
Abstract
Derivation of bone forming cells (osteoblasts) from human embryonic stem cells (hESCs) is a prerequisite for their use in clinical applications. However, there is no standard protocol for differentiating hESCs into osteoblastic cells. The aim of this study was to identify the emergence of a human stromal (mesenchymal and skeletal) stem cell (hMSC)-like population, known to be osteoblastic cell precursors and to test their osteoblastic differentiation capacity in ex vivo cultures and in vivo. We cultured hESCs in a feeder-free environment using serum replacement and as suspension aggregates (embryoid bodies; hEBs). Over a 20 day developmental period, the hEBs demonstrated increasing enrichment for cells expressing hMSC markers: CD29, CD44, CD63, CD56, CD71, CD73, CD105, CD106, and CD166 as revealed by immunohistochemical staining and flow cytometry (fluorescence-activated cell sorting) analysis. Ex vivo differentiation of hEBs using bone morphogenic protein 2 (BMP2) combined with standard osteoblast induction medium led to weak osteoblastic induction. Conversely, subcutaneous in vivo implantation of day 20 hEBs in immune deficient mice, mixed with hydroxyapatite/tricalcium phosphate (HA/TCP) as an osteoconductive scaffold, revealed bone and cartilage, and fibrous tissue elements after 8 weeks. These tissues were of human origin and there was no evidence of differentiation to nonmesodermal tissues. hEBs implanted in the absence of HA/TCP formed vacuolated tissue containing glandular, fibrous and muscle-like tissue elements. Conversely, implantation of undifferentiated hESCs resulted in the formation of a teratoma containing a mixture of endodermal, mesodermal, and ectodermal tissues. Our study demonstrates that hMSC-like cells can be obtained from hESCs and they can be induced to form skeletal tissues in vivo when combined with HA/TCP. These findings are relevant for tissue engineering and suggest that differentiated hEBs can provide an unlimited source for functional osteogenic cells.
Introduction
M
Several studies have demonstrated the possibility of inducing differentiation of hESCs through an intermediate 3-dimensional (3D) cell spheroid formation called human embryoid bodies (hEBs). hEBs mimic the structure of the early embryo and recapitulate many of the early embryonic developmental events, including gastrulation [9], which is important for proper specific germ line lineage differentiation. Thus, previous reports have demonstrated that hEBs create a suitable microenvironment to induce differentiation of cells to all 3 germ layers [5,10]. Additionally, both hematopoietic and mesenchymal tissues have been reported to develop in hEBs demonstrating enriched expression of lineage specific markers for example, hematopoietic CD34 and mesenchymal CD73 positive cells [6,11]. Using these markers, lineage progenitor cells were isolated and employed in further differentiation studies. However in these studies, early progenitor cells did not demonstrate lineage progression and required additional differentiation signals from murine bone marrow-derived OP9 stromal cells to allow further maturation into an osteogenic lineage. The ability of hEBs to support the development of osteogenic lineage cells has previously been reported, however, assessment of the phenotype of the resultant osteogenic cells was based on a limited number of in vitro differentiation markers [12,13].
In the present study, we examined the ability of long-term culture of hEBs to support the emergence of a MSC-like cell population and compared their differentiation capacity with bone marrow-derived MSC, in ex vivo cultures and in vivo implantation assays.
Materials and Methods
Cell culture
The hESC lines HUES-1 and HUES-9, generously gifted by D. Melton, Harvard Stem Cell Institute, [14] were maintained undifferentiated on mitomycin C (Sigma, M-4287) treated primary mouse embryonic fibroblast feeder cells seeded at 20 K/cm2 in dishes or plates (NUNC) precoated with 0.1% gelatin (Sigma, G1393). Undifferentiated hESCs were cultured in 85% KO-DMEM (Invitrogen, 10829-018) supplemented with 15% knockout serum replacement (SR; Invitrogen, 10828-028), 1% glutamax (Invitrogen, 35050-038), 1% MEM nonessential amino acids (Invitrogen, 11140-035), β-mercaptoethanol (0.1 mM) (Sigma-Aldrich, M7522), penicillin/streptomycin (5000 U/mL/5000 μg/mL, Invitrogen, 15070), 0.5% Human Serum Albumin (SSI, 8409), and 10 ng/mL basic fibroblast growth factor (bFGF; Invitrogen, 13256-029). Cells were passaged at a 1:6 ratio every 5 to 6 days using trypsin/EDTA (0.1%/1 mM, Invitrogen). The passages used in this study were between 19–30. Colony formation was visible within 2 to 3 days of passage. Daily half medium change was performed after the first 48 hours in culture. Human stromal (skeletal) stem cell line-telomerase reverse transcriptase (hMSC-TERT) that has been created and extensively characterized in our lab was included as a positive control [15,16]. The hMSC-TERT was cultured as described previously [15,16].
EB formation
To induce differentiation, hESCs were disaggregated using trypsin/EDTA (0.1%/1 mM, Gibco, 25300-0549), into small clumps containing 5–10 cells, and transferred to low adhesion plastic Petri dishes (Costar Ultra Low Attachment; Corning Life Sciences) in culture media without bFGF and β-mercaptoethanol (designated EB media). EBs formed after 3 days in culture, and media change was performed every 3 days using gravity sedimentation for 10 min at 37°C, 5% CO2. In addition, in selected cultures BMP-2 (10 ng/mL; R&D) in presence or absence of osteogenic induction mixture (for content see below) from day 0 of EB formation and for 20 days with media changes every 3 days.
Ex vivo differentiation into osteoblastic cells
hEBs at day 10 and day 20 were collected and trypsinized for 4–5 min to gain a single cell suspension. When hEBs were dissociated and plated very little cell death was noted. Few cells did not plate following trypsin-ethylenediaminetetraacetic acid (EDTA) treatment; it was believed that they died due to the duration of trypsin-EDTA treatment and mechanical dissociation of the cells. Fibronectin-coated 6-well culture plates were prepared in advance, and the single cell suspension was plated onto the fibronectin-coated plates and grown in EB media. For osteogenic differentiation, cells were seeded at a density of 104 cells/cm2 in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS). After 24 h (or when the cells were between 70%–80% confluent), osteogenic induction mixture containing 10 mM β-glycerophosphate, 50 μg/mL L-ascorbic acid 2-phosphate, and 10 nM dexamethasone was added. The media was renewed every 3 days throughout the study period of 20 days. A concentration of ascorbic acid of 100 μg/mL was also tried but we did not detect significant differences between this concentration and the standard concentration of 50 μg/mL.
Immunohistochemical- and cytochemical staining
Human EBs were collected at day 3, 5, 10, 15, and 20, washed once with phosphate-buffered saline (PBS), fixed for 10 min in 4% formaldehyde, washed thrice in PBS+/+, and pelleted (280 g, 5 min) in polystyrene conical tubes. The final pellet was aspirated dry before preparing a cell block using Shandon Cytoblock (Thermo Electron Corporation) according to the manufacturer's instructions. Cell blocks were embedded in paraffin and sectioned (4 μm) using conventional histopathological methods. Sections were stained for hematoxylin and eosin (H&E). Immunohistochemical staining was performed on hEBs and implants using DAKO En Vision+ and PowerVision according to the manufacturer's instructions. In brief, paraffin sections were incubated for 1 h at room temperature with primary antibodies, diluted in ChemMate Antibody diluent (S2022, DAKO), against Anti-human CD29 (Integrin beta-1 subunit; BioGenex; 1:20), CD31 (DAKO; 1:50), CD34 (DAKO; 1:20), CD44 (DAKO; 1:100), CD56 (Novocastra; 1:20), CD71 (Novocastra; 1:50), CD63 (Novocastra; 1:100), CD106 (Novocastra; 1:20), CD166 (Novocastra; 1:100), CD105 (DAKO; 1:100), Anti-human Ki67 (Dako; 1:400) β-III-tubulin (AH-diagnostic; 1:200), Oct-4 (Santa Cruz; 1:100), anti-human Desmin (Novocastra; 1:25), anti-human ASMA (1:200), anti-human OPN (1:100), anti-human vimentin (1:100), anti-human osteonectin (1:100), and anti-human collagen I (Chemicon; 1:1000). Sections were subsequently washed thrice in Tris buffered saline (TBS; 0.05 M, pH 7.4), then incubated for 30 min with secondary anti-mouse Ig/HRP-conjugated polymers (K4001; En Vision+, DAKO) and visualized with 3,30-diaminobenzidine tetrahydrochloride (DAB, S3000; DAKO) according to the manufacturer's instructions. Control experiments were performed without including the primary antibodies under identical conditions; they showed no evidence for staining (data not shown).
EB cells were grown to 80% to 90% confluence on 2-, 4-, and 8-well Permanox chamber slides (Nunc) coated with fibronectin (Sigma-Aldrich). The slides were fixed in 4% formaldehyde for 10 min and then washed thrice in PBS−/−. Nonspecific immunoglobulin G binding sites were blocked for 20 min with 5% bovine serum albumin (BSA) in PBS or serum-free block solution (Zymed). The cells were labeled with primary antibodies (mentioned above), incubated for 1 h, then incubated for 30 min with secondary anti-mouse Ig/HRP-conjugated polymers (K4001; En Vision+, DAKO), and visualized with 3,30-diaminobenzidine tetrahydrochloride (DAB, S3000; DAKO) according to the manufacturer's instructions.
Flow cytometry (fluorescence-activated cell sorting) analysis
Single cells from hEBs, at different time points, were harvested using 0.05% trypsin-0.53 mM EDTA (Invitrogen) and, after neutralization, resuspended in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS +/+, Invitrogen] with 0.5% BSA (Sigma, fraction V) at a concentration of 106 cells per mL. For each antibody, 105 cells were stained. Preconjugated antibodies for CD29FITC (ABCAM), CD44PE, CD63FITC, CD73PE and CD166PE, and isotype controls: IgG1-FITC and IgG1-PE, were obtained from BD Biosciences (San Diego,
Polymerase chain reaction
Total RNA from cell samples was isolated using the TRIZOL method according to the manufacturer's instructions. RNA quantitation was determined using a NanoDrop ND-1000 Spectrophotometer (NanoDrop). cDNA was synthesized from 1–2 μg of total RNA using a commercial revertAid H minus first strand cDNA synthesis kit (Fermentas) according to the manufacturer's instructions. Polymerase chain reaction (PCR) was performed at 60°C in HYBAID multiblock system from VWR. Primers used were Oct4: forward GAAGGTATTCAGCCAAAC, reverse CTTAATCCCAAAAACCCTG, Sox2: forward GTATCAGGAGTTGTCAAGGCAGAG, reverse TCCTAGTCTTAAAGAGGCAGCAAAC, MEOX1: forward AATCATCCAGGCGGAGAAA, reverse AAGGCCGTCCTCTCCTTG, Twist-1: forward GGGCCGGAGACCTAGATG, reverse TTTCCAAGAAAATCTTTGGCATA, OPN: forward CCAAGTAAGTCCAACGAAAG, reverse GGTGATGTCCTCGTCTGTA, Col-1: forward TGA CGA GAC CAA GAA CTG, reverse CCA TCC AAA CCA CTG AAA CC Runx2/cbfa1: forward CACCATGTCAGCAAAACTTCTT, reverse ACCTTTGCTGGACTCTGCAC, and housekeeping gene β-actin: forward ATTGGCAATGAGCGGTTCCG, reverse AGGGCAGTGATCTCCTTCTG.
Cytochemical staining
Staining for alkaline phosphatase (ALP): Cells undergoing osteoblast differentiation in osteogenic medium (os) containing (b-glycerophosphate (10 mM), L-ascorbic acid 2-phosphate (50 to 100 mg/mL), and dexamethasone (10 nM; Sigma-Aldrich)) for 21 days were stained for ALP as described previously [7] using incubation in an ALP substrate solution containing naphthol AS-TR phosphate (0.2 mg/mL in water) mixed with fast red TR (0.417 mg/mL in 0.1 M Tris buffer, pH 9.5) for 1 h at room temperature.
Alcian blue staining for chondrocytes
Sections of paraffin-embedded implants were stained with Alcian blue (Sigma) solution, pH 2.5, at this pH all the glycoproteins (neutral and acidic) will be stained blue.
Transplantation into NOD/SCID mice
Twenty-day-old hEBs were subcutaneously implanted under the dorsal surface of 8-week-old female NOD/SCID mice (NOD/LtSz-Prkdcscid). Equal amounts of hEBs were mixed with hydroxy-apatite/tricalcium phosphate ceramic powder (HA/TCP, 40 mg; Zimmer Scandinavia) and incubated overnight before transplantation on either side of the dorsolateral area of NOD/SCID mice. For each mouse, one implant contained hEBs in combination with HA/TCP and a second implant, on the contralateral side of the spine, contained the same amount of hEBs but without any scaffold. The transplants were recovered 8 weeks after transplantation, fixed in 4% paraformaldehyde, dehydrated in a series of alcohol gradients (70%–100%), and embedded in paraffin. Sections of 4 μm were used for histological examination.
Results
Patterning of mesoderm during hEB differentiation
To explore the possible induction of MSC-like cells during hESC culture, we characterized the surface marker profile of differentiating hESC during hEB formation. Undifferentiated hESC colonies were detached and grown in suspension for up to 20 days leading to the formation of EBs. During a 20 day period, the EBs exhibited morphological changes from small loose cell aggregates to densely compact bodies together with very few cyst-like structures, thus mimicking the developing fetal membranes. The vacuolated cysts were not included in further experimentation.
Immunohistochemical staining of paraffin sections of hEBs for the pluripotency marker Oct-4 and a number of mesodermal-lineage markers including endothelial, muscle and MSC markers were examined throughout the 20-day period. We observed that Oct-4 expression gradually decreased from hEB day 5 to a negligible expression at day 20 (Fig. 1A). The presence of Oct-4+ cells within 20-day-old hEBs (Fig. 1A) suggests heterogeneous differentiation within hEBs.
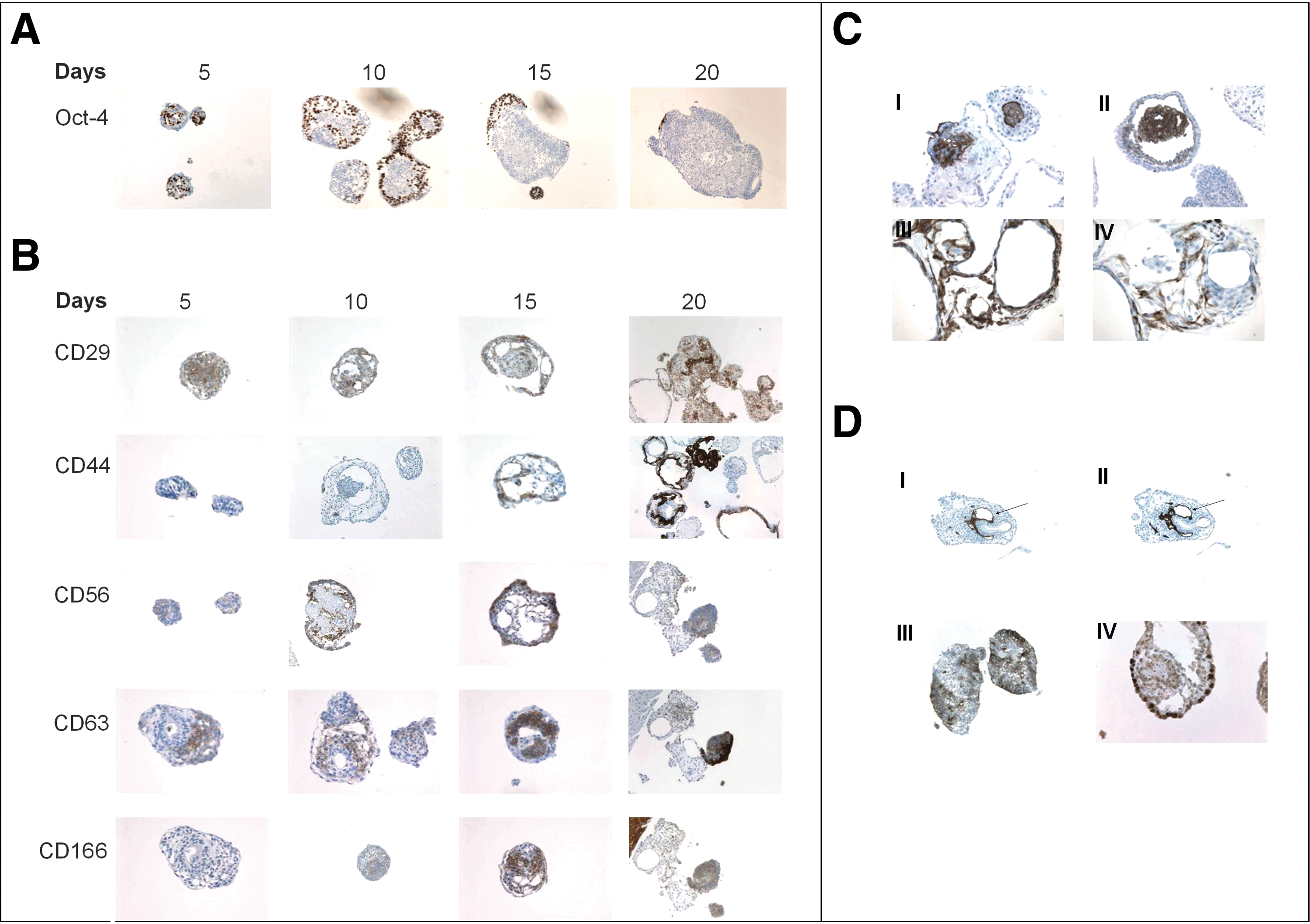
Differentiation of human embryonic stem cells (hESCs) as human embryoid bodies (hEBs) for 20 days.
We examined the expression of known characteristic MSC surface markers such as CD29 (Integrin beta-1 subunit), CD44 (hyaluronic acid receptor), CD56 (NCAM), CD63, CD105 (endoglin, SH2), CD106 (vascular cell adhesion molecule-1 (VCAM-1)), CD166 (AL-CAM), and vimentin; additionally, we employed a human bone marrow-derived MSC cell line (hMSC-TERT) as a positive control [16] (Fig. 1B). Human EBs stained positive for CD29, CD44, CD56, and CD71 (transferrin receptor) in day 10 hEBs and the number of positively stained cells significantly increased by day 20 hEBs (Fig. 1B). Staining for CD166 was weak prior to day 10, however, expression levels had increased by day 15 and day 20 (Fig. 1B). Further, we examined the presence of hematopoietic and endothelial markers that included CD31, CD34, CD45, CD105, and CD106 (Fig. 1C). The hEBs were found to be negative for CD45 while staining for CD34 (a marker of primitive human hematopoietic stem cells, endothelial and satellite stem cells) revealed few positive cells in day 15 hEBs, but the expression decreased at day 20. Positive staining for CD105 and CD106 was only detectable in day 20 hEBs (Fig. 1C, I and II). In day 15 hEBs, we observed the appearance of vessel-like structures that stained positive for the endothelial marker CD31 [platelet endothelial cell adhesion molecule (PECAM-1)] and co-localized with CD34 (Fig. 1D, I arrow, II). The staining for CD31 and CD34 did not co-localize with other MSC markers. To further verify that the hEBs contained stromal cells, staining for vimentin (the intermediate filament protein characteristically expressed in cells of mesenchymal origin; Fig. 1C, III) and desmin (a muscle-specific cytoskeletal protein found in smooth, cardiac, and skeletal muscles) were performed; and hEBs exhibited high expression levels of both markers at day 20 (Fig. 1C, IV). During the same time period, hEBs did not reveal any positive staining for osteoblastic markers including osteopontin, osteonectin, and osteocalcin (data not shown).
We observed that hEBs also contained very few cells derived from other germ-layers. Neural tube-like formation was observed in day 15 hEBs as evidenced by positive staining for β-III-tubulin (a neuroectoderm cell marker) that was detected in the small compact hEBs (Fig. 1D, III). Small amount of staining for HNF-1β (an endoderm lineage specific marker) was apparent in day 10 and day 15 hEBs, and it was restricted to few cells in day 20 hEBs (Fig. 1D, IV). When we stained day 20 hEBs with Ki67 a proliferation marker it revealed that almost all cells in the hEBs were proliferating (Supplementary Fig. S1; Supplementary Data are available online at
Quantitative expression analysis of mesenchymal cell surface markers by flow cytometry (FACS)
We quantified the number of MSC-like cells in differentiating hEBs using FACS analysis. Undifferentiated hESCs and whole hEBs were disaggregated into single cells and analyzed by FACS. A representative analysis performed on undifferentiated hESCs and hEBs at 10 and 15 days of culture is presented in Fig. 2A. Undifferentiated hESCs stained negative for most of the MSC markers except CD29 that was positive in 62%±15% of the cells. During 15 days of hEB differentiation the percentage of CD29, CD44, CD63, CD73, and CD166 positive cells increased significantly from an average of 62%, 2%, 8%, 0%, and 3% respectively in undifferentiated hESC to an average of 88%, 23%, 48%, 2%, and 78% (in all cases the standard deviation was±5%), respectively, based on 3 independent experiments carried out on 2 different cell lines (Fig. 2A). We included hMSC-TERT used as positive control (Fig. 2A). FACS analysis at day 20 hEBs was difficult to perform due to the high cellular density of hEBs and some cyst formation, making it difficult to maintain the cell surface epitopes due to enzymatic and mechanical load.

Surface antigen expression of hESCs and hEB
Effects of in vitro osteogenic induction on the differentiation of hESC-derived MSC-like cells
To enrich MSC-like cells evolving in culture, we incubated hEBs with 10 ng/mL BMP-2 and osteogenic induction mixture containing ascorbic acid, beta-glycerophosphate, and dexamethasone. Immunohistochemical staining did not show any significant difference between the treated and nontreated hEBs when we stained with MSC cell surface markers (CD44, CD56, CD63, CD106, and CD166) (Supplementary Fig. S3). However, induction with BMP-2 did increase the number of Brachyury (early mesoderm) and Sox-17 (early to definitive endoderm) positive cells at hEB day 5 indicating higher level of primitive streak formation (Supplementary Fig. S4B), thus suggesting recapitulation of a mesendoderm lineage rather than specifying for mesoderm alone.
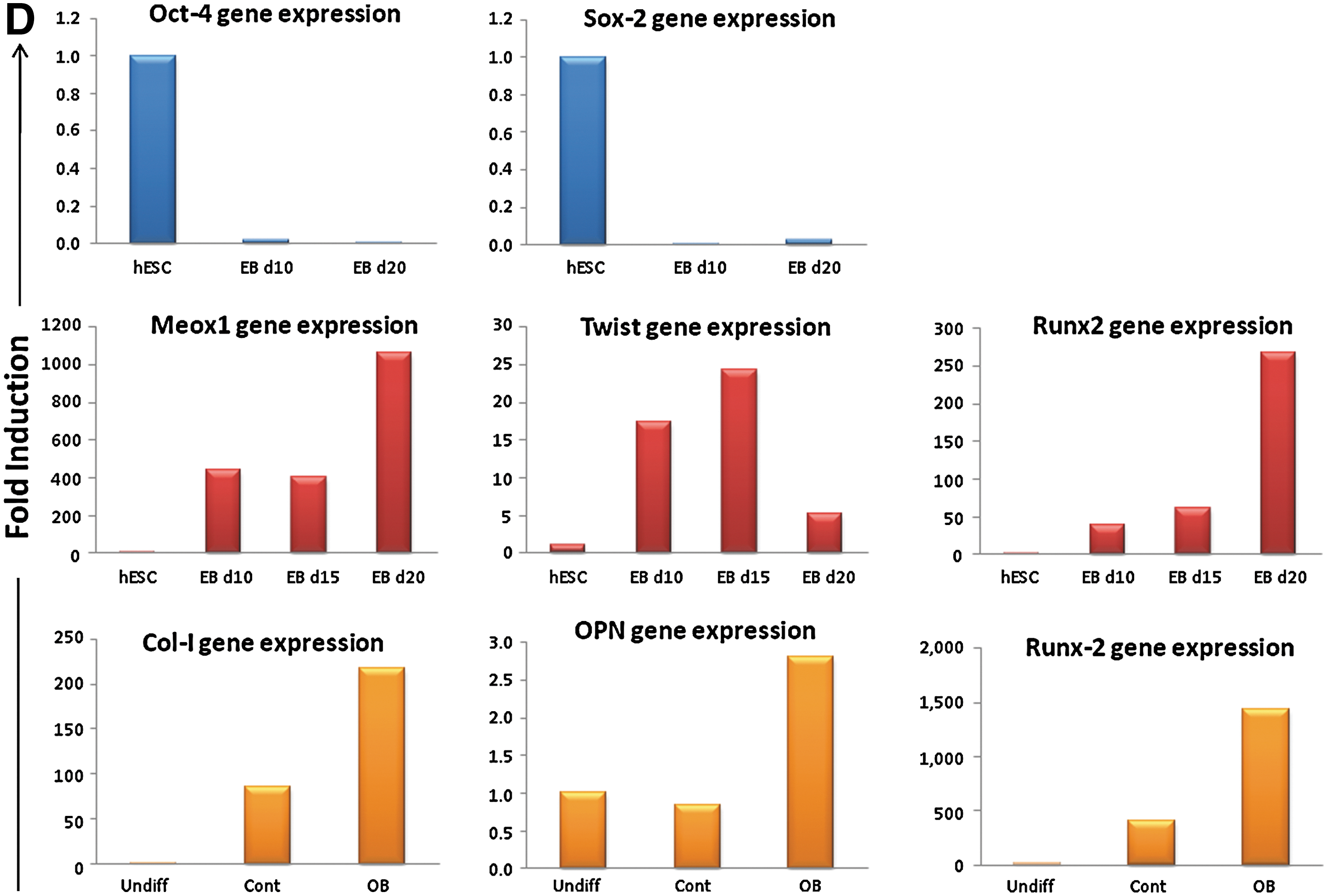
To test the ex vivo differentiation potential of hEB-derived MSC-like cells, day 10 and day 20 hEBs were trypsinized and cells plated onto fibronectin-coated cell culture dishes. After one passage the monolayer cells were tested for expression of mesenchymal cell surface markers. While grown as monolayer, the explanted cells maintained expression of MSC marker expression when cultured in growth media containing 10% FBS. Immunohistochemical staining demonstrated that the monolayer cells were more than 90% positive for CD29, CD44, and CD63, but they became negative for CD166 at this stage (Fig. 2B). Ex vivo standard osteoblastic induction medium did not lead to clear osteoblast differentiation and only cells obtained from day 20 hEBs exhibited positive staining for ALP activity indicating commitment to the osteoblastic lineage (Fig. 2C, III-IV). To verify the EBs differentiated phenotype, gene expression for Oct-4 and Sox2 was examined by reverse transcriptase-PCR (Fig. 2D, upper panel), and demonstrated that EBs at day 10 and day 20 lost the gene expression of pluripotent markers. Further, we examined gene expression of mesoderm specific markers such as Meox1, Twist, and Runx2, which were all highly upregulated (Fig. 2D, middle panel). Gene expression analysis of osteoblast differentiation markers showed that collagen I, osteopontin, and Runx-2 were also highly upregulated during differentiation in ex vivo culture (Fig. 2D, lower panel). Thus, indicating osteoblast differentiation induction. However, cellular outgrowths from EBs under osteogenic induction did not form ex vivo mineralized matrix (data not shown).
In vivo differentiation of hESC-derived MSC-like cells
MSCs are functionally characterized by their ability to form bone in vivo in the presence of the osteoconductive scaffold such as hydroxyapatite/tricalcium phosphate (HA/TCP) [17]. To test for the functional ability of hESC-derived MSCs, we implanted day 20 hEBs mixed with HA/TCP, subcutaneously in immune-deficient mice (NOD-SCID) for 8 weeks; cyst-like EBs that were observed in 20 day hEBs were not employed for in vivo implantation. As a control, hEBs from the same culture were implanted, subcutaneously, without HA/TCP scaffold into the contralateral side of the same mouse. In HA/TCP implants bone formation was evident and we observed the presence of calcified bone and bone marrow (Fig. 3, I). Osteoid and mineralized lamellar bone with embedded osteocytes were formed and stained positive for human-specific collagen type I (Fig. 3, III). The lamellar bone was covered by cuboidal osteoblasts and the mesenchymal tissue was infiltrated with hematopoietic cells (Fig. 3, II). The tissues stained positive for human specific osteopontin (OPN) (Fig. 3, IV). Staining for the cytoskeletal protein vimentin (Fig. 3, V) showed heavily positive areas around and close to the HA/TCP. In addition, hypertrophic chondrocytes and cartilage tissue, which stained positive for Alcian blue, were evident in implants with HA/TCP with prominent lacunae (Fig. 3, VII). Implants injected without HA/TCP formed big fluid-filled cysts and histological analysis of these cysts demonstrated that they contained differentiated cell types, mostly from the mesoderm germ-layer (Fig. 3, IIIV-IX). The cysts were generally well demarcated from the surrounding muscle and exhibited organized clusters of cells and primitive tissue structures, including fibrous tissue, smooth muscle, and cardiac muscle like cells (data not shown). The different tissue types formed by the cells implanted with or without HA/TCP are listed in Table 1. In contrast, hMSC-TERT formed only bone and bone marrow supporting stromal cells and no cartilage formation was observed (data not shown).

In vivo implantation of human EBs (hEBs) in immune deficient mice. hEBs grown in suspension for 20 days were mixed with hydroxyapatite/tricalcium phosphate (HA/TCP) scaffold and subcutaneously implanted into immune-deficient mice for 8 weeks
One+scored as 1/5 of the section of the implant is positive morphologically and or immunohistochemically, (+) scored for less than 1/5 positive staining,−scored for no staining seen.
ASMA, alpha smooth muscle actin; EB, embryoid body; HA, hydroxyapatite; H&E, hematoxylin and eosin; PAS, periodic acid Shiff.
Discussion
Skeletal tissue is composed of various types of cells, for example, osteoblasts, chondrocytes, adipocytes, and stromal cells. These cells originate, during embryonic development, from the mesoderm and, in the postnatal organism, from a stem cell population present among the stroma in bone marrow and are termed MSCs [18 –20]. Due to their ability to form skeletal tissues in vitro and in vivo, MSCs are being tested as a cell-based treatment for repair of nonhealed bone and cartilage defects. However, harvesting MSCs for autologous transplantation from bone marrow requires an invasive procedure with only a limited proportion of cells collected having the capacity of differentiation to the osteoblastic lineage [18]. Other restricting factors for the successful use of hMSCs in therapeutic applications are the limited number of MSCs that can be obtained in culture from a single donor due to poor, long-term proliferation caused by an in vitro replicative senescence phenotype [21]. Thus, there is a need to identify alternative sources of hMSCs. A TERT gene modified version of human bone marrow MSCs is established in house (hMSC-TERT) and hMSC-TERT was utilized as a model for primary MSCs due to its stable phenotype and similarity to primary MSCs [16]. Telomerase expression extends lifespan and prevents senescence-associated impairment of osteoblast functions [15]. In the present study, we demonstrated that MSC-like cells can be obtained from hESCs during 20 days culture as 3D-EB aggregates, the aim of long time in EB stage was to test the possibility of enhancing differentiation in 3D-microenviroment. The resultant cells expressed characteristic surface markers and gene expression of MSC and when implanted in vivo in combination with HA/TCP, the cells formed a mixture of mesodermal tissues including; bone, cartilage, and stromal cells.
We employed the 3D EB assay, which allows undirected differentiation of hESC, and has been traditionally utilized to initiate differentiation of ESC in a biologically relevant 3D context [9]. When mouse ESCs (mESCs) are allowed to differentiate into mouse EBs (mEBs), the cells become organized into an outer epithelial layer of primitive endoderm surrounding a population of epiblast-like cells [22 –24]. This inner population often forms cavities lined with polarized epithelium reminiscent of gastrulation [25]. However, EBs exhibits no evidence for polarized axis formation, and morphogenesis appears to proceed in a nonorchestrated fashion. Despite this limitation, differentiation of embryonic stem (ES) as EBs induces a physiological, well-conserved cascade of genes that govern the earliest events of gastrulation and germ layer formation is activated [26].
We have employed the hEB model for hESC differentiation to obtain hMSC population. Several different techniques have been reported that induce differentiation of hESC toward osteogenic lineages including EB formation [27 –29], monolayer differentiation [30 –33], through coculture with OP9 cells [3,6,31,32,34] or fibroblasts [35]. Although monolayer and coculture methods may increase homogeneity of cells during differentiation, they fail to recapitulate the morphological changes occurring in a more “natural” context that is provided during EB differentiation [33,36]. It is plausible that hMSC obtained through EB formation retain more “stemness” characteristics compared with MSC-like cells obtained through monolayer or coculture methods.
Previous studies have reported hESC differentiation into skeletal cells [5,7,37,38] and other lineage cells [39 –41] based on initial differentiation of hESCs as hEBs. For example, it has been reported that early mesoderm markers are expressed during hEB formation and that osteogenic cells can be induced by treating hEB with osteoblastic induction mixture [12,33,36,42]. Some studies of murine (m) ESCs have employed members of the transforming growth factor (TGF)-β superfamily (i.e., BMP's, Activin's, and TGF-β3) for induction and enrichment of stromal (mesenchymal) and osteogenic cell populations in mEBs [38,43,44] or high serum concentration (20%) as reported in a study by Käner et al. [45]. Our study demonstrates that using standard culture conditions, hEB assays are permissive for the development of an MSC-like phenotype. Interestingly, treating the hEB cultures with exogenous BMP2 or standard osteoblastic induction mixture did not increase the number of MSC-like cells based on surface marker expression that may be related to the low concentration employed. Kärner et al. [45] employed a higher concentration of 50–100 ng/mL BMP2 that resulted in enhanced osteoblastic differentiation of hESC-MSC. The possibility of enhancing hESC differentiation into osteoblastic cells through manipulation of ex vivo cellular microenvironment has been recently demonstrated, where a number of different combinations of media and serum gave rise to MSC-like cells that formed bone and cartilage upon in vivo implantation [46].
We have employed a number of surface markers characteristic for hMSCs, to monitor the development of hMSC-like cells within hEBs. Currently, there is no single marker that defines the identity of hMSCs [15,47]. Additionally, the surface markers, defining the phenotype of MSCs, exist in a number of other cell types including differentiated endothelial, pericytes, and muscle cells and thus it is plausible that the cells identified within the EBs as CD29+, CD44+, CD63+, CD56+, CD71+, CD73+, CD105+, CD106+, and CD166+ represent a mixture of MSCs and other mesodermal-lineage precursor cells including endothelial, pericytic, and myocytic lineage cells.
During human development, the first hematopoietic and endothelial precursors arise from extra-embryonic mesoderm and differentiate to form the blood islands in the secondary yolk sac. The close spatial and temporal development of these lineages within the blood islands provide the basis for the hypothesis that these cells arise from a common progenitor, a cell known as the hemangioblast [48,49]. It is thus plausible that the cells stained positive for CD34+, CD31+, and CD45−, may represent earlier hematopoietic and mesenchymal progenitors [5,6,34,50,51]. Further studies are needed for prospective isolation of these cells and further phenotypic characterization to determine their exact lineage.
Few studies have demonstrated the ability of hESC-derived osteogenic cells to form bone in vivo. However, markers defining hMSC phenotype in ex vivo cultures are not predictive for their in vivo phenotype [15]. Also, hESC-MSC positive for CD73, and STRO-1 exhibited weak differentiation capacity into osteoblasts or adipocytes [52]. We implanted hESC-derived MSC in a standard heterotopic bone formation assay [17,21,53] and demonstrated that hEBs, enriched for MSC, were able to form normal bone tissues. Moreover, we verified that the bone formed was of human origin since it stained positive with human specific extracellular matrix antibodies, such as human desmin, alpha smooth muscle actin, and collagen I. Our results demonstrate that the conditions of the current ex vivo differentiation assays are not optimal and thus further studies are needed to create an ex vivo microenvironment conducive to a more efficient differentiation.
HA/TCP scaffolds induced bone and cartilage formation in combination with hESC-derived stromal cells that is different from bone marrow-derived hMSCs where only bone is formed. Our data suggest that hESC-derived EB MSC cultures contain an osteo-chondroprogentior cell population. In support of this hypothesis, our group has recently isolated a chondroprogenitor population of hESC-derived MSC based on dlk1/FA1 as a novel surface marker [54]. Alternatively, hESC-derived MSC may form bone in vivo through endochondral bone formation-like mechanism that is different from the intramembraneous bone formation assumed to take place in case of bone marrow-derived MSC [7,55].
Differences exist in the tissue composition of the implants obtained from bone marrow-derived hMSC and hEBs. hMSC formed bone and bone marrow supporting stroma, whereas hESC-derived MSCs formed a number of tissues including bone, cartilage, and muscle cells and undifferentiated stroma. Additionally, some differentiated elements of epithelial and neuronal cells were present. While this is expected, due to the incomplete differentiation nature of EBs, it may also suggest that local microenvironment does not completely abolish the intrinsic differentiation capacity of the implanted hESCs and corroborates our previous study showing that implantation of undifferentiated hESCs in different in vivo locations did not affect the prevalence of differentiated tissues within the teratomas formed [56].
There is a concern that transplanting undifferentiated hESCs may result in uncontrolled cell growth and tumor formation [57,58]. In an implantation study where hESCs were differentiated to neuronal cells, the implanted cells formed tumors in mice due to the presence of undifferentiated cells [59]. In our study, no tumor formation was observed in spite of the fact that the implanted day 20 hEBs contained some Oct-4+ cells suggesting that the presence of pluripotency markers is not predictive of in vivo tumor formation and thus the teratoma formation potential is significantly reduced following in vitro differentiation of hESC.
While EB formation provides a biologically relevant context to induce hESC differentiation, the use of EBs as a method to study lineage specific differentiation has its limitations. EB-based differentiation is undirected and leads to incomplete differentiated phenotype. Thus, further studies for standardizing EB differentiation using different morphogens [38], combining EB formation and EB explants to obtain a homogenous population of cells [7] or to prospectively isolate EB lineage committed cells [60] should be explored as ways for obtaining homogenous, well-defined, differentiated, and biologically relevant osteogenic cell populations for clinical studies.
Footnotes
Acknowledgments
The authors wish to thank Ms. Lone Christiansen for excellent technical assistance and Mr. Nicholas Ditzel for animal handling and cell transplantation. This work was supported by grants from the Lundbeck Foundation and from the National Plan for Science Technology and Innovation (NPST) with grant ID: 10-BIO1304-02.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.