
Abstract
Throughout every pregnancy, genetically distinct fetal microchimeric stem/progenitor cells (FMCs) engraft in the mother, persist long after delivery, and may home to damaged maternal tissues. Phenotypically normal fetal lymphoid progenitors have been described to develop in immunodeficient mothers in a fetus-treats-its-mother paradigm. Since stem cells contribute to muscle repair, we assessed this paradigm in the mdx mouse model of Duchenne muscular dystrophy. mdx females were bred serially to either ROSAeGFP males or mdx males to obtain postpartum microchimeras that received either wild-type FMCs or dystrophin-deficient FMCs through serial gestations. To enhance regeneration, notexin was injected into the tibialis anterior of postpartum mice. FMCs were detected by qPCR at a higher frequency in injected compared to noninjected side muscle (P=0.02). However, the number of dystrophin-positive fibers was similar in mothers delivering wild-type compared to mdx pups. In addition, there was no correlation between FMC detection and percentage dystrophin, and no GFP+ve FMCs were identified that expressed dystrophin. In 10/11 animals, GFP+ve FMCs were detected by immunohistochemistry, of which 60% expressed CD45 with 96% outside the basal lamina defining myofiber contours. Finally we confirmed lack of FMC contribution to statellite cells in postpartum mdx females mated with Myf5-LacZ males. We conclude that the FMC contribution to regenerating muscles is insufficient to have a functional impact.
Introduction
D
Persistent fetal microchimeric stem/progenitor cells (FMCs) have been implicated in both protective [3,4] and pathogenic [5] roles in a range of diseases, yet most evidence to date is circumstantial. FMCs selectively home to damaged tissues where they are found in a higher frequency than in uninjured control tissues [6,7]. They can either be a part of the inflammatory infiltrate or integrated into the surrounding maternal tissue, displaying niche-appropriate phenotypes [4,8]. It is these end-point phenotypes that have been used to argue that FMCs have tissue regenerative properties. Although evidence for the latter is strong, there are few data demonstrating that FMCs functionally benefit the affected maternal tissue.
A number of features indicate that FMCs are likely to be stem and/or progenitor cells: their longevity, apparent homing capacity, fetal source, and phenotypic diversity. Transfer of a more primitive pluripotent progenitor, however, cannot be excluded [9], since transplacental traffic of fetal cells begins soon after implantation in both humans and mice [8,10]. There is evidence for the transfer and persistence of diverse multi- and unipotent fetal progenitors, including hematopoietic [1,3], endothelial [4,11], mesenchymal [12], and even neuronal [14] lineages. This suggests that FMCs could act as cell therapy agents delivered naturally through pregnancy, and that FMCs from a normal fetus might potentially ameliorate maternal genetic disease in a fetus-treats-its-mother paradigm. The effect of fetus-to-mother transfer of stem/progenitor cells might arguably be expected to produce a greater effect, given the developmentally immature, but translationally advantageous, characteristics of more primitive stem/progenitor cell populations [14,15].
Muscular dystrophies are inherited myopathies characterized by cycles of skeletal muscle degeneration and regeneration. The commonest, Duchenne muscular dystrophy, is associated with mutations in the dystrophin gene, which encodes a protein integral to muscle fiber stability [16]. In the main mouse model of Duchenne muscular dystrophy (mdx), transplantation of various stem cell populations has shown therapeutic promise [17 –19]. Muscle-resident stem cells or satellite cells have been obvious therapeutic candidates for muscular dystrophies [20], as they are activated during normal muscle repair [20,21]. Progenitors from distal sites such as bone marrow can also repair damaged muscles, although their contribution to repair is relatively small in comparison to the resident satellite cell population. We have previously shown that human fetal mesenchymal stem cells (MSCs) delivered to mdx fetuses in utero systemically engraft all muscle tissues displaying myogenic differentiation [17]. Moreover, allogeneic bone marrow transplant recipients have small numbers of muscle fibers that contain the donor nuclei [18,22].
In this study, we tested whether FMCs could act as muscle regenerative stem cells to improve dystrophin levels in maternal muscles that are dystrophin deficient. To maximize FMC recruitment to a specific muscle site and exaggerate the relatively mild phenotype of the mdx mouse, we generated an acute muscle insult in tibialis anterior (TA) muscles of parous mice using the myotoxin, notexin. We hypothesized that wild-type FMCs naturally acquired during gestation would preferentially target the more damaged muscles and elaborate sufficient functional dystrophin to ameliorate the muscle pathology in their mother.
Methods
Animals
C57BL/10ScSn-Dmd mdx (mdx) females (Animal Resources Centre) were serially bred with males hemizygous for the enhanced green fluorescent protein (GFP) inserted into the ROSA26 locus Tg(Gt(ROSA)26sor-EGFP)IIAble (ROSAeGFP) (JAX®) or Myf5nlacZ/+ (Myf5-LacZ) males [23]. After each delivery, we ensured that at least 1 GFP mouse and 1 female mouse could be identified in the litter. For functional controls, we used mdx females serially bred with mdx males. This control group allowed us to examine exclusively the effect in mdx of harboring wild-type FMCs. Never-pregnant or virgin animals were used as technical controls for qPCR assays. All mice were treated in accordance with institutional guidelines and ethical approvals for the care of experimental animals.
Notexin injury
Notexin treatment was performed as described by others [24] to amplify the mild pathology in the mdx mouse. In brief, 4 weeks after their last delivery, mdx mice received an injection of 10 μL of 10 μg/mL notexin in 0.9% saline (Latoxan) down the length of the TA. The contralateral TA was injected with 10 μL of saline. Seven days later when muscle regeneration peaks and new correctly localized dystrophin protein is seen in this model [24 –26], mice were sacrificed and TA muscles dissected.
DNA extraction and qPCR
gDNA was extracted from each TA using a QIAamp DNA mini kit (Qiagen) according to the manufacturer's instructions. For qPCR assays, we used previously described primers and a probe to amplify a specific 75-base-pair region of the GFP transgene [27] or a region of the SRY gene located on the Y chromosome (Applied Biosystems). In our model, the GFP transgene or SRY is present in a single copy in positive FMCs. To measure total gDNA in each reaction, we amplified the murine genomic sequence of the apolipoprotein b (apob) gene, present in 2 copies per cell. Amplification and detection were performed using an ABI 7700 Sequence Detection System with SDS v1.9 software (Applied Biosystems). Relative amounts of fetal DNA were calculated using the comparative ΔΔCT method with apob as the reference sequence.
Tissue processing
Dissected TA muscles were fixed for 2 h in 4% PFA and subsequently infused with 30% sucrose before cryoembedding. Infused muscles were mounted in an optimal cutting medium and frozen rapidly in isopentane cooled with liquid nitrogen. Frozen 7-μm sections were used for immunohistochemistry studies.
Immunohistochemistry
For staining of specific antigens, cryosections were permeabilized in 0.5% Triton-X-100 before blocking with 10% normal goat serum. Primary antibodies included 1 in 400 rabbit anti-dystrophin (Abcam, Cambridge, United Kingdom), 1 in 500 rabbit anti-GFP (Abcam) or 1 in 100 chicken anti-GFP (Invitrogen), 1 in 100 rat anti-mouse CD45 (Becton Dickinson), 1 in 200 chicken anti-mouse vimentin (Abcam), and 1 in 50 rabbit anti-laminin (Sigma-Aldrich). Secondary antibodies conjugated with Alexa-Fluor 568 or 488 (Invitrogen) were used for fluorescence detection. Nuclear staining was revealed in specimens mounted with a ProLong® Gold mounting medium containing DAPI (Invitrogen). Images were captured using an Axio Imager M1 (Zeiss) or an LSM 710 confocal microscope (Zeiss).
Scoring and quantification
To eliminate bias, all slides for dystrophin quantification and FMC engraftment were scored blind to the gestational history of the animals. Background green-specific autofluorescence, emitted by oxidative skeletal muscle fibers [28], was factored into the stringent criteria for analyzing engraftment of FMCs to prevent identification of false-positive cells. Slides were first scanned using a dual GFP/dsRED filter and selected cells emitted only green-specific fluorescence. To include these as FMCs, the selected cells also had to contain a nucleus and manifest a regular cell membrane. Cells considered as FMCs also had to emit GFP fluorescence at comparable levels to the mononuclear cells in muscle from the control ROSAeGFP animals. Finally, to be counted as true FMCs, cells had to have homogenous GFP staining under a high magnification (40×objective).
Statistical analysis
Analyses were performed using GraphPad Prism v5c software. Normally distributed data were evaluated by standard t-tests, while nonparametric tests, such as Mann–Whitney, were used on skewed data, and Fisher exact testing was used for categorical analyses. A P value<0.05 was considered significant.
Results
FMCs are specifically recruited to muscles after notexin injury
To investigate the role of FMCs in muscle repair, we generated a cohort of mdx female mice that had been serially pregnant with GFP+ve fetuses. The breeding strategy and pregnancy history are outlined in Fig. 1. To allow engraftment of pregnancy-acquired FMCs, mothers were housed for further 4 weeks after their last delivery. A local muscle insult using the myotoxin, notexin, was then employed [24] to amplify the continuous muscle regeneration that occurs in this mouse mutant. In addition to satellite cell-mediated regeneration, notexin injection also induces the rapid recruitment and differentiation of bone marrow-derived stem cells to the muscle, substantially increasing the proportion of newly formed fibers [18]. Notexin-induced damage was confirmed by hematoxylin and eosin staining (Supplementary Fig. S1; Supplementary Data are available online at

mdx breeding strategy and pregnancy history.
To examine whether FMCs are recruited specifically to notexin-damaged muscles, we amplified the paternally inherited GFP transgene [29]. GFP gDNA was detected in all postpartum mdx females that had produced GFP transgenic fetuses. GFP gDNA was detected more often in TA muscles exposed to notexin compared to saline injection (9/11 vs. 4/11, P=0.04 Fisher exact test). In addition, more GFP gDNA was detected in TA muscles of mdx females regenerating after notexin insult compared to their contralateral saline-injected limbs (P=0.02). Of note, although GFP gDNA could be detected in some saline-injected contralateral limbs, this did not reach statistical significance compared to never-pregnant notexin-treated control animals (Fisher exact=0.1) (Fig. 2A). The number of GFP+ve pups delivered by mdx females did, however, correlate linearly with the amount of the GFP sequence detected in notexin-treated limbs (Pearson's r P=0.01, R2=0.6) (Fig. 2B). The presence of fetal gDNA in mdx muscles was also confirmed using a qPCR strategy directed specifically against the Y-chromosome (Supplementary Fig. S2).
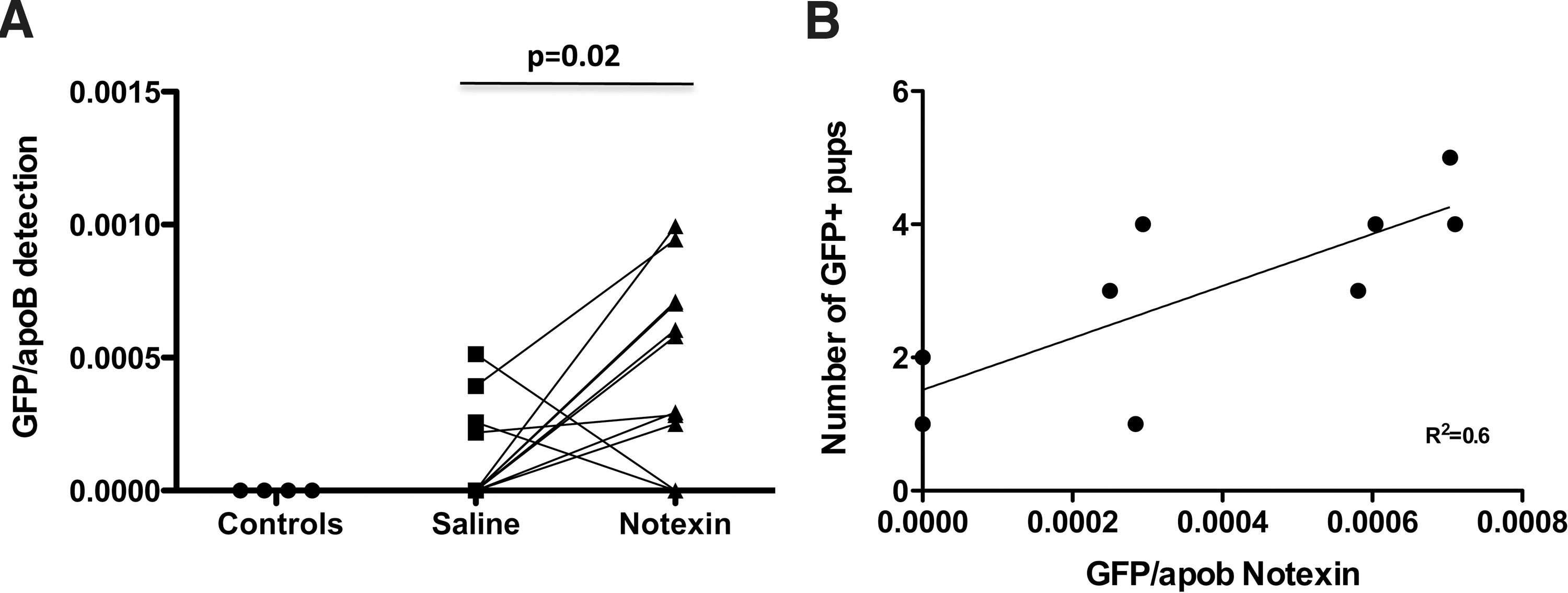
More fetal gDNA is specifically detected in mdx TA muscles after treatment with notexin.
FMCs do not contribute to the percentage of dystrophin+ve fibers
We next sought to evaluate whether naturally acquired FMCs could actively contribute to maternal tissue repair. Dystrophin is central for maintaining muscle fiber integrity during contraction, and without it, mechanical stress associated with contraction leads to muscle fiber necrosis, and eventually fibrosis. Idiosyncratic exon skipping produces multiple in-frame dystrophin transcripts that are translated into a functional protein [30]. These so-called revertant fibers are, however, too few in proportion (ranging from 1% to 6% of total myofibers) [31] to overcome the phenotype in either the human or murine disease (Fig. 3A). We therefore evaluated the efficacy of wild-type FMCs as opposed to dystrophin-deficient FMCs to contribute to dystrophin production in mdx females. Notexin-treated TA muscles were chosen for this investigation, as the day-7 timepoint reflects the expected peak for the production of new appropriately localized dystrophin protein [26,32], and mdx muscles previously transplanted with young satellite cells have been shown to have donor-derived dystrophin+ve fibers 7 days after notexin treatment [33]. Individual dystrophin+ve fibers were counted and normalized to the total number of fibers present in each field of view. The average percentage of dystrophin+ve fibers was calculated for entire sections at 2-sectional depths so that ∼3,000 muscle fibers were counted in each mouse. Since FMCs could potentially contribute to the satellite cell pool, we also compared the number of revertant fiber clusters between the 2 groups as a possible influence of FMC engraftment.
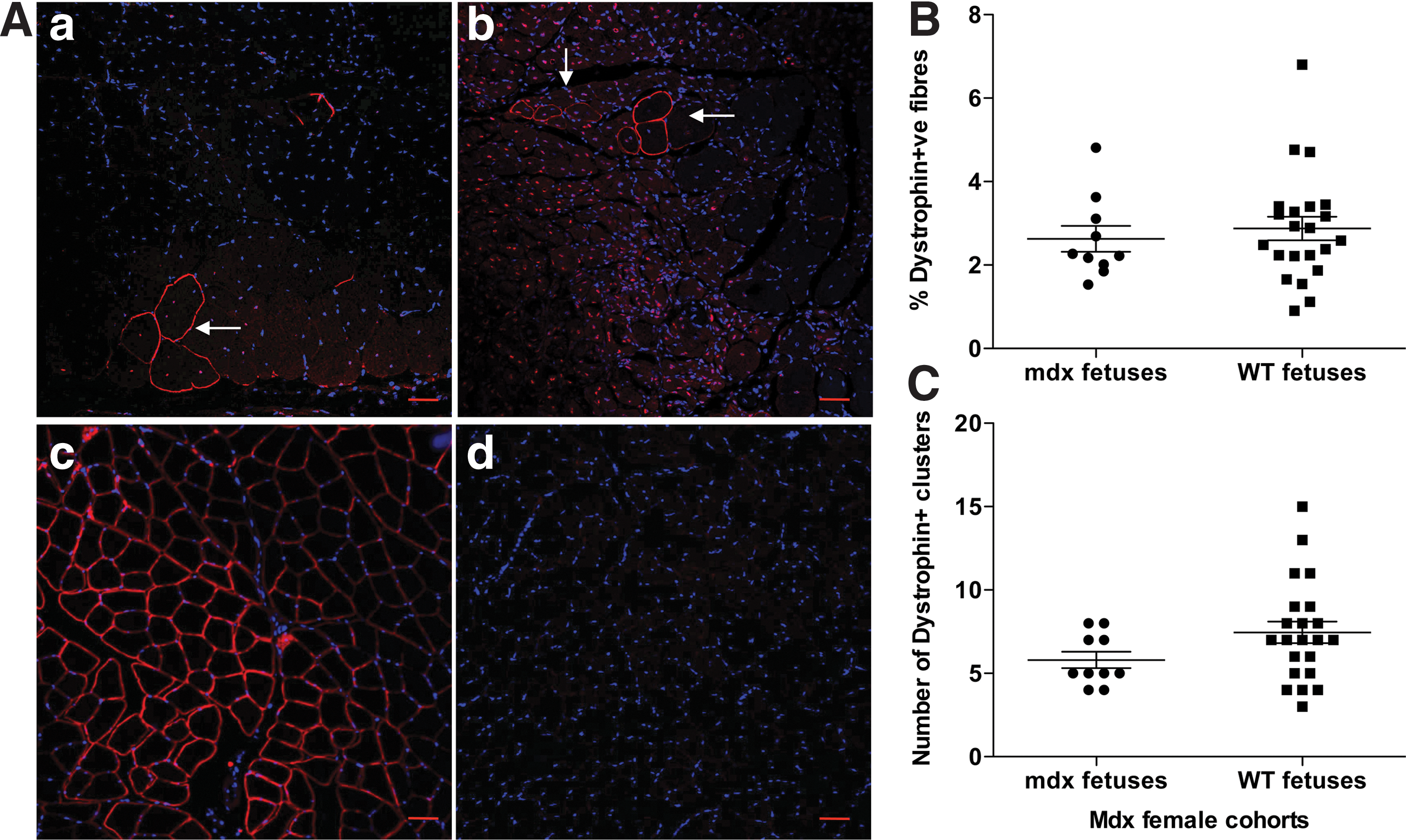
FMCs do not contribute substantially to dystrophin levels in regenerating TA muscles.
The percentage of dystrophin+ve fibers (2.6% vs. 2.9% P=0.5 Mann–Whitney) (Fig. 3B) or the number of dystrophin+ve clusters (5.8 vs. 7.5 P=0.12) (Fig. 3C) did not differ significantly between the 2 cohorts.
Dystrophin+ve fibers cannot be attributed to delivery of female offspring
The single-base mutation in mdx is X-linked, and as a consequence, loss of functional dystrophin occurs in homozygous females and hemizygous males. Under our breeding strategy, only FMCs from female fetuses had the possibility of producing wild-type dystrophin. Because our qPCR (Fig. 2B) and other recent data have linked the proportion of FMCs with the litter size [34], we undertook a subanalysis to explore any link between dystrophin+ve fibers and delivery of female fetuses. Linear regression analysis of dystrophin percentage to either the number of female fetuses (Fig. 4A) or the percentage of female fetuses (Fig. 4B) showed no correlation with dystrophin levels. Animals that cannibalized their pups were excluded for this analysis. There was also no correlation between the dystrophin percentage and qPCR detection of GFP gDNA (Fig. 4C), suggesting that these dystrophin+ve fibers identified were likely revertant and not the product of even small numbers of wild-type FMCs that engrafted.

Levels of dystrophin+ve fibers in regenerating TA muscles cannot be attributed to female progeny. Linear regression analyses comparing dystrophin percentage to the number of female fetuses delivered among those of known gender
FMCs recruited to notexin-treated TA muscles are part of the inflammatory infiltrate and do not express dystrophin
Although there were no significant outcomes to dystrophin levels from wild-type FMC in mdx muscles, we sought to examine FMC properties by analyzing their engraftment and immunophenotype. First, multiple sections of notexin-treated TA muscles were analyzed for engraftment. GFP+ve muscle fibers were never found, indicating a lack of myogenic capacity by FMCs (minimum of 12 sections per animal were scanned). However, single, mononuclear GFP+ve FMCs were detected in the inflammatory exudate using two different anti-GFP antibodies (Fig. 5A). Although few in number, GFP+ve FMCs were found in 10 of 11 animals (Fig. 5B), and in 36%, they were present in at least 2 consecutive slides (Fig. 5C). Approximately 60% (17/28) of GFP+ve FMCs identified expressed the pan-hematopoietic marker, CD45 (Fig. 5D, F), whereas costaining with GFP and vimentin revealed that 25% of GFP+ve cells (3/12) identified expressed vimentin (Fig. 5E G). Additional costaining with GFP and laminin revealed that GFP+ve FMCs were nearly always found outside the basal lamina (23/24) (Fig. 5F-a, F-b, G). However, an example of a single GFP+ve FMC contained within the basal lamina was identified and confirmed by confocal microscopy (Supplementary Fig. S3), suggesting their capacity to integrate in the appropriate stem cell niche. However, GFP+ve FMCs coexpressing dystrophin were never found (0/16) (Fig. 5F-c, F-d, G). To explore further whether FMC could definitively contribute to satellite cells, we repeated the experiments but instead crossed mdx females with Myf5-LacZ males that express the LacZ reporter in satellite cells. Using this approach, only Myf5+ FMC and therefore pregnancy-acquired satellite cells would be labeled. Myf5+ FMC could not be found in either saline or notexin-treated TA muscles from post-partum mdx females (Supplementary Fig. S4). These data suggest that FMCs in regenerating muscle do not have significant myogenic propensity.
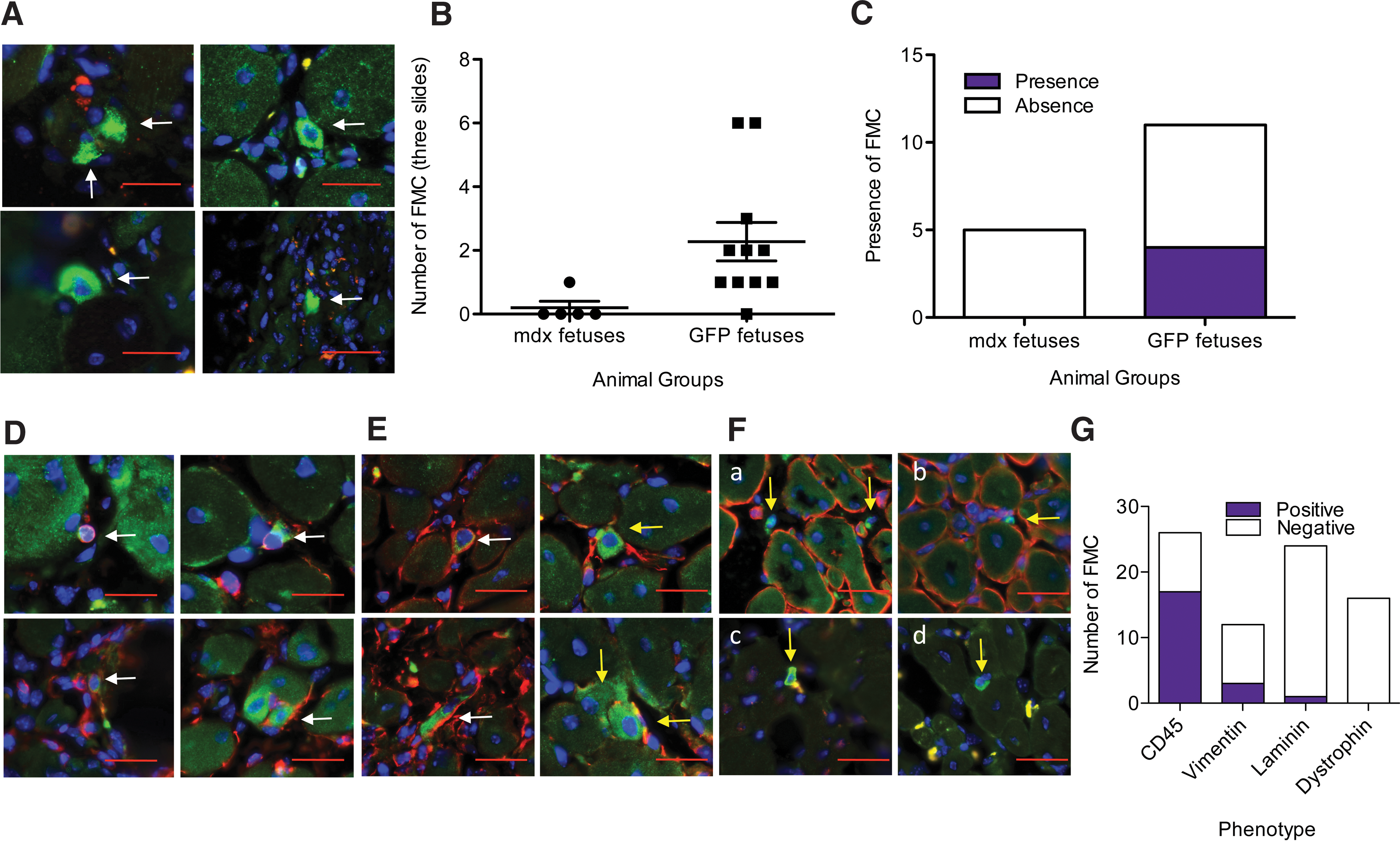
GFP+ve FMCs detected among maternal muscle fibers are mostly CD45+ve and outside the basal lamina.
Discussion
The contribution of FMCs to injured maternal tissue has been investigated in maternal heart muscle [35] and skin [4], but not as yet in skeletal muscle. Indeed, very few studies have explored the active contribution of persistent FMCs to tissue repair in postpartum mothers. Our data demonstrate that long-lived FMCs are selectively recruited to damaged muscle, in particular, the muscle undergoing acute inflammation in response to a myotoxin. The recruited FMCs found in damaged, but regenerating, muscles were associated with an inflammatory exudate, and 60% expressed the pan-hematopoietic marker, CD45. Moreover, ∼96% of recruited FMCs were found outside the basal lamina. FMCs expressing dystrophin were also never detected. Contrary to our original hypothesis, the acquisition of wild-type FMCs through multiple pregnancies did not significantly impact dystrophin expression in deficient mothers.
The lack of significant impact by FMCs was surprising, given that the mdx mouse undergoes continuous muscle regeneration, as indicated by the high proportion of centrally nucleated myotubes [36]. It is thought that these constant cycles of muscle degeneration–regeneration eventually lead to premature satellite cell exhaustion and consequent demise in this animal [37]. Since a local notexin insult is known to recruit bone marrow populations to aid in this repair process [38], we postulated that persistent FMCs (likely from the bone marrow compartment [8,29]) would be similarly recruited and participate in this repair process. Our results however cannot exclude the possibility that FMCs below detectable levels reside locally, proliferate, and become detectable in response to injury. Since most FMCs found in situ were not part of the muscle syncytia and were predominantly either CD45+ or vimentin+ (Fig. 5), we conclude that FMCs do not contribute to the fibrosis observed in the mdx mouse [39].
Although we observed a selective recruitment of what were likely bone marrow-derived FMCs (based on their expression of CD45), they had little functional impact. Despite chronic FMC therapy, the lack of functional contribution can arguably be attributed to the reduced disease severity characteristic of mdx mice. Their relatively mild phenotype can be partly explained by the compensatory effects of the dystrophin-like gene, utrophin, as administration of utrophin alone is sufficient to restore the muscle function in double-dystrophin/utrophin-deficient mice [40,41]. The presence in mdx of a ubiquitous sugar residue, Neu5Gc, the synthesis of which has been lost in human cells, also reduces mdx muscle pathology. The introduction of this human-specific mutation in mdx animals results in a more severe phenotype that emulates better the human disease [42]. It is thus conceivable that the mdx muscle pathology may not be severe enough to facilitate engraftment of FMCs.
An alternative, and more likely, explanation for the limited impact we observed could be the relative infrequency of persistent FMCs among maternal cells [29]. Despite maximizing the dose of FMCs acquired by mothers through serial breeding [29], their proportion was small (0.001%) compared to maternal cells. Moreover, the dystrophin mutation in mdx is X-linked [43], limiting potential benefits to FMCs acquired from female fetuses, and thus 50% of the progeny (regardless of their GFP expression). Notwithstanding this, we found no correlation between the number of female progeny and either the presence of fetal gDNA or dystrophin levels, rendering this a less likely reason for the little to no effect from FMCs.
Transfer and engraftment of FMCs that are functional lymphopoeitic [3], endothelial [4,11] and neuronal [13] progenitors have been demonstrated previously. Whether FMCs include a functional myogenic precursor remains unclear. Consistent with FMC phenotypes reported in other studies [2,44], the majority of FMCs identified in this study were hematopoietic. Despite previous reports where hematopoietic cells have beneficially contributed to muscle differentiation during repair [18], they did not produce this outcome in our study.
Bone marrow from postpartum women contains a small population of fetal mesenchymal stem cells (MSCs) with a known myogenic differentiation capacity [12]. Although we successfully found FMCs expressing the mesenchymal marker, vimentin, they were probably too few in proportion (3/12 FMCs) to have a significant influence on muscle pathology. Even 0.7% engraftment of fetal MSCs after intrauterine transplantation did not ameliorate disease severity in mdx [17]. Moreover, we did not find FMCs robustly expressing proteins associated with muscle differentiation or integration. Despite using notexin to amplify the muscle regeneration and stimulate production of dystrophin [32], FMCs expressing dystrophin were never found (0/16). Furthermore, only one FMC (1/24) was detected within the basal lamina. The basal lamina is important for coordinating satellite cell and fibroblast positioning and differentiation during muscle regeneration [45]. Although we found an example of a GFP+ve FMC in this area, which is a known satellite cell niche [20], this was undeniably rare. Additionally, we also demonstrated the absence of Myf5+ FMC in post-partum mdx, suggesting a lack of contribution by FMC to satellite cells. Together these data suggest that FMCs do not include a functional myogenic precursor.
Conclusion
The functional contribution of FMCs to women's health has long-generated debate. In this study, the transfer, persistence, and preferential engraftment of FMCs did not ameliorate disease manifestations in murine mothers with muscular dystrophy. This may reflect model specific limitations of muscular dystrophy or that FMC numbers at least in these experiments were insufficient to contribute actively to tissue repair. Notwithstanding this, we report the use of repeated mobilization of FMCs into the maternal circulation during pregnancy as a proxy method to test the effect of chronic stem/progenitor cell administration to treat maternal disease.
Footnotes
Acknowledgments
This work was funded by the National Health & Medical Research Council in Australia, project grant no. 569822. The authors are also grateful for the Myf5-LacZ transgenic mice, kindly provided by professor Peter Zammit.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.