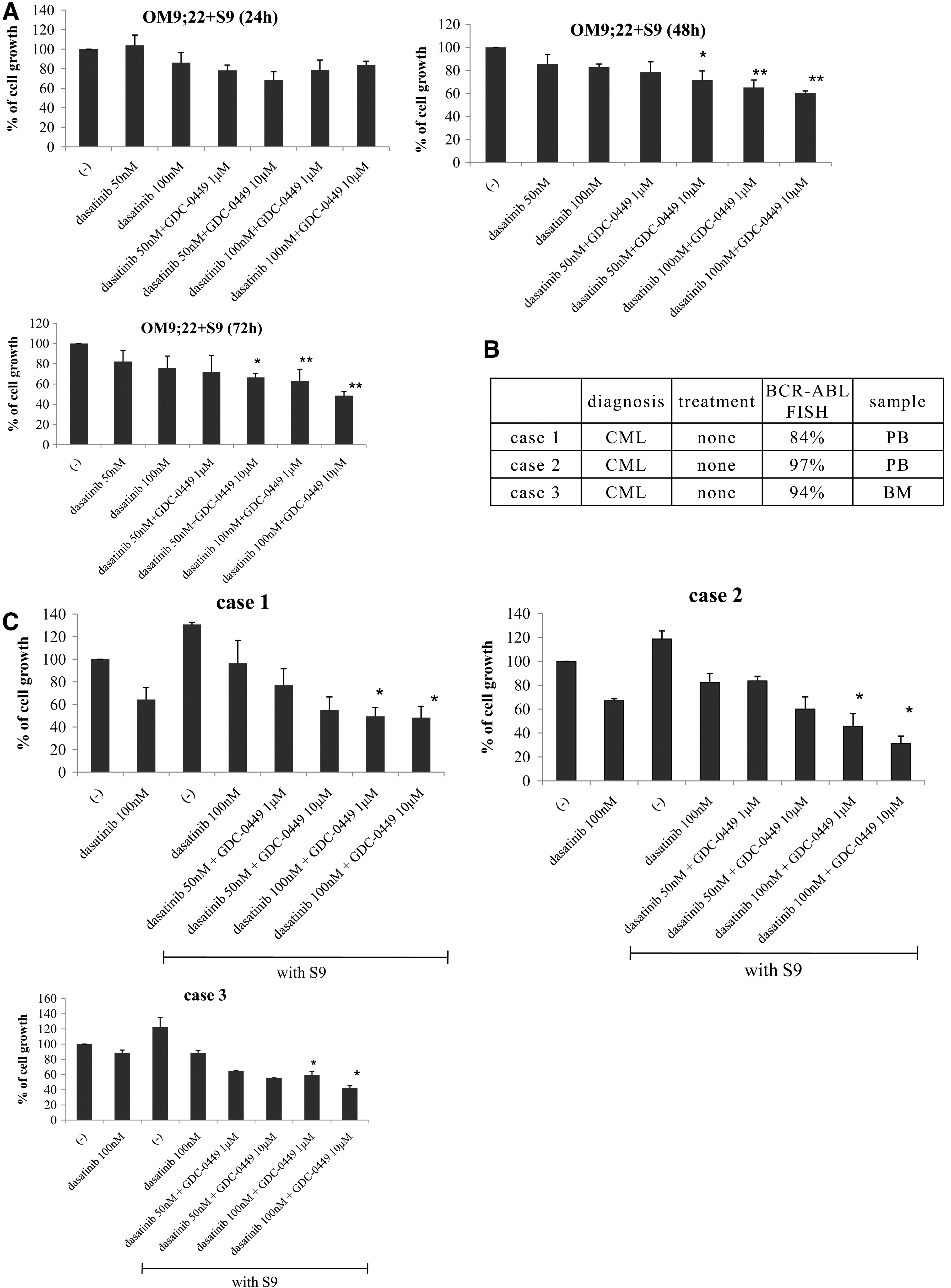
Co-treatment with GDC-0449 and dasatinib inhibits Ph-positive leukemia cell proliferation.
Antiproliferative activity after siRNA and shRNA of Gli1 and Gli2 transfection
To determine the effects of Gli1 and Gli2 in Ph-positive cells, we transfected the Gli1, Gli2, and control siRNA and shRNA into K562 cells. Immunoblot analysis revealed that Gli1 and Gli2 siRNA transfection caused >40% reduction of Gli1 and Gli2 protein levels after 48-h transfection. We then examined the intracellular signaling. In an earlier report, c-myc and cyclin D were downstream targets of Hh signaling [15]. The expression of c-myc and cyclin D was decreased after Gli1 and Gli2 siRNA transfection (Fig. 5A). The inhibition rate of c-myc and cyclin D was 30%–40%. The cell growth inhibition by dasatinib was significantly increased compared to the control siRNA transfectant cells (Fig. 5B). We found that Crk-L phosphorylation was not decreased; however, PARP activation by dasatinib was increased after cells were transfected with Gli1 and Gli2 siRNA (Fig. 5C). We next examined colony growth using the re-plating assay and found that the number of colonies was significantly reduced in the Gli1- and Gli2-shRNA-transfected K562 cells (Fig. 5D).

Antiproliferative activity after siRNA and shRNA of Gli1 and Gli2 transfection.
Discussion
In this study, we demonstrated that a Smo inhibitor, GDC-0449, induces cell growth inhibition in Ph-positive leukemia cells. Moreover, combined treatment with GDC-0449 and dasatinib significantly inhibited proliferation of the BCR-ABL-positive cell line OM9;22 and Ph-positive primary samples, respectively.
Recent studies suggest that the bone marrow niche is converted into an environment containing dominant signals that favor leukemia cell proliferation and growth [16]. Leukemia cell growth disrupts this normal bone marrow niche of hematopoietic progenitor cells and creates a tumor microenvironment [17]. Hematopoietic cells, including leukemia cells, are strictly regulated by bone marrow stromal cells. Intracellular signaling of leukemia cells is initiated by cellular adhesion and cytokine production by stromal cells [16]. Furthermore, Hh is secreted by bone marrow stromal cells and maintains progenitor cell populations.
The paracrine signaling in stromal cells might be critical to the Shh-mediated effects on Ph-positive leukemia cell progression. The present data suggest that Shh inhibits Ph-positive leukemia cell proliferation. Moreover, we demonstrated that the efficacy of dasatinib against Ph-positive cell proliferation decreased in the presence of Shh and feeder cells (S9). Furthermore, the Shh protein of S9 cells was not inhibited by dasatinib treatment, suggesting that Ph-positive leukemia cells were still supported by Shh in the presence of dasatinib. Several models denote the Hh pathway systems that exist in tumor environments [9]. Type I is ligand-independent, and type II is autocrine ligand-dependent, and type III is a paracrine or reverse paracrine signal system. Hh ligand is produced by stromal cells derived from the bone marrow, the spleen, or lymph nodes and maintains B-cell lymphoma and multiple myeloma cells [10,18]. Regarding the paracrine mechanistic (type III) actions of Shh, our results are consistent with previous findings that Shh paracrine signaling is required for Ph-positive cell proliferation.
Imatinib treatment significantly improves survival in patients with Ph-positive leukemia. However, ABL TKI does not alleviate Ph-positive leukemia. Cancer stem cells that propagate leukemia cells are resistant to ABL TKI. Bone marrow stromal cells also provide structural and physiological support for hematopoietic cells, including leukemia cells. Recent evidence demonstrates that aberrant reactivation of the Hh-signaling pathway contributes to tumor initiation and progression in various human malignancies, such as BCR-ABL-positive leukemia, and maintains cancer stem cells [11,19,20]. In particular, the loss of Smo, an essential component of the Hh pathway, impairs hematopoietic stem cell renewal and decreases CML induction by the BCR-ABL1 oncoprotein. Smo loss also depletes CML stem cells. The expression of constitutively active Smo increases the frequency of CML stem cells and accelerates CML development, suggesting that Hh signaling is involved in CML cell maintenance [11].
Hh signaling regulates the self-renewal and survival of cancer stem cells in different tumor types. Thus, targeted Hh pathway inhibition might have beneficial effects against tumors. In particular, an Hh-Gli inhibitor, that is, cyclopamine, restricts tumor growth and prevents recurrence without any side effects in mouse xenograft models [21]. GDC-0449 is reported to be a more potent and specific Smo inhibitor and is currently used in human clinical trials. The optimal use of Hh pathway inhibitors such as GDC-0449, alone or in combination with dasatinib, has not been examined until now. In this study, we demonstrated that GDC-0449 is a novel agent that induces antileukemic responses by inhibiting components of the Hh pathway such as cyclin D and c-myc.
The combined administration of an Hh inhibitor and dasatinib effectively inhibits Ph-positive leukemia cell proliferation, which is supported by feeder cells. Moreover, we have shown that combining Hh inhibitor with Abl TKI is very effective in overcoming the drug resistance of Ph-positive leukemia cells.
Footnotes
Acknowledgments
This work was supported by a High-Tech Research Center Project for private universities—a matching fund subsidy from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)—and by the University–Industry Joint Research Project for private universities—a matching fund subsidy from the MEXT. This work was also supported by Grants-in-Aid for Scientific Research from the MEXT.
Author Disclosure Statement
The authors declare no conflicts of interest.