
Abstract
The increasing use of mesenchymal stromal cells (MSC) in clinical cellular therapy requires a safe and controlled production process compliant with Good Manufacturing Practice guidelines. Pooled blood group AB human serum (HS) has been used to replace fetal bovine serum (FBS), critically rated by the regulatory agencies, since it can support the expansion of adipose tissue-derived mesenchymal stromal cells (ASC). However, it remains unknown whether the choice of serum affects application-relevant characteristics of ASC. A microarray-based screen has revealed differentially expressed adhesion and extracellular matrix-associated molecules in HS- and FBS-ASC. Since cell therapy relies on the cells' efficacy to home and engraft, HS- and FBS-ASC were compared by analyzing adhesion, migration, and transmigration as well as short-term homing in vivo. HS-cultivated ASC demonstrated a higher adhesion to plastic, but reduced adhesion to extracellular matrix molecules, that is, laminin, and to endothelial cells both under static and flow conditions. Migration and transmigration assays confirmed the attraction of ASC by the tumor conditioned medium irrespective of the supplement. Coinjecting differently labeled HS- and FBS-ASC into nonobese diabetic, severe combined immunodeficiency mice revealed reduced numbers of HS-ASC in lungs and liver. This has been interpreted as reduced capillary entrapment. Our data indicate that varying the serum supplement may alter application-relevant characteristics of ASC, such as adhesion, as well as lung entrapment after infusion. Appropriate injury models and further molecular analyses are required to provide mechanistic insight into the differential effects of HS versus FBS on ASC cultures.
Introduction
The translation of experimental protocols into clinical manufacturing protocols for the therapeutic use of MSC demands a defined production process according to the Good Manufacturing Practice principles [6,7]. Fetal bovine serum (FBS) containing growth factors, nutrients, and attachment factors is commonly used to supplement MSC cell culture media [8]. As MSC are known to internalize xenogenic proteins, the use of FBS-containing media has been discussed critically regarding previous diseases and immunological reactions [9 –12]. Moreover, clinical failures could be attributed to FBS-specific antibodies [9,13].
In an attempt to define surrogates for FBS, bridging the time until a fully chemically defined medium has been developed, we have previously identified pooled human AB serum (HS) as an appropriate alternative supplement to FBS to expand ASC [14 –16]. However, reduced cell size, different growth pattern, and adhesion of ASC cultivated in HS-containing media have been evident [14]. We consistently detected a reduced expression in a cluster of adhesion and extracellular matrix-associated molecules in HS-ASC [15]. Since migration, adhesion, and interaction with extracellular matrix molecules are prerequisites for efficient cell therapy, we asked whether the cell culture supplement differentially affects homing of ASC.
For the therapeutic use of MSC, their capacity to home into the desired target tissue is pivotal, especially when cells are injected into the circulation [17 –19]. Thereby, cells have to arrest within the vasculature and extravasate into the target tissue. Adhesion to endothelium, transmigration, interaction with the local extracellular matrix and chemotactic migration are mechanisms involved in this process, though the key players involved in MSC homing have not been yet identified in every aspect [19 –22]. Mimicking these steps in vitro, we compared ASC cultivated in FBS and HS with respect to adhesion to plastic, the extracellular matrix molecule laminin, and endothelial cells. Migration and transmigration toward tumor conditioned medium (TCM) as a potent MSC chemoattractant were addressed as well as short-term in vivo homing after infusion into nonobese diabetic, severe combined immunodeficiency (NOD/SCID) mice.
Design and Methods
Isolation and culture of adipose tissue MSC
After approval of the local ethics committee (2006-192N-MA, 2009-210N-MA) and having received informed consent, human adipose tissue of 12 female donors, aged 25 to 64, was obtained by elective tumescence liposuction of the abdomen and hip/thigh region. Lipoaspirates were processed as described using the FBS- or HS-supplemented medium [4,14]. ASC were maintained under these two culture conditions with twice-weekly media changes. At a confluence of 70–80%, cells were passaged using trypsin-ethylenediaminetetraacetic acid (EDTA) (PAA) and either replated at a density of 200 cells per cm2 for all passages or used for experiments. Cells from passages two to five were analyzed. Cell numbers, viability, and cell size were determined using a CASY® cell counter. Cells were characterized as MSC as described in detail in [4,14]. Within each of the subsequently described experiments, ASC pairs, thus being cells from the same donors and cultivated in both supplements, were directly compared with each other.
Preparation of supplements and media
ASC were cultured with either 10% FBS (preselected to favor outgrowth and expansion of ASC meeting the following criteria: plastic adherence, immune phenotype, and mesodermal differentiation potential [23]; Sigma-Aldrich) or 10% HS in the Dulbecco's modified Eagle's medium–low glucose (DMEM-LG; Lonza) with 4 mM L-glutamine (PAA) and 0.05 U penicillin/0.05 μg streptomycin (PAA). HS was prepared as previously described [14] from voluntary healthy regular blood donors, with blood type AB, tested for the presence of irregular antibodies. Blood samples were tested for sterility and absence of viral infections as specified by the German authorities. In brief, serum from whole-blood donations was centrifuged at 2000g for 15 min, and then aliquoted and frozen at −30°C. After thawing, HS of 5 donors was pooled and sterile filtered through 0.2-μm pores (Nalgene filtration device; Nunc). Different HS pools have been used.
Human umbilical vein endothelial cells
Human umbilical vein endothelial cells (HUVEC) were isolated according to ref. [24]. T25 culture flasks (Greiner) and the Endothelial Cell Growth Medium (EGM-2) (Lonza) were used for cultivation.
Flow cytometric analysis
FBS- and HS-ASC pairs were harvested using trypsin/EDTA for flow cytometric analysis at a confluence of 70–80%, unless otherwise stated. To analyze growth factor receptors, ASC were detached using 0.5 mM EDTA on ice. After washing with PBS, 1×105 FBS- and HS-ASC per tube were incubated with the human FcR blocking reagent (Miltenyi Biotec) for 15 min, and then stained in a cell wash (BD Pharmingen) with antibodies listed in Table 1 and incubated for 20 min at 4°C. After two washing steps, cells were incubated for 10 min with 7-AAD to exclude dead cells (7-aminoactinomycin; Beckman Coulter). Stained cells were analyzed using a fluorescence-activated cell sorting (FACS) Canto II (BD). Intracellular staining of CXCR-4 and CXCR-7 has also been checked in 3.5% formaldehyde-fixed (Roth) and 10% saponin-permeabilized (Sigma-Aldrich) ASC.
The data represent the percentage of positive cells and mean fluorescence intensity (MFI) for the indicated cell surface marker, determined by flow cytometry.
Significant differences (P<0.05) in % positivity marked in bold.
n=12, except a n=4; b n=3.
Photo multiplier tube (PMT) settings differ between fetal bovine serum (FBS) and human serum (HS) due to differing autofluorescence. MFI values have been corrected against unstained controls. Isotype controls have been checked to yield similar fluorescence values like unstained controls. Significance levels have not been calculated due to the different PMT settings.
Adhesion under static conditions
Adhesion to plastic, laminin, and HUVEC, either not stimulated or stimulated with tumor necrosis factor alpha (TNFα) or histamine was assessed. The test was performed in a 96-well plate (hydrophilic plastic for adherent cells, CellStar, Greiner), each condition in triplicate. For investigating the adhesion to laminin, wells were coated with 5 μg/cm2 laminin (from mouse Engelbreth-Holm-Swarm sarcoma, Roche Diagnostics) and incubated for 45 min at 37°C. For investigating the adhesion to a HUVEC monolayer, wells were coated with 10 μg/mL human fibronectin (Sigma-Aldrich) for 45 min before plating HUVEC (2×104 cells per well). After at least 48 h, HUVEC monolayers were optically checked for confluence (Zeiss Axiovert 200; Carl Zeiss). To enhance endothelial adhesiveness, HUVEC were either stimulated for 16 h with 100 ng/ml TNFα (Sigma-Aldrich) or for 15 min with 50μM histamine (Sigma-Aldrich). ASC pairs were trypsinized, washed, and stained with 5 μM Cell Tracker Green-5-chloromethylfluorescein diacetate (CMFDA; Molecular Probes). After staining, 1×105 ASC/mL in Roswell Park Memorial Institute (RPMI) 1640 (w/o phenol red) with 0.5% bovine serum albumin (BSA) was prepared and 100 μl were added per well. All wells were washed with warm RPMI before adding ASC. Wells were incubated at 37°C, 5% CO2 for 30, 60, or 90 min (60 min in subsequent assays). Confluence-dependent expression of integrins, including the changed adhesion capacity to laminin was assessed for 3 ASC samples trypsinized at 50%, 80%, and 100% confluence. Additionally, integrin binding sites were blocked in RPMI containing 0.5% BSA with 10 μg/μl anti-integrin α6 (CD49f; GoH3, BD), 40 μg/μl anti-integrin β1 (CD29; 6S6, Chemicon), and 20 μg/μl anti-integrin β4 (CD104; ASC-3, Chemicon) for 30 min at 37°C and 5% CO2 before applying the cells to laminin-coated wells.
Total and retained cell fluorescence before and after washing with warm RPMI 0.5% BSA was measured in triplicate in a fluorescent plate reader (Infinite 200; Tecan) at 485 nm excitation and 520 nm emission. Titration curves verified the linear correlation between fluorescence intensity and cell concentration. Wells filled with RPMI 0.5% BSA, but without fluorescent cells were used as the blank. Results were calculated based on the percentage of retained to initial total fluorescence.
Integrin signaling: immunoblot and immunofluorescence
Integrin-mediated adhesion to extracellular matrix molecules is known to activate a signaling cascade increasing tyrosine phosphorylation of focal adhesion kinase and paxillin recruited to focal adhesions [25]. For immunoblot analyses, 10 cm2 dishes (Falcon; BD) and for immunofluorescence 8-well chamber slides (Nunc) were coated with 5 μg/cm2 laminin and 1×106 or 1×104 ASC, respectively, were added (n=3). After 10, 20, 30, and 60 min, nonadherent cells were discarded by aspiration. For immunoblot, adherent cells were lysed on ice in a complete radioimmuno-precipitation assay (RIPA) buffer (phenylmethanesulfonyl fluoride [PMSF], sodium orthovanadate solution and protease inhibitor cocktail solution added, Santa Cruz). The protein concentration was determined using Pierce BCA Protein Assay (ThermoScientific). Protein samples (10 μg total protein, confounded by the laminin coating) were prepared in the Sample Loading Buffer (Fermentas), separated on 10% Bis-Tris gels, and transferred onto a polyvinylidene fluoride membrane (GE Healthcare). Anti-FAK (rabbit polyclonal, clone C-20; 1:200, Santa Cruz), anti-p-FAK (mouse monoclonal, clone 2D11, 1:400, Santa Cruz), and anti-p-Paxillin (mouse monoclonal, clone A-5, 1:200, Santa Cruz) was dissolved in the same buffers used for 2h blocking: Tris-buffered saline (Roth), 0.05% Tween-20 (Merck) with either 5% skim milk powder (Roth), 5% BSA (Applichem), or 1% BSA, respectively, and incubated overnight. Secondary antibodies, affinity-isolated polyclonal goat anti-mouse/HRP (P0447; 1:1000) and goat anti-rabbit (P0448; 1:2000, both Dako), were incubated for 2 h. All procedures were performed at 4°C. Bands were detected using an enhanced chemiluminescence reagent (Amersham™ ECL Select Western Blotting Detection Reagent, GE Healthcare). Each blot was normalized to glyceraldehydes 3-phosphate dehydrogenase expression (GAPDH, sc-47724, 1:100000, Santa Cruz) using the ImageJ gel analysis software tool (ImageJ 1.43u). As a positive control, Hela cells were serum starved for 2 h, and then the FBS-supplemented medium and 100 μM pervanadate added (Applichem, data not shown)
For immunofluorescence, adherent cells were fixed 3 min in 3.7% formaldehyde, permeabilized 3 min in 0.5% Triton-X-100 (Sigma-Aldrich), washed 3 times, blocked in 1% BSA/PBS/0.1% Tween 20, and stained overnight at 4°C using the antibodies listed above at a dilution of 1:50. After three washes, Alexa Fluor 488 goat anti-mouse IgG (H+L), highly cross-adsorbed (1:1000, Invitrogen) or Cy3-Affini Pure goat anti-rabbit polyclonal IgG (H+L) (1:500, Dianova) were incubated for 90 min at room temperature, followed by three washes, one wash in Aqua dest, nuclear stain with 300 nM 4',6-diamidino-2-phenylindole (DAPI) 3 min, and mounting (Fluorescence Mounting medium; Dako). Fluorescent microscopy was performed with Axio Imager.D1 (Carl Zeiss) using AxioVision software.
Electric cell–substrate impedance sensing
To detect adhesion and cell spreading via bioelectrical impedance, the electric cell–substrate impedance sensing (ECIS) model 1600 with 8W10E+electrode arrays (40 electrodes per well, each with a diameter of 250 μm and connected in parallel, Applied Bio Physics) and appropriate software were used. Wells were preconditioned with the respective medium (FBS or HS) and 5×104 cells were placed on the wells for 14 h at 37°C, 5% CO2. Each condition was performed in duplicate, testing ASC pairs from four different donors. Data are depicted as mean±standard deviation of the four donors normalized to the initial value.
Adhesion under perfusion conditions
Since mechanical shear stress has a major impact on adhesion, an electro pneumatically controlled flow system (BioFlux 200; Fluxion) was used to analyze adhesion under flow conditions. Flow channels were coated with gelatine and HUVEC were seeded on top. After two days, HUVEC monolayers were checked for confluence. ASC were labeled with Cell Tracker Green as described above. A cell suspension of 1×106 cells/mL in the HEPES-buffered Ringer solution (140 mM NaCl, 5 mM KCl, 1 mM MgCl, 1 mM CaCl, 5 mM glucose, 10 mM HEPES, adjusted to pH7.4 at 37°C) was prepared. HUVEC were prestimulated for 15 min with 50 μM histamine (Sigma-Aldrich). TNFα has been pretested to cause an overproportional activation under flow conditions compromising HUVEC monolayer confluence. ASC pairs from 4 individual donors were tested in 5 independent experiments. FBS- and HS-ASC pairs from the same donor were tested simultaneously in 2 proximate observation channels. They were subjected to a shear stress of 1, 2, and 10 dyne/cm2 for 5 min in each case. Video recordings were performed using Visitron “Spot Insight 4MPixel monochrome” (Visitron Systems) using maximal definition and taking three frames per second on an inverted microscope Zeiss Axiovert 200. Pictures were processed with AxioVision 4.7 software. Events of rolling and adhesion were counted under several frame rates by two independent observers.
Migration assay
Preparation of chemotactic media
Among other, the tumor milieu is known to chemoattract MSC [26,27]. To prepare TCM, 1.5×106 SF188 cells (human glioma cell line) were plated in a T75 flask. The medium was changed after 2 days. Before collection of the medium on day 4, cells were serum starved by washing twice with a minimal adhesion medium (MAM: DMEM-high glucose (Lonza), 0.5% BSA, 4 mM L-glutamine, 0.05 U penicillin/0.05 μg streptomycin (both PAA)), and then the MAM medium was added. After 18 h, TCM was collected, centrifuged at high speed, and filtered through a 0.22-μm filter; aliquots were frozen at −80°C. MAM was used as control.
Migration Assay
Migration of ASC was evaluated using a Transwell system (Sigma-Aldrich 6.5-mm with 8.0-μm pore size) in 24-well plates. 2×104 cells per well were seeded in the upper Transwell compartment in the MAM medium. MAM, TCM, the stromal cell-derived factor 1α (SDF1α, 100 ng/mL; R&D Systems), the insulin-like growth factor (IGF, 100 ng/mL, Millipore), and the hepatocyte growth factor (HGF, 40 ng/mL; Millipore) were added to the lower compartment to create a chemotactic gradient. Each condition was performed in triplicate and incubated at 37°C, 5% CO2 for 16 h. Then, the incubation was stopped by removing the cell suspension from the upper compartment of the insert, cleaning it with a cotton swap, and finally fixing the insert in ice cold methanol. After washing with H2O, cells on the lower surface of the insert were stained with DAPI, the membrane was removed and embedded on a slide. On the whole, membrane nuclei were counted with AxioVision Rel 4.7. Results are given in number of cells per mm2.
To analyze transendothelial migration induced by TCM, HUVEC were seeded in the EGM-2 medium on fibronectin-coated Transwell membranes, 8.0-μm pore size, and grown till confluence. Before proceeding as previously described, one representative monolayer was verified for intactness and confluence by coomassie brilliant blue staining. 2×104 ASC in the MAM medium were added in triplicate on top of the preformed HUVEC monolayer. The lower chamber was filled with either MAM or TCM. After 24 h, the transmigration was stopped and analyzed as described before. To account for migrating HUVEC, in each experiment, 3 inserts were processed without ASC on top. These migrated cells were deducted from nuclei, counted in the transmigration experiments. Results are given in number of ASC per mm2.
Short-term in vivo homing
HS- and FBS-ASC pairs were alternatively labeled with PKH 26-red or PKH 67-green fluorescence dyes (Sigma-Aldrich) according to the manufacturer's instruction before infusion. Viability, assessed by trypan blue, was>95%. NOD/SCID mice aged 10–12 weeks were irradiated with 2.5 Gy using a Cs-γ source. Then, each 3×106 ASC of the same donor, cultivated in either FBS or HS, labeled with either PKH 26 or PKH 67, were mixed at a 1:1 ratio and coinfused intravenously through the tail vein. In total, 5 animals were analyzed; in 2 animals, ASC of the same donor were injected applying a dye swop. To set the examination gates for flow cytometry analysis, 1 nontransplanted control mouse was included in each experiment. Two hours after injection, mice were sacrificed and blood, bone marrow, liver, lung, and spleen of each mouse independently explanted and prepared for flow cytometry. Samples were analyzed, acquiring at least 105 events per forward scatter/sideward scatter (FSC/SSC) gated samples at a BD FACS Canto II running FACS DIVA software.
Medium switch
To analyze the direct effects of the medium supplements, FBS-ASC were cultivated for 7 days in FBS and vice versa. Population doubling was determined using the formula: [log10(NH)–log10(N1)/log10(2), where N1 is the number of inoculated cells and NH the number of harvested cells. Cell size and flow cytometry analyses were performed as described before.
Statistical analysis
Statistical tests were performed using SigmaPlot 11.0 (Systat Software). Data were tested for normality (Kolmogorov–Smirnov test) and equal variance (Levene Median test) before statistical analysis. Differences between more than 2 groups were calculated using one-way analysis of variance (ANOVA) or ANOVA on ranks in case equal variance testing failed. Two groups were compared using paired, where applicable, or the unpaired t-test or the Mann–Whitney rank sum test if normality and equal variance testing failed. P-values>0.05 were regarded as significant and those >0.005 as highly significant.
Results
Adhesion under static conditions
Previous results suggested that the culture in HS may alter the adhesion of ASC since adhesion and extracellular matrix-interacting molecules in ASC cultivated in either FBS or HS were differentially expressed [15]. We performed different assays to test whether the choice of supplements indeed affects adhesion. We assessed adhesion to the hydrophilic tissue culture plastic surface, to laminin, and to endothelial cells, either unstimulated or activated with TNFα or histamine to enhance adhesiveness.
Adhesion to plastic surfaces
It appeared that HS-ASC adhered more strongly to plastic surfaces, although due to large donor variance differences to FBS-ASC were not significant at any time point (Fig. 1A). Adhesion proceeded remarkably fast, increasing from a mean value of 8% (FBS)/14% (HS) after 30 min to an average of 27% (FBS)/31% (HS) with no further increase after 60 min adhesion time.
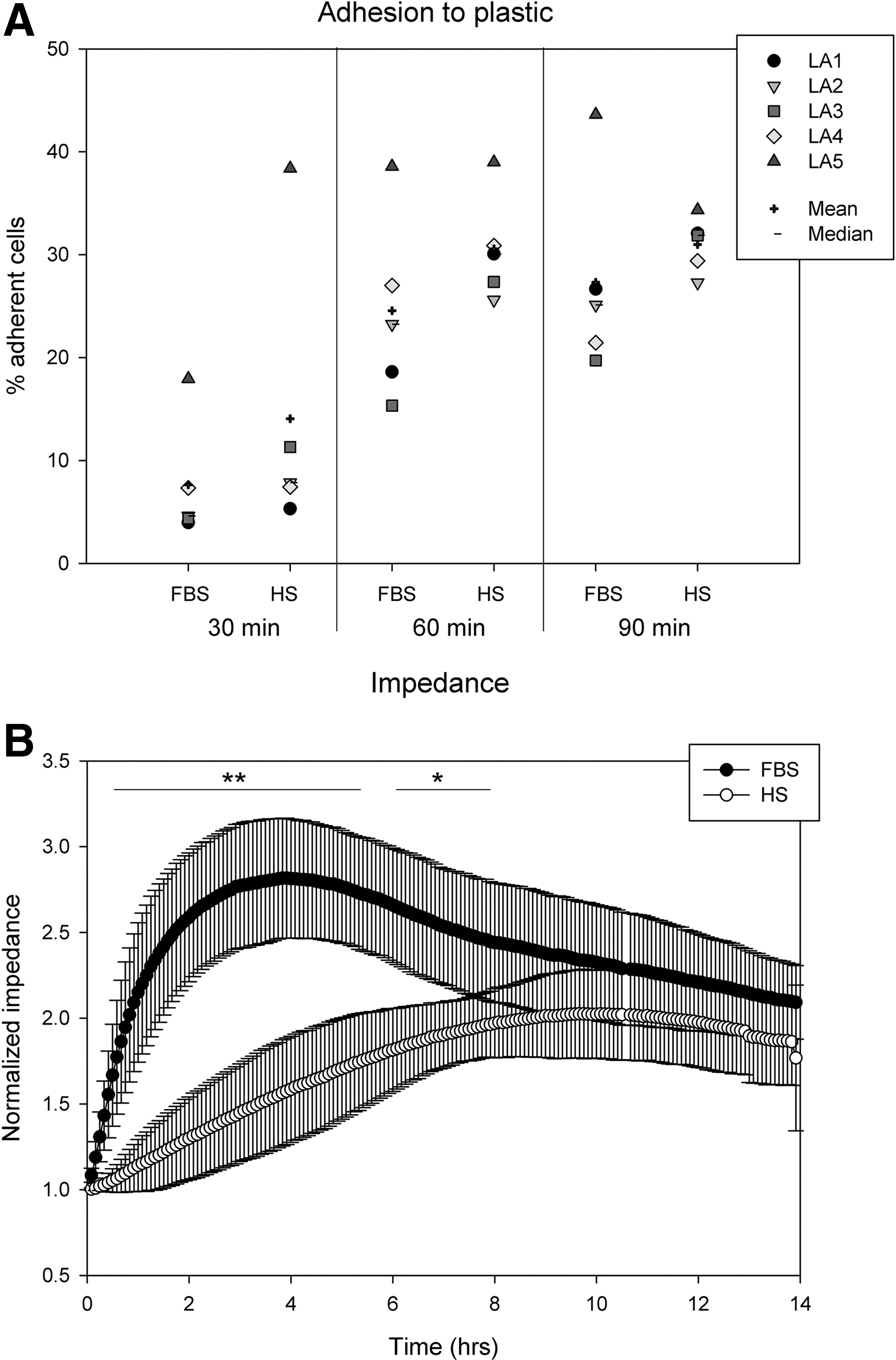
Adhesion under static conditions.
Electric cell–substrate impedance sensing
Based on the differing adhesion profiles of HS- and FBS-ASC, we performed a noninvasive real-time monitoring of adhesion and spreading kinetics by means of ECIS. Electric cell–substrate impedance sensing is known as a noninvasive and sensitive technique to detect cell attachment and spreading [28,29]. When cells adhere to the basal electrode surface, the insulating properties of the cellular membranes cause a rise in alternating current impedance. Current is impeded in a manner related to the number and surface of cells, covering the electrode, their morphology, and nature of cell attachment. We analyzed impedance profiles over a period of 14 h (Fig. 1B). Cell membranes insulate the current and cause a rise in impedance. Cells from 4 donors assessed revealed similar profiles: Impedance caused by FBS-ASC rose rapidly especially within the first 2 h and reached a maximum 2.8-fold higher than the initial value after 4 h. Then, impedance of FBS-ASC decreased almost linearly. In contrast, HS-ASC produced a slower, approximately linear increase of impedance to reach the maximum after 9.5 h. Differences between FBS- and HS-ASC impedance profiles were significant over the period of 8 h (highly significant until 6 h). Thereafter, the impedance profiles of HS and FBS-ASC ran in parallel.
Adhesion to laminin
Since we detected a significantly reduced expression of integrin α6 (CD49f) in HS-ASC in our previous study [15], we proposed a reduced adhesion to laminin. Thus, we analyzed the potential beta subunits of the laminin receptor: CD29 (integrin β1) and CD104 (integrin β4) forming the VLA-6 or α6/β4 complex, respectively. Because variance of distribution of mean fluorescence intensity (MFI) values was too high to calculate significant differences, we decided to compare% positivity instead, although expression was homogenously distributed within the cell population marked by a peak (bottom and top percentile) change, indicating that it was not a subpopulation of cells expressing the integrin subunits, but rather the whole population (Fig. S1). As presented in Figure 2A, CD49f expression was reduced in HS-ASC, but not statistically significant. Whereas CD29 was highly expressed, CD104 staining intensity was comparably low. HS-ASC expressed CD104 to a significantly higher extent (HS: 5.99±4.07% compared to FBS: 1.57±1.42%, P<0.01; Fig. 2A and Table 1). Consistent with a lower CD49f expression intensity of HS-ASC, adhesion to laminin was significantly reduced compared to FBS-ASC (Fig. 2B, C). Blockage of CD29 and CD49f, but not of CD104, reduced the adhesion to laminin, pointing at VLA-6 as the functional laminin receptor on ASC. No blockage (inhibition −10% to +5%) was obvious to uncoated plastic surfaces (data not shown). We speculated that the level of integrin expression may relate to the stage of cell confluence; thus, we checked the expression and adhesion in three ASC pairs at different stages of confluence: 50%, 80% (general cell passage), and 100%. At 100% confluence, indeed, the expression intensity of CD49f was reduced (Fig. 2D). Similarly, CD104 positivity became reduced. Adhesion to laminin, however, resulted in different data: both FBS- and HS-ASC at 100% confluence depicted a similar adhesion capacity like those at 50% confluence, and ASC at 80% adhered to a lower degree (Fig. 2E). Adhesion of HS-ASC to laminin was significantly lower than the adhesion of FBS-ASC. In all cases, laminin-specific adhesion was reduced by blocking CD29 (50%: 89.43% reduction in FBS and 90.2% for HS-ASC). Whereas CD104 blocking had no effect, CD49f blockade reduced adhesion to laminin (64.6% for FBS and 61.6% for HS at 50% confluence). Fitting the lower receptor expression, adhesion blocking effects of CD49f were reduced at 100% confluence (FBS 42.6% and HS 18.4%).
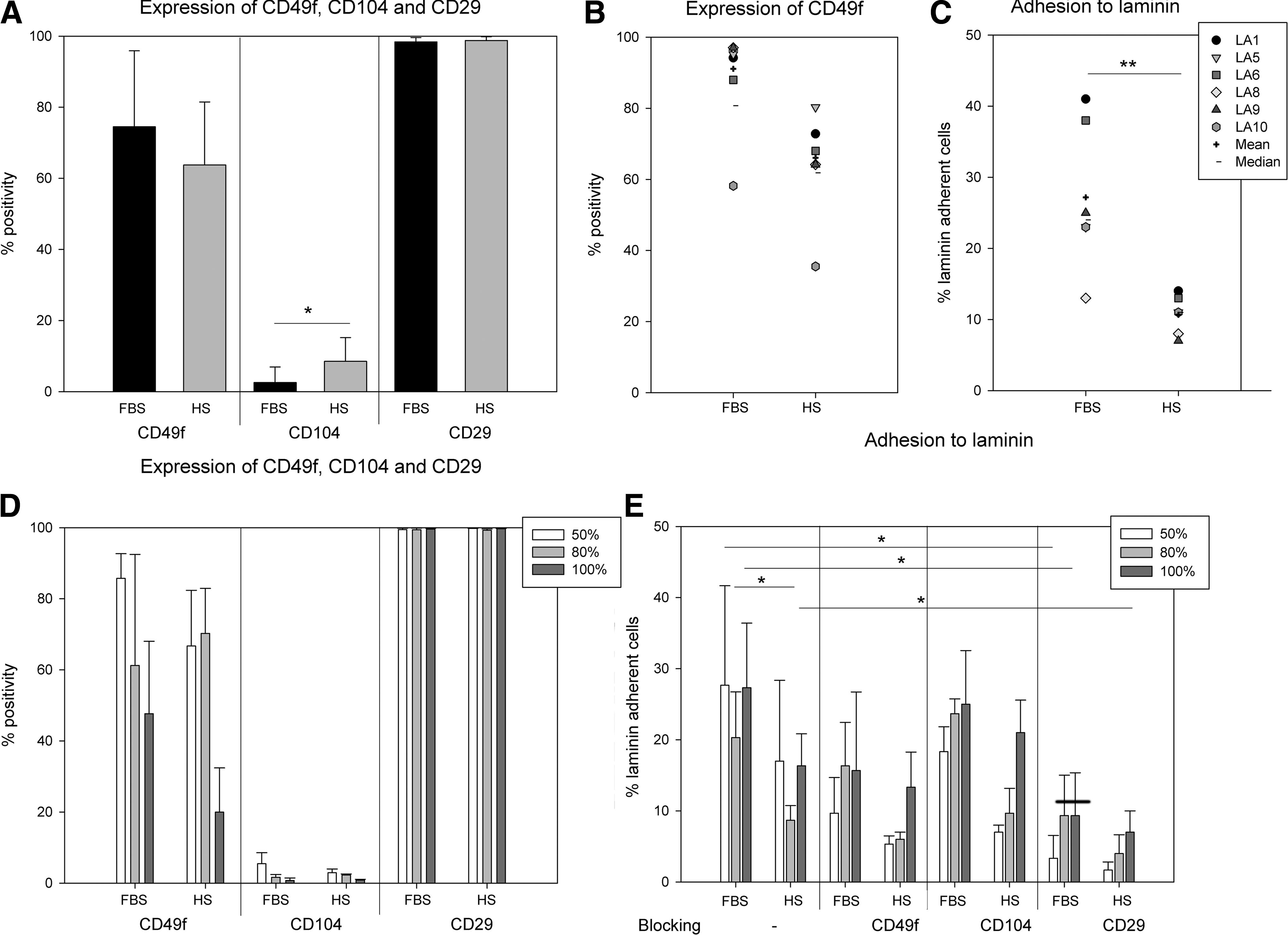
Expression of integrin receptor subunits and adhesion to laminin.
Integrin signaling
Upon adhesion to laminin, within 60 min, cell spreading caused very rapid changes of the cell shape, characterized by lammelipodia formation as depicted in Figure 3A and B. Within the 60-min period, an increasing number of cells adhered, as apparent in Figure 3A and B and the increasing GAPDH bands in 3C. Downstream signal transducers of integrin, p-FAK and p-PAX were expressed predominantly at these focal adhesion sites (Fig. 3A, B, respectively). As the cells spread on the substrate, especially FAK tyrosine phosphorylation occurred mainly at the cell protrusions, in contrast to FAK, which was evenly distributed in the cytoplasm. Expression of p-PAX appeared to be fiber-associated and to be gradually reduced within the 60-min period analyzed (Fig. 3C, D), whereas p-FAK and FAK signals increased within the increasing number of membrane ruffles. From the 3 donors analyzed, integrin signaling appeared to be faster in FBS-ASC, marked by the faster increase in FAK phosporylation and the faster decrease in p-Pax expression; however, no statistically significant differences were calculable. Both methods indicate a delayed integrin signaling of HS-ASC compared to FBS-ASC, marked by both FAK and Pax phosphorylation.
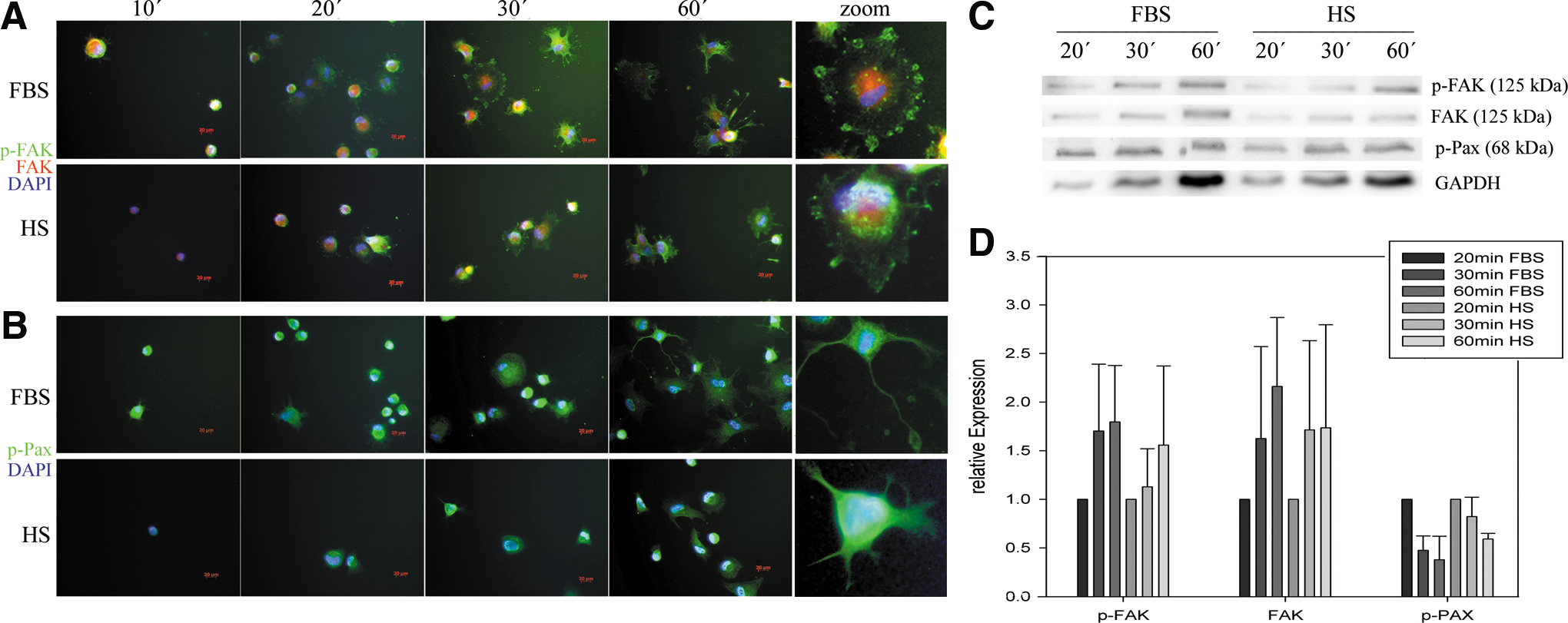
Integrin signaling upon ASC adhesion to laminin immunofluorescence analysis of
Adhesion to endothelial cells
Compared to the adhesion to plastic surfaces, the adhesion on the HUVEC monolayer was in general lower and apparently less fast (Fig. 4A). Interestingly, HS-ASC demonstrated significantly lower adhesion to HUVEC than FBS-ASC. Both TNFα and histamine activation enhanced ASC adhesion. However, adhesion of HS-ASC on TNFα-stimulated HUVEC was still significantly reduced compared to FBS-ASC.
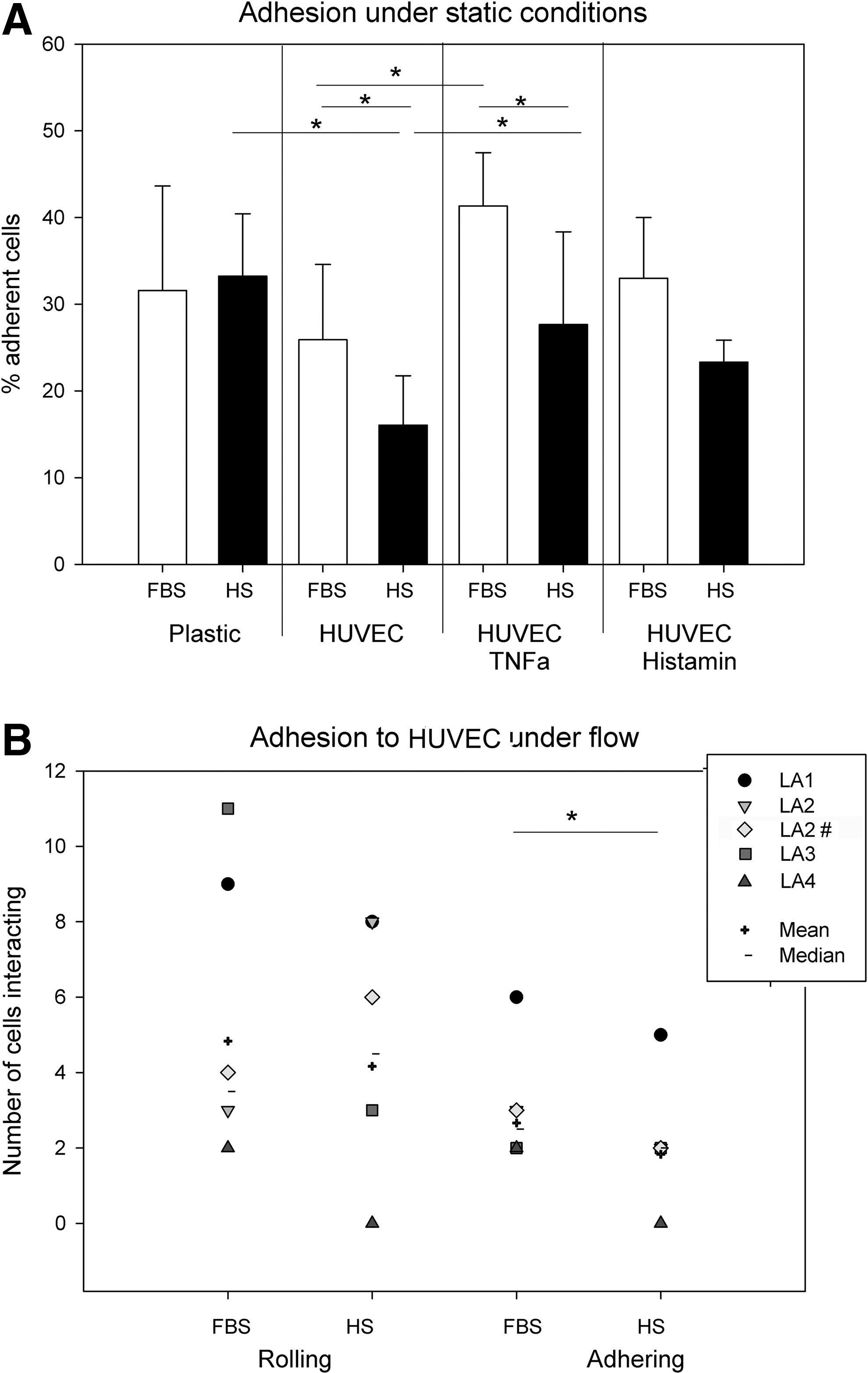
Adhesion to endothelial cells under static and shear flow conditions.
Adhesion and rolling under shear flow conditions
The differing adhesion profiles of FBS- and HS-ASC to HUVEC prompted us to investigate adhesion under shear flow conditions. Shear flow minimizes nonspecific adhesion often occurring in static conditions, and mimics the effect of the circulation in a better way [30]. Shear rates were increased in 2 steps from 1 to 10 dynes/cm2. FBS- and HS-ASC pairs were tested simultaneously in 2 proximate observation channels of an electropneumatically controlled flow cell. Under shear stress of 1 dyne/cm2, all ASC demonstrated only a low interaction with the histamine-activated endothelial monolayer (Fig. 4B and supporting video, TNFα was not used because of compromised HUVEC confluence under flow conditions). Other cell types, e.g., melanoma cells tested with the same system under equal conditions interacted far more intensely with the histamine-activated monolayer depicting rolling and tethering (unpublished data).
From the few cells interacting, HS-ASC revealed even less rolling and less adhesion than FBS-ASC, supporting the adhesion data under static conditions. Again, a high donor-dependent fluctuation was obvious. Increased shear rates to 10 dyne/cm2 completely abolished the interaction with the endothelium causing detachment of ASC (Supplementary Video; Supplementary Data are available online at
The reduced adhesion to endothelial cells prompted us to investigate the expression of few cell surface antigens associated with adhesion next to other markers defining MSC. Regarding % positivity, none of the antigens was differentially expressed (Table 1).
Migration and transmigration
To assess whether the supplements affect the migratory activity of ASC, TCM, known as an efficient chemoattractant for MSC, but also different cytokines like SDF1α, IGF, and HGF, described to coordinate MSC migration [31], were assessed compared to control MAM [26].
Examining ASC migration with the chemotaxis assay demonstrated that only TCM significantly induced ASC migration (Fig. 5A). HS-ASC appeared to migrate less than FBS-ASC toward TCM. However, HS-ASC of 2 out of 8 donors analyzed exerted a higher migratory activity than their counterparts cultivated in FBS. All ASC failed to migrate toward SDF1α (Fig. 5A). Corresponding extra- and intracellular flow cytometric analyses demonstrated that the majority of ASC lacked expression of CXCR4 and CXCR7 (C-X-C chemokine receptor type 4, 7) as SDF1α receptors (Table 1). Only LA11 cultivated in FBS was positive for CXCR4 and CXCR7 (CXCR4: 26.64% and CXCR7: 32.14%); in HS LA2 and LA11 were positive for CXCR7 (24.43 and 12.06%, respectively), but these ASC showed no migration either. Using U937 cells expressing CXCR4, SDF1a was validated to be functional. Related to the higher proportion of HS-ASC expressing the respective growth factor receptors (Table 1), IGF and HGF induced slightly more HS-ASC to migrate than FBS-ASC.
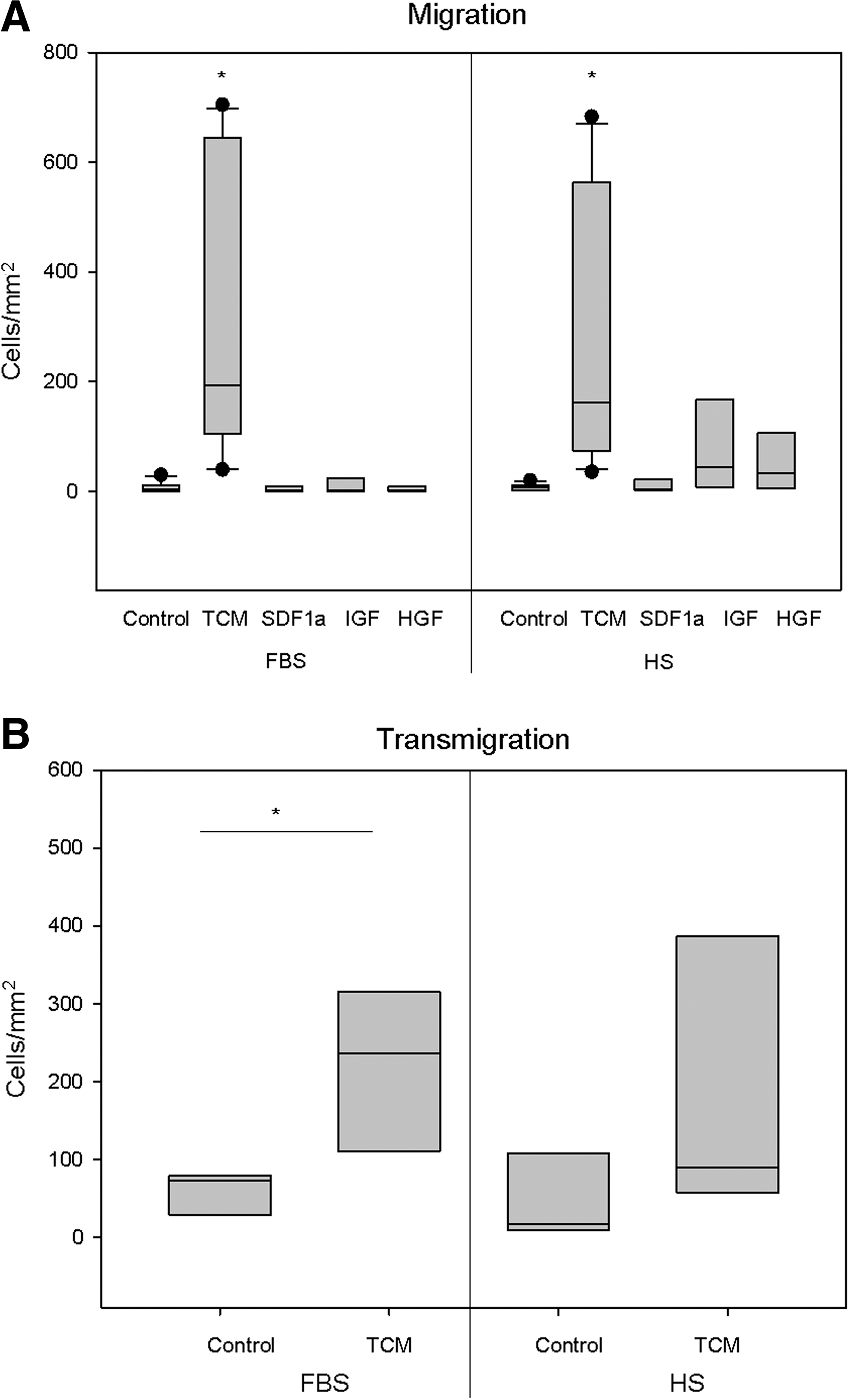
Chemotactic migration.
Given the differing adhesion propensity toward endothelial cells, ASC in the 2 culture conditions were also tested for their transmigratory capacity through an endothelial monolayer to mimic the capacity to egress from the circulation into the tissue. Here, transwells were seeded with HUVEC. Since the growth factors promoted only minor chemotactic activity, only TCM was used as an attractant and MAM as the control medium. Four of the 5 donors assessed showed a reduced transmigration of HS-ASC compared to the counterparts cultivated in FBS.
In summary, neither migration nor transmigration assays indicated significant differences in FBS- or HS-ASC behavior.
Short-term in vivo homing
Finally, to observe if there is any difference within the homing process in vivo, ASC in the 2 different supplements were labeled with 2 different fluorescent dyes (red and green), and then coinfused into one NOD/SCID mouse. After 2 h, mice were sacrificed and organ cell suspensions analyzed by flow cytometry to quantify the proportion of red/green labeled cells (Fig. 6). All fluorescent signals were backgated ruling out the possibility that some cells were not detected due to their smaller size. As observed in other studies [32,33], only a low percentage of the total transplanted cells were detectable in the investigated organs, predominantly in blood, liver, and lung. No events were detected in nontransplanted control mice in the search gates. The number of animals that could be analyzed, that is, 5, may not be very high; however, we could prove in all animals and organs that there is a tendency of a higher number of FBS-ASC. In blood, cell numbers appear to a similar extent but FBS-ASC seem to be entrapped at much higher rates in the lung and to a lesser extent in the liver than HS-ASC. Again, this may be due to the smaller size of HS-cultivated ASC (18.99±0.28 μm for HS- and 20.98±0.32 μm for FBS-ASC; t-test: P<0.001).

Short-term in vivo homing of ASC. ASC pairs were labeled alternately with either PKH 26-red or PKH 67-green. Cells were then coinjected into irradiated nonobese diabetic, severe combined immunodeficiency mice and allowed to home for 2h. Then, organ preparations were analyzed by flow cytometry for the proportion of green- and red-labeled cells and given as number of labeled cells per 105 events (see figure insert representing a dot plot with red cells=FBS-ASC in P2 and green cells=HS-ASC in P3). In the chart, each symbol represents 1 mouse and 4 different ASC donors: mouse 2 and 3 received ASC of the same donor applying a dye-swop. Color images available online at
Medium switch
To analyze which direct effects the medium supplements exert, FBS-ASC were cultivated for 7 days in HS and vice versa. It was apparent that HS significantly promoted ASC proliferation and caused the smaller cell size previously described (Supplementary Fig. S2). Regarding antigen expression, the transfer from HS-ASC to the FBS medium led to a slight increase of CD49f (% and MFI), yet failed to reach the high expression values observed in FBS-ASC. Fitting to the slightly higher expression of CD104 on HS-ASC, MFI values increased upon transferring FBS-ASC to HS and decreased vice versa. Both fibroblast growth factor receptor 2 (FGFR-2) and on a larger proportion of ASC, transforming growth factor receptor (TGFR) was induced in FBS-ASC switched to HS.
Discussion
Major changes in the manufacturing process, such as changing the source of supplement from FBS to HS, can have a major impact on cellular functions. Since homing is a prerequisite for efficient MSC therapy, we asked whether the culture supplements impact adhesion, migration, and homing of ASC. Differences in gene expression, especially in gene clusters associated to adhesion and extracellular matrix, prompted us to address these issues further by directly comparing ASC pairs from the same donor cultivated in FBS and HS. Despite obvious donor-specific variances, our data indicate that HS may affect the adhesion to extracellular matrix molecules and endothelial cells.
Considering that ASC are either injected locally or infused systemically, the therapeutic efficacy may be modified: When injected locally, HS might reduce homing and engraftment due to the slightly reduced interaction with ECM, that is, laminin tested within this study. When infused systemically, HS-ASC might home and engraft to a lesser extent as the interaction with the endothelium appears slightly reduced. It is still not known whether MSC actively home, thus actively arrest in the circulation, and then transmigrate or whether they get entrapped in capillaries [19]. At present, we have indications that both mechanisms may be operative. Compared to other cells, ASC demonstrate a low interaction with endothelial cells, either nonactivated, activated with TNFα or histamine, under static and flow conditions. Previous analysis on MSC, however, revealed coordinated rolling and adhesion mediated by P-selectin and VCAM/VLA-4, indicating that at least some players of the classic leukocyte homing cascade are involved [30]. PSGL-1 as a ligand for P-selectin was not expressed in the ASC pairs. Other studies demonstrated the involvement of CXCR4, integrins, and matrix metallo proteases [19,32,34 –36]. Some data, however, indicated that cells get entrapped in capillaries, causing either risky or beneficial effects: to increase the risk of pulmonary embolism [37,38] or to improve myocardial infarction by release of immunomodulatory TSG-6 (TNF-stimulated gene 6 protein) [39]. Our in vivo data suggest that HS-ASC get less entrapped in the lung and liver, probably due to their smaller size. On the other hand, the slightly reduced interaction with the endothelium might also contribute to this effect. Published data indicate a specific tropism of MSC/ASC to sites of injury, ischemia, and also tumor growth and much higher engraftment rates in mice receiving total body irradiation [32,33,40,41]. Since we have used a short-time homing model assessing homing after 2 h, whereas most other models examine organs after 20–24 h, with only few animals to be analyzed, we do not dare to speculate on the target tissues yet, and whether FBS- or HS-cultivated cells differ in this aspect. The reduced numbers in lung and liver, however, correspond to the reduced cell size. We speculate that the smaller size is also well reflected by the ECIS data producing a gently inclined rise in the impedance profile compared to FBS-HS. FBS-ASC are larger in size and upon settling and spreading cover a larger surface area of the electrodes causing a steeper slope in impedance.
The adhesion data presented herein correspond to data presented by Semon et al. analyzing integrin expression and adhesion to various ECM molecules and different endothelial cell preparations [35]. The authors also mention the great variability in integrin expression and, in consequence, in adhesion, migration, and homing. This is supported by our data especially regarding CD49f expression, which demonstrate donor-specific expression and reduced expression as function of confluence relating to the degree of adhesion to laminin. HS-ASC expressed CD49f to a lower extend resulting in reduced adhesion to laminin. Signaling upon Integrin-mediated laminin adhesion was demonstrated revealing only minor differences in the time course of FAK and paxillin phosphorylation.
The migratory capacity of ASCs is very poorly defined. There are limited data on transmigration indicating a significantly reduced transmigratory capacity compared to leucocytes [42], but again engaging VCAM/VLA-4 [43]. The significant deviance to leucocytes is discussed by Karp and Leng Teo to be related to the nonactivated endothelium [19]. In another study, we analyzed that TCM, similar to TNFα, upregulates homing molecules on HUVEC (unpublished data). Thus, in our system the endothelial monolayer used for transmigration can be regarded as activated. TCM is a crude mixture of chemotactic substances. Various growth factors and chemokines have been addressed to promote migration also toward tumors, especially SDF1α acting on CXCR4 [19,36,44]. CXCR4 is known to be rapidly induced on the MSC surface from intracellular pools [32] and to increase migration and homing [36]. However, in our studies, CXCR-4 was not expressed (neither extra nor intracellular) and neither stimulation with TNFα or SDF1α, nor TCM-induced expression of CXCR4 or CXCR7 (not shown). Fitting to this, SDF-1α failed to induce ASC migration. In contrast, IGF and HGF triggered at least a few ASC to migrate. Corresponding to the higher receptor expression, HS-ASC exerted a slightly enhanced chemotactic activity. Quite a low migratory capacity of ASC toward a variety of chemokines and growth factors compared to 30% FBS has already been described [45 –47], fitting to the data presented herein.
To explain the differences observed, we analyzed a number of cell surface molecules known to promote adhesion, but failed to yield statistically significant differences between the ASC pairs. Comparing these to published data on human ASC revealed significant discrepancies. Maijenburg et al. indicate that only 8.4±11.5% ASC express CD49f and approximately 22% CD49b and CD49d, whereas all ASC are positive for CD49e [45]. Baek et al. indicate that approximately 55% of human ASC express CXCR4 and CXCR7 [46]. For bone marrow MSC, Chamberlain et al. reviewed the variable results regarding chemokine receptor expression [48]. Honczarenko et al. analyzed that CCR1, CCR7, CCR9, CXCR4, CXCR5, and CXCR6 were expressed and functional [49]. The authors also indicate a loss of chemokine receptor expression upon cell expansion.
Our previous microarray and supporting polymerase chain reaction data confirmed significant differences for osteoprotegerin, osteoactivin, fibromodulin, elastin, and the elastin microfibril interfacer 2 (EMILIN2), the cartilage oligomeric protein, aggrecanase 2, shroom family member 3 embryonal Fyn-associated substrate, collagen type 8α2, SOCS2, CXCL5, GDF15, and the insulin-like growth factor 2 receptor [15]. Data were tested for differences in protein expression of osteoprotegerin, fibromodulin, and cartilage oligomeric protein. Preliminary analysis of candidates and literature research so far failed to explain the observed differences in adhesion and migration. Osteoprotegerin released by endothelial cells has been demonstrated to neutralize the chemotactiv activity of TNFα-induced TNF-related apoptosis-inducing ligand (TRAIL) toward bone-marrow MSC [50]. Osteoprotegerin in our hands, however, was expressed significantly higher in FBS- than in HS-ASC.
Karp and Leng Teo highlighted that numerous variables contribute to the heterogeneous results concerning ASC/MSC homing behavior [19]. In our study, in all assays, pairs of ASC have been investigated at similar passages and stages of confluence. Because extended culture and increased confluence appear to negatively influence the migratory propensity, cells need to be compared within defined settings and standardized assays. However, although we follow fully standardized protocols, strong effects of the donor, and possibly the site of isolation, hamper to establish accurate assays capable of predicting potency in vitro. Taking everything together, although most of our current data indicate that the culture in HS at least not significantly alters ASC features, the herein observed data regarding ECM and endothelial adhesion, migration, and homing advise to further validate this in well-defined models to ensure efficiency and safety of significantly altered culture conditions.
Footnotes
Acknowledgments
The authors would like to thank Michael Angstmann and Christian Maercker, the Mannheim University of Applied Sciences, Biotechnology, Mannheim, Germany for providing the ECIS platform and Olga Roeder for valuable help in the in vivo experiments. We would like to thank Daniela Griffiths for editing the manuscript and Andrea Hecker, Maximilian Nick, and Stefanie Uhlig for excellent technical assistance.
This work was supported by research funds of the German Federal Ministry of Education and Research (START-MSC: 01GN0531 and 01GN0939) to KB and a project commissioned by the European Community (CASCADE: FP7-223236) to KB and RH.
Prior conference presentation of the submitted material:
1. Bieback K, L Dreher, S Elvers-Hornung, et al. (2010). Does the culture in human supplements affect the migration and homing of mesenchymal stromal cells? Vox Sanguinis 99:43–44.
2. Abstracts of the XXXIst International Congress of the International Society of Blood Transfusion in joint cooperation with the 43rd Congress of the DGTI, Berlin, Germany, 26 June–1 July 2010.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.