
Abstract
Human embryonic stem cells (hESCs) are regarded as a promising approach to generate transplantable cells for the treatment of several diseases. These cells offer an immense potential as a source of cells for regenerative medicine, but the possible ability of these cells to produce tumors in vivo presents a major impediment for the achievement of this potential in clinical reality. hESCs can obtain growth advantages in vitro by acquired mutations, a phenomenon called culture adaptation. The most common chromosome modifications involve chromosomes 12, 17, and X. The mechanisms that may influence chromosome modification in hESCs are not well known. We have performed a comparative in vitro and in vivo study on 3 hESC lines produced in our laboratory to see if there are changes also during in vivo growth. In vivo differentiated cells and in vitro cultured hESCs were analyzed by using a high-resolution Affymetrix SNP 6.0 array revealing DNA copy number variations. We were able, for the first time, to identify chromosomal aberrations that had occurred in vivo in one out of the 3 hESC lines. In the hESC line HS364 differentiated in vivo, an amplification of the whole X chromosome was detected, possibly due to mosaicism of XY and XX cells. In the hESC line HS366, array results showed small amplifications and gains. The third hESC line (HS368) was less altered, but contained also a new gain verified by fluorescent in situ hybridization in a teratoma in 21% of the cells. These results indicate that mutations occur during the in vivo differentiation process as well as in vitro. The potential of precancerous mutations in in-vivo conditions is important to consider for safety measures, and underlines the necessity to remove all pluripotent stem cells from the differentiated cell population that will be transplanted.
Introduction
H
A challenge today is to produce patient-specific stem cells to avoid rejection after transplantation. The immunogenicity of such cells remains a problem [4, 5]. This could be overcome by somatic cell nuclear transfer [6] or by using human induced pluripotent stem cells (hiPSCs) [7] and immunosuppressive drugs [5]. However, the chromosomal instability of pluripotent stem cells is a major obstacle for a safe clinical use. All hESC lines in long-term culture suffer from mutations in response to the environmental conditions. They obtain growth advantages by acquired mutations, a phenomenon called culture adaptation [8]. The most common chromosome alterations involve chromosomes 12, 17, and X [9 –12], but there is also a sporadically chromosome modification as well reported by other groups [13, 14]. In a previous study, we identified an isodicentric X chromosome in one of our hESC lines (HS237) at passage level 61, and it proved to be a key mutation in culture adaptation [15].
Consequently, tumorigenicity remains a big concern [16, 17], and the first experiments with induced mouse adult fibroblasts resulted in tumors in the offspring [7]. There are obviously line-specific variations in the tendency to undergo genetic changes [18]. It is of importance to identify the nature of hESC- and hiPSC-derived tumors, and especially their malignant potential, to evaluate the risk involved in their clinical use. Diploid hESCs from low passages produce benign teratomas with fully differentiated tissues, so-called mature teratomas, in which no cells present pluripotency markers or markers for undifferentiated cells [1, 19 –21]. On the other hand, there are hESCs that can acquire genetic changes associated with malignant tumor formation; they form teratocarcinomas [22, 23] in which still incompletely differentiated cells are found among fully differentiated tissues.
Single-nucleotide polymorphism (SNP) microarrays have been used recently to identify duplications, deletions, and losses of heterozygosity in hESC lines cultured in vitro [23, 24]. Genetic changes are direct responses to the microenvironment. The mechanisms that may influence cell modifications and culture adaptation in hESCs are not well known, and therefore more in vitro and in vivo studies are needed in this field.
We have studied the chromosomal stability of 3 hESC lines in in-vitro and in vivo conditions using a high-resolution Affymetrix SNP 6.0 array DNA–copy number variations (CNVs) and fluorescent in situ hybridization (FISH) analyses. The hESC lines were analyzed by karyotyping at passages 23, 40, and 21. Array analyzes of the cell lines were performed at passages 27, 42, and 28, and at that point, cells were also introduced in severe combined immunodeficiency (SCID) mice to obtain teratomas. After 8 weeks, the teratomas were analyzed with SNP arrays and interphase FISH. The long-term culturing of the cell lines was continued in parallel with the teratoma formation, and the cell lines were analyzed again by SNP arrays at passages 34, 49, and 35. We found that additional mutations occurred in vivo and not only in vitro, suggesting that in vitro methods may not suffice to monitor the safety of stem cells for clinical use.
Material and Methods
hESC maintenance and culture
Three hESC lines were used in this study: HS364, HS366, and HS368, which all had been derived from the inner cell mass of 6-day-old preimplantation blastocyst-stage embryos that had not been used for infertility treatment. They were donated by couples undergoing in vitro fertilization at the Fertility Unit of Karolinska University Hospital, Huddinge, Sweden [25, 26]. The Ethics Committee of the Karolinska Institutet had given approval for derivation, expansion, banking, and differentiation of hESC lines. These hESC lines have been fully characterized. For characterization, the expression of pluripotency markers such as Oct-4, Nanog, SSEA-4, TRA-1-60, and TRA-1-81 had been analyzed using immunocytochemistry and real-time polymerase chain reaction (RT-PCR). For the testing of pluripotency, the hESCs had been differentiated into embryoid bodies by culturing colonies of hESCs as spheres in low-adhesion culture dishes in an serum replacement-containing medium, without the addition of basic fibroblast growth actor (bFGF), for 3 weeks. The expression of components of all 3 germ layers has been observed. Pluripotency had also been tested in vivo by the formation of teratoma in SCID/beige mice as described earlier [27]. The presence of tissue components of all 3 embryonic germ layers has been seen.
Karyotype analyses were performed by G-banding with normal results, that is, the line HS364 having the karyotype 46, XY, HS366 46, XX, and HS368 46, XY.
The hESC lines were cultured and maintained on a feeder layer of human foreskin fibroblasts (CRL-2429; ATCC) mitotically inactivated by irradiation (40 Gy). The cells were cultured in a Knockout Dulbecco's modified Eagle's medium supplemented with 20% Knockout Serum Replacement, 2 mM Glutamax, 0.5% penicillin–streptomycin, 1% nonessential amino acids (all from Gibco Invitrogen Corporation), 0.5 mM 2-mercaptoethanol (Sigma-Aldrich Co.), and 8 ng/mL bFGF (R&D Systems) at 37°C in 5% CO2, as previously described by Ström et al. and Skottman et al. [25, 26, 28]. The hESCs were mechanically passaged every 5–7 days, and transferred onto fresh feeder cells.
Immunocytochemical characterization
Immunocytochemical characterization was carried out on all 3 hESC colonies obtained between passages 22 and 34. The primary antibodies were specific for Oct-4 (Chemicon), Nanog, TRA-1-60, TRA-1-81, and SSEA-4 (all from Santa Cruz Biotechnology, Inc.), all diluted at 1:100. Human foreskin fibroblasts were used as negative control cells. Immunostaining was performed by fixation of the cells with 4% paraformaldehyde in PBS for 10 min at room temperature, followed by blocking with 10% fetal bovine serum for 2 h for surface markers. For nuclear markers, a blocking solution of 0.1% Triton-X and 0.1% bovine serum albumin supplemented with 2% donkey serum was used. The primary antibodies were added in an appropriate blocking buffer overnight at 4°C. Binding of the primary antibodies was detected with a fluorochrome-conjugated secondary antibody, diluted 1:500 in a blocking buffer, and added to cells for 60 min at 4°C in the dark. After 3 washes with PBS, the cell nuclei were counterstained with Hoechst B2261 (Sigma-Aldrich) for 10 min.
In addition to negative control cells, hESCs were also incubated without primary antibodies. Stained cells were analyzed using an Olympus IX71 inverted microscope (Olympus Sverige AB), and the images were acquired with an Olympus DP71 camera and Soft Imagine Cell F version 2.6 Software (Olympus Sverige AB).
Real-time quantitative PCR
The hESC colonies were analyzed for the expression levels of Oct-4 and Nanog mRNA.
In brief, total RNA from hESCs was extracted (QIAGEN; RNeasy Mini kit, according to the manufacturer's recommended protocol) and converted to cDNA (Invitrogen; SuperScript™ III First-Strand Synthesis System for RT-PCR, Cat. No 18080-051, also according to the manufacturer's recommended protocol). The cDNA was then analyzed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Cat. No 4368814) for probes Nanog (HS02387400_G1) and Oct-4 (HS03005111_G1).
The data were analyzed using MxPro™ QPCR analysis software version 3.0 (STRATAGENE®). The assays were performed in triplicates. To provide negative controls and exclude contamination by genomic DNA, reverse transcriptase was omitted in the cDNA synthesis step, and the samples were subjected to the PCR assay in the same manner with each primer pair.
Teratoma formation analysis
The pluripotency of these 3 cell lines in vivo was tested as previously described [15, 27]. In brief, exponentially growing hESCs of 3 different cell lines from passages 27, 42, and 28 were harvested from the culture plate using dispase and mechanical treatment. Five colonies (103 to 104 hESCs) were washed twice in phosphate-buffered saline and subsequently implanted beneath the testicular capsule or subcutaneously of a young (7-weeks old) SCID/beige male mouse (C.B.-17/GbmsTac-scid-bgDF N7; M&B). Teratoma growth was determined by palpation every week, and the mice were killed by cervical dislocation 8 weeks after implantation.
The teratomas that were generated from the hESC lines HS364, HS366, and HS368 were dissected after 8 weeks (corresponding to 7 passages in vivo). Imprints for the interphase FISH analysis were generated by sectioning the teratomas and pressing the sliced solid teratoma onto a glass slide. A number of imprints from different sections were generated for each teratoma. DNA was extracted from the remaining of the solid teratoma and was analyzed by a high-resolution Affymetrix SNP 6.0 array.
All animal experiments were performed at the infection-free animal facility of the Karolinska University Hospital in accordance with the ethics committee's approval.
Karyotyping
The cell lines HS364, HS366, and HS368 were karyotyped as described earlier by Inzunza et al. [27] at passages 23, 40, and 21. Samples of cells were treated with colcemid KaryoMAX (0.07 μg/mL; GIBCO) overnight. After washing, the cells were incubated in a 0.4% trypsin solution (GIBCO) for 2–3 min. Cells were treated with collagenase (1,400 IU/mL; Worthington) at 37°C for 20 min and harvested using standard procedures. The metaphases were analyzed after Q-banding. A total of 14–24 metaphases were analyzed for each cell line.
High-resolution Affymetrix SNP 6.0 array DNA-CNVs
The genotyping was performed using the Affymetrix Genome-Wide Human SNP Array 6.0. Labeling and hybridization were performed following the protocols and kits provided by the manufacturer (Affymetrix Genome-Wide Human SNP Nsp/Sty Assay Kit 5.0/6.0).
As a copy number neutral reference sample, we used a set of 50 noncancerous whole-blood DNA arrays previously hybridized and genotyped in the same laboratory. CNVs and losses of heterozygosity (LOHs) were extracted with Affymetrix proprietary software Genotyping Console Software, using the standard setup recommended by the producer. Variations were annotated with the annotation libraries supplied by Affymetrix (version 29).
Fluorescent in situ hybridization
The imprints from the teratomas generated from the 3 hESC lines were analyzed by interphase FISH using DNA probes specific for the X centromere (CEPX- Aqua; Vysis) and the SHOX gene on Xp/Yp (LLN04C03’M’15D10 and LLN04C03’M’34F5) to detect X chromosome aberrations and the Y chromosome. The SHOX probe was labeled with Spectrum Orange by nick translation (N5500; GE Healthcare), and after precipitation, the probe was resolved in a hybridization solution (Vysis), and 2 μg Cot-1 DNA (BRL) and the CEP X probe were added. The slides with teratoma cells were incubated in 1% formamide for 45 min and then treated with 2× saline-sodium citrate (SSC) at 60°C for 20 min. The slides were then incubated at 37°C in 100 mL 0.01 M HCl containing 10 μg pepsin, and then fixed in 1% formamide. The probes and slides were co-denatured at 73°C for 3 min, and hybridizations were then performed in a moist chamber at 37°C overnight (Hybride; Vysis). The slides were washed in 2× SSC at 72°C for 5 min. After dehydration, the slides were mounted in glycerol containing 2.3% 1,4-diazabicyclo-(2,2,2)octane (DABCO) as an antifade and 4,6-diamino-2-phenyl-indole (DAPI) at 0.5 μg/mL as a counterstain. The signal was visualized using a Zeiss Axiophot fluorescence microscope equipped with a cooled CCD camera (Photometrics). Gray-scale images were captured, pseudocolored, and merged using SmartCapture software (Digital Scientific). For each teratoma, 100 interphase cells were analyzed for each probe mixture.
Results
The 3 analyzed hESC lines had been derived from the inner cell mass of 6-day-old preimplantation blastocyst-stage embryos, cultured, and maintained on a feeder layer of human foreskin fibroblasts culture. During long-term culture, they have remained karyotypically stable. The karyotype analyzes of these hESC lines were performed after passage 23 for HS364, passage 40 for HS366, and passage 21 for HS368. All 3 cell lines showed normal karyotypes, male 46 XY (HS364), female 46 XX (HS366), and male 46 XY (HS368) (Fig. 1). Colonies of these 3 hESC lines expressed immunocytochemical surface markers SSEA-4, TRA-160, TRA-1-81, and the nuclear marker Nanog, and Oct-4 (Fig. 2).

Normal 46 XY Karyotype of HS 364 (passage 23), 46 XX of HS366 (passage 40), and 46 XY of HS 368 (passage 21).
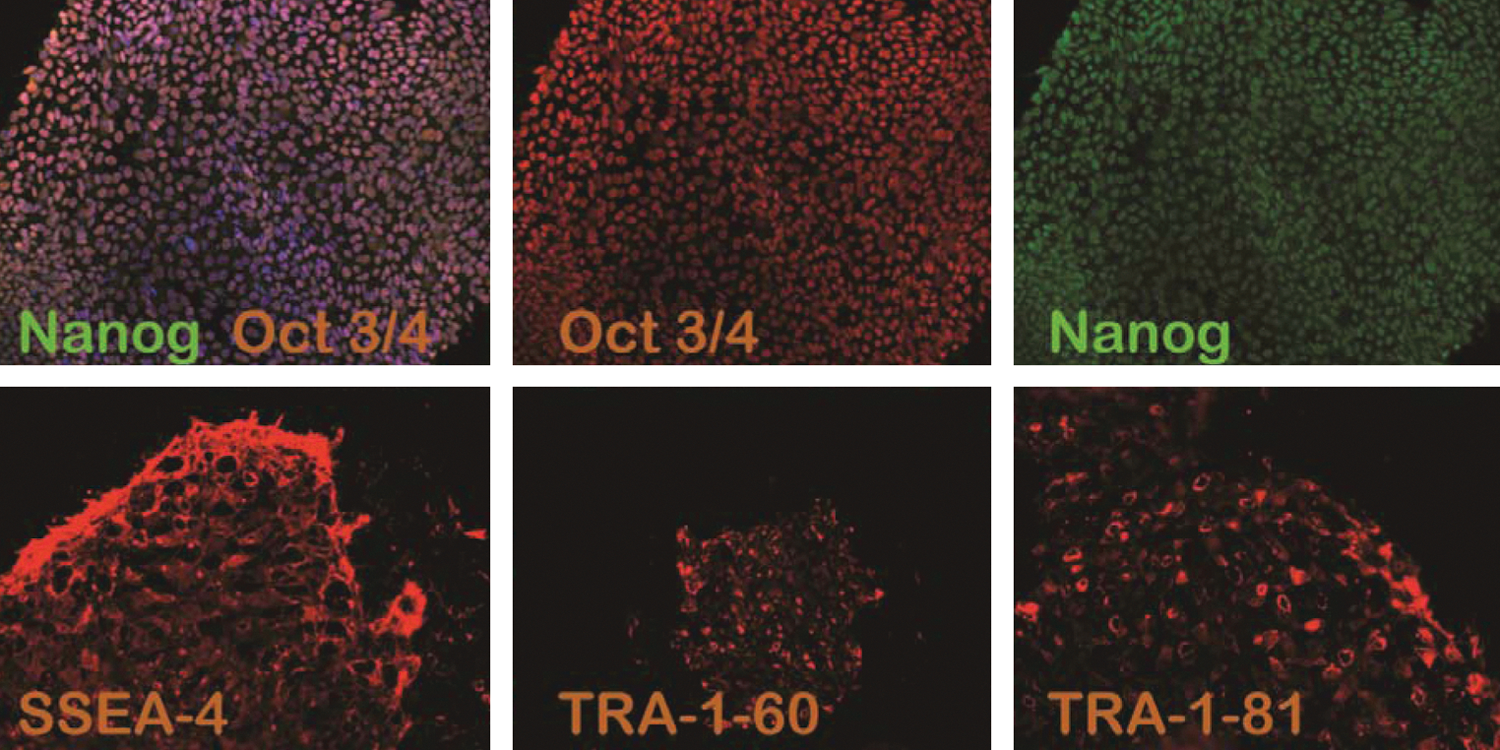
Immunohistochemical characterization of undifferentiated colonies of human embryonic stem cell (hESC) line HS 368 (passage 22) transcription factors and surface markers. Merged staining of Oct4 red and Nanog green, Hoechst staining blue, and red staining for SSEA-4; red staining for TRA-1-60; red staining for TRA-1-81.
The mRNA expression of Oct-4 and Nanog of these 3 cell lines was also revealed by quantitative RT-PCR and showed the maintenance of the undifferentiated state.
Subsequently, pluripotent cells from these 3 cell lines were implanted beneath the testicular capsule or subcutaneously of a young SCID mouse. The implanted hESCs were injected at the following passages: passage 27 (HS364), passage 42 (HS366), and passage 28 (HS368). From HS364, 3 teratomas were generated, 2 from testis and one from subcutaneous injection of cells (testis 1A, testis 2A, and SC 1). From line HS366, 4 teratoma samples were generated, 3 from testis and one from subcutaneous injection (testis 1A, 2A, 3A, and SC1). For HS 368 line, 5 teratoma samples were generated (testis 1A, 2A, 3A, and SC1, SC2).
All 3 hESC lines formed solid teratomas with components of the 3 primitive embryonic germ layers as expected (Fig. 3).
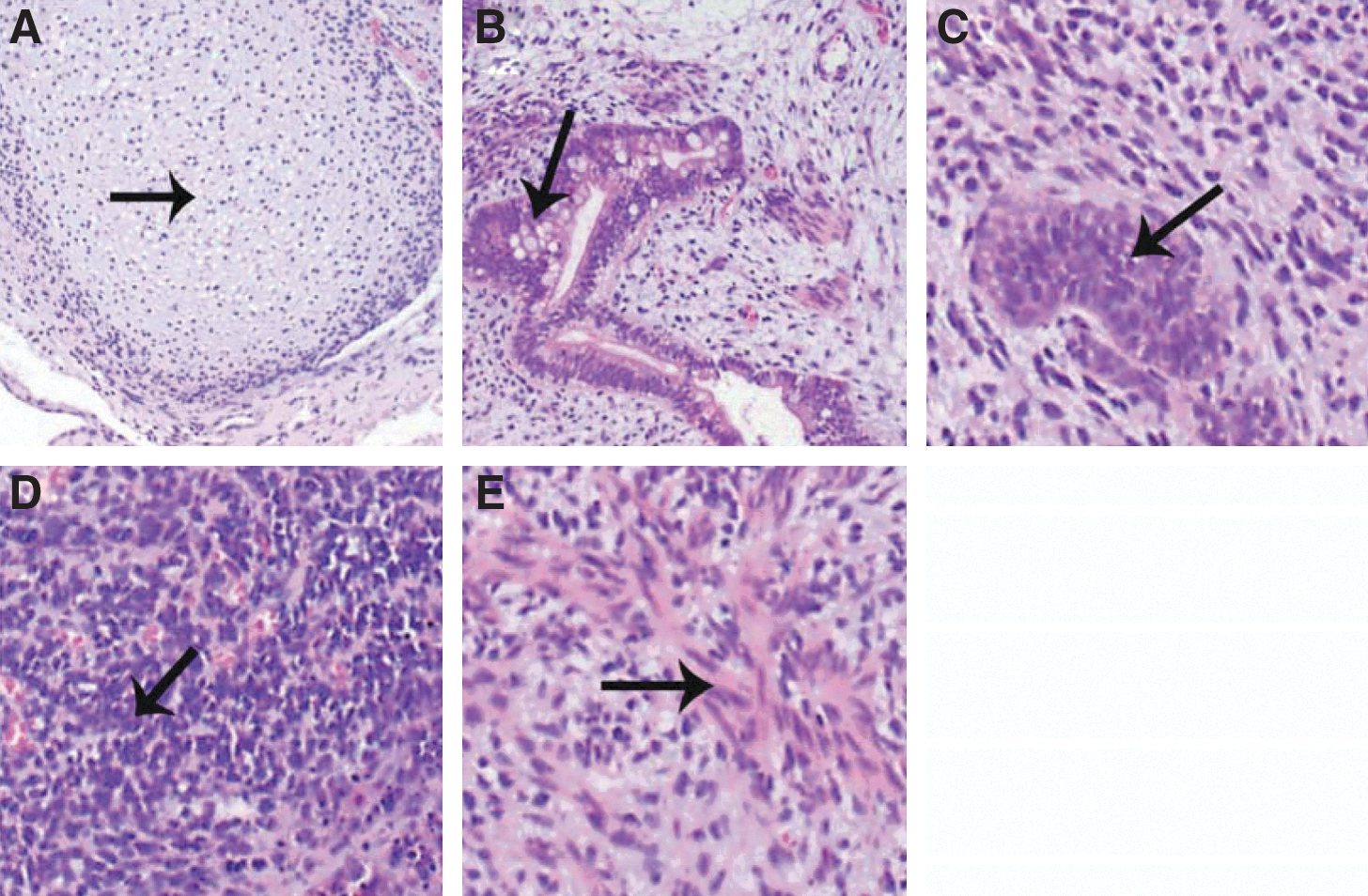
Teratoma, containing tissue component of all 3 embryonic germ cell layers from the cell line HS 368,
High-resolution Affymetrix SNP 6.0 array DNA-CNVs
DNA extracted from in vivo cultured hESCs prior and after teratoma formation and the teratomas were run on the SNP 6.0 genotyping platform. Genotypes and probe intensities were combined to estimate structural variations, that is, CNVs and LOHs.
Results for the HS364 cells line are shown in Fig. 4. Gains in DNA copies are reported with blue arrows and losses with red ones, alongside banded chromosomal representations. Gains and losses are estimated in comparison to a copy number neutral baseline data set composed of 50 noncancerous whole-blood DNA arrays genotyped in the same laboratory.
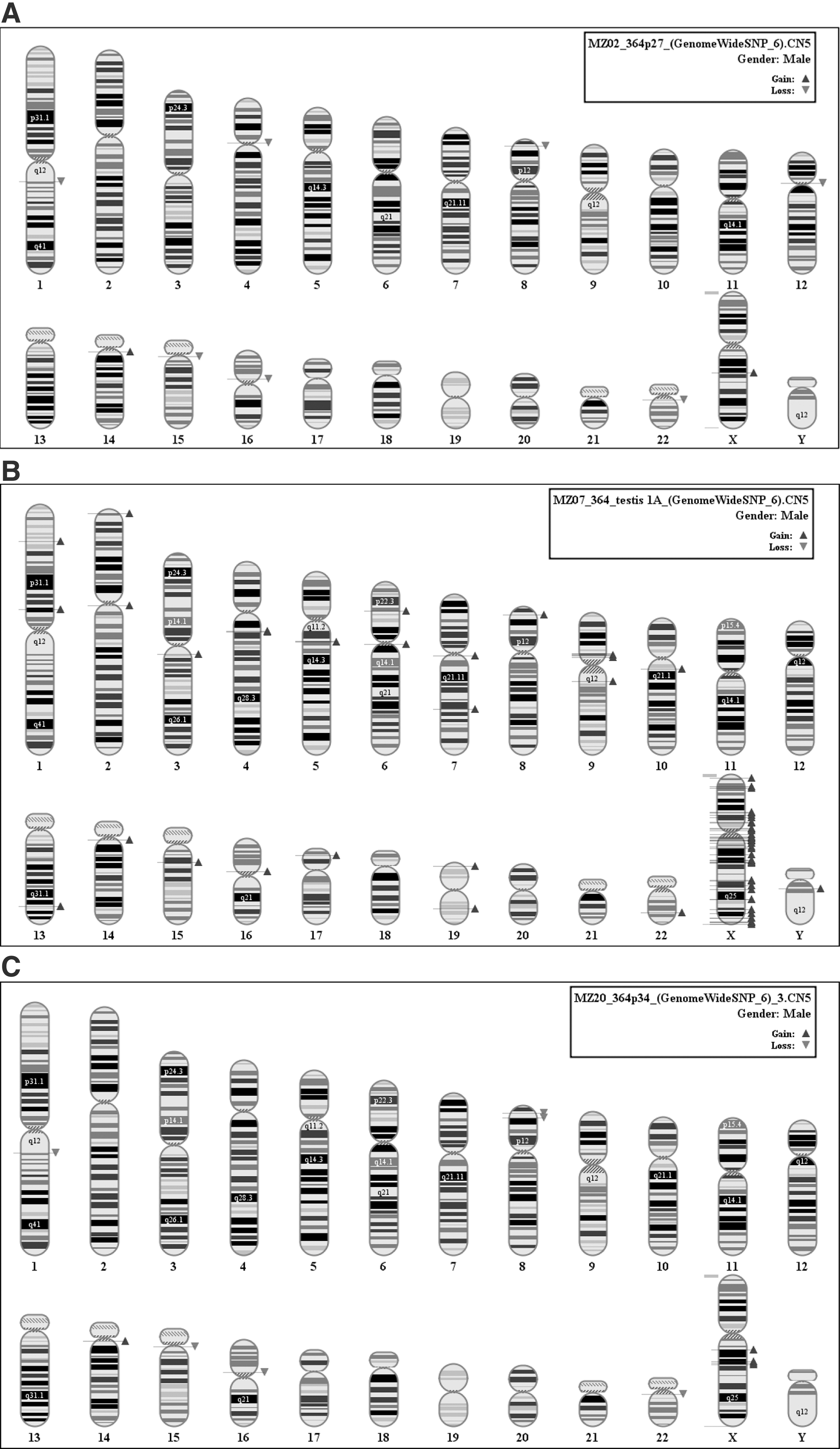
The HS364 hESCs at starting time passage 27 showed a total of 9 variations on different chromosomes with total genome coverage of 3.7 Mb (Fig. 4A). Cells from the same sample were both injected in mouse to form teratomas and grown in parallel in vitro. At passage 34, teratoma cells and in vitro grown stem cells were genotyped to compare the time evolution of structural variations (Fig. 4B, C). The in vitro grown cells still showed a low mutation rate with a number of variants in common with passage 27 (chr1, chr14, chr15, chr16, chr22, and chr X, 10 variations for a coverage of 3.6 Mb). Notably, 2 gains on chromosome X appeared at q21.1 and q21.31 that do not have any match in the Toronto Database of Genomic Variants (DGV;
Teratoma cells from HS364 showed a higher degree of mutations with a total of 67 variations with a genomic coverage of 17.3 Mb in the teratoma testis 1A. All the variations were gains and mostly clustered on chromosome X, suggesting that the cells have an extra X chromosomal material (Fig. 4B). Out of 67 variants, 21 did not have any match in the DGV database. Gene Ontology enrichment analysis showed a significant duplication of MIR genes.
Interphase FISH analysis of same teratoma (testis 1A) formed from HS364 showed the pattern of normal XY in 58% of the cells (one X centromere and 2 SHOX signals), whereas 24% showed 2 X centromere signals and 2 SHOX signals. This pattern is consistent with the duplication of the X chromosome. This result suggests mosaicism for XY and XX (with lost Y chromosome) cells and confirms the results of an extra X chromosome material in the teratoma sample analyzed with an SNP array.
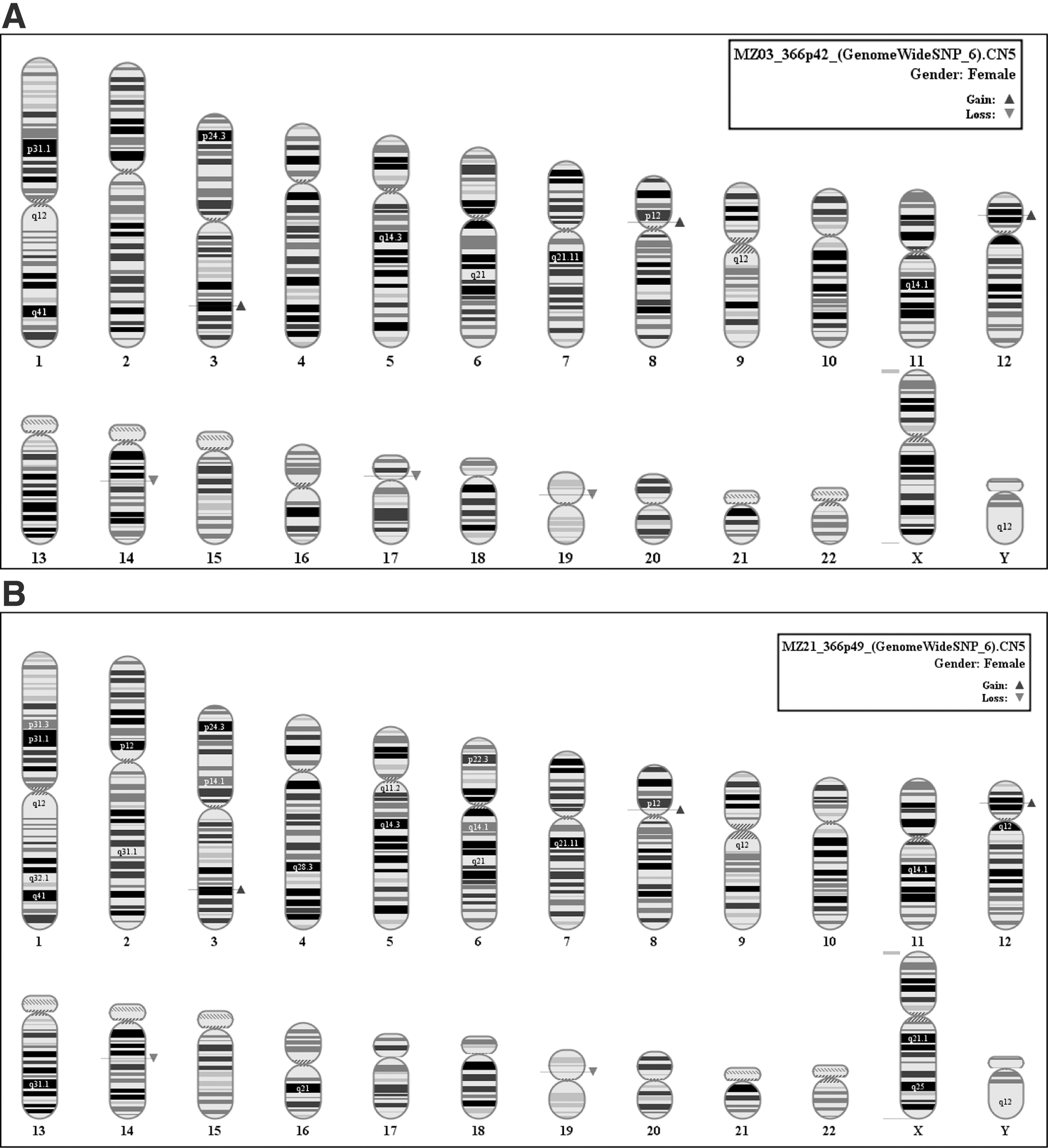
In a similar experimental layout, the cell line HS366 at passage 42 showed a total of 6 alterations with a coverage of 835 kb (Fig. 5A). At passage 49, a loss registered on chromosome 17p11.2 had disappeared from the cells (Fig. 5B).
The cell line HS 368 at passage 28 showed 10 variants with coverage of 3 Mb.
After 7 passages in the teratoma (testis 2A), a new gain appeared on chromosome X encompassing ca. 150 kb of the pseudoautosomal gene-rich region PAR1 (ZBED1, which may have a role in cell proliferation, and CD99, which may act as an oncosuppressor) (Fig. 6A). In one of the teratomas, we found new mutations on 1p36.13, 4.q13.2, 7p22.3, 7q11.21, 9p11.2, 10q11.22, Xq25, and Yq11 (Fig. 6B)
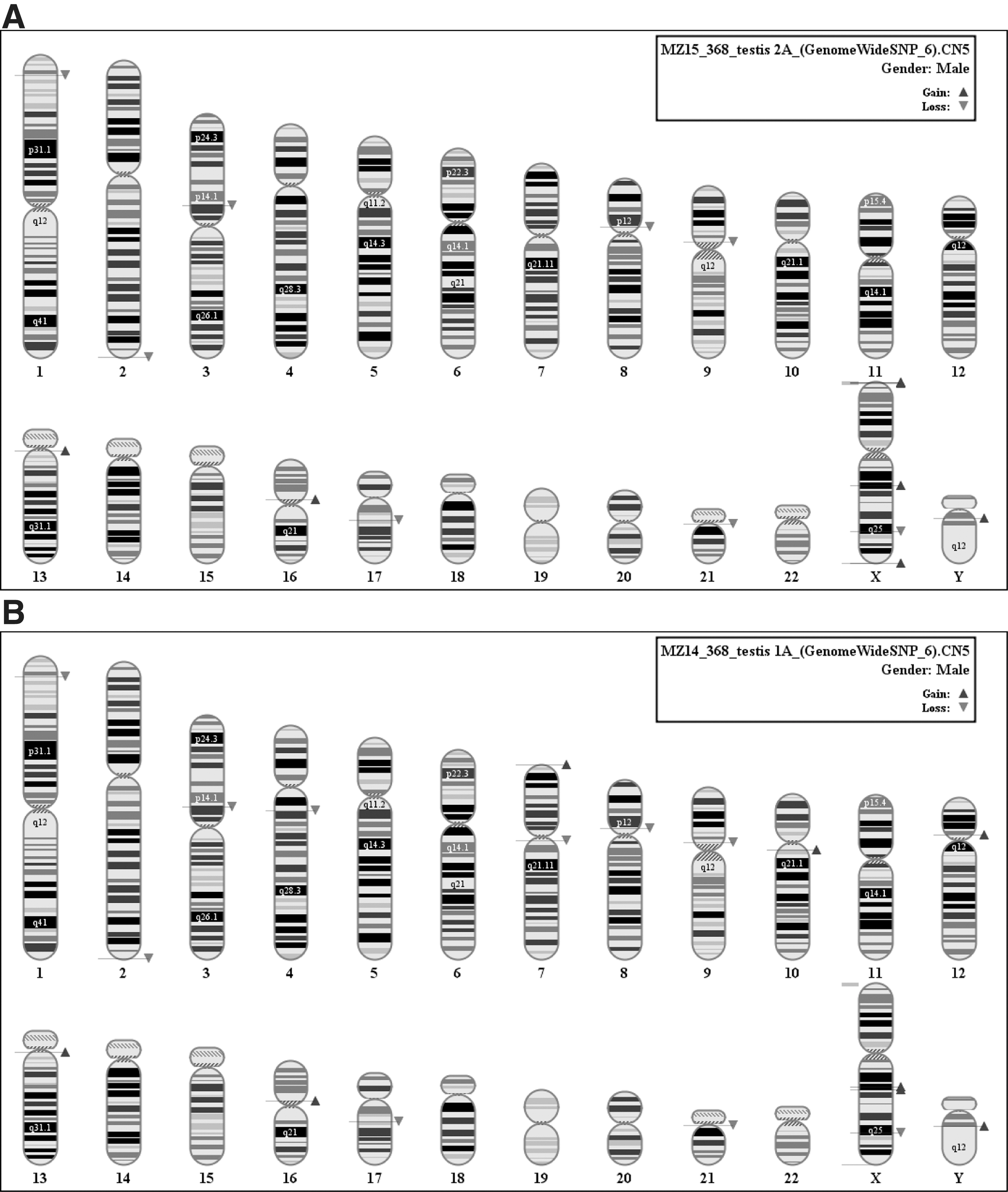
In total, 3 hESC lines were used in this study, namely HS364, HS366, and HS368. Cells from each line were injected in young SCID male mice both in testis and subcutaneous tissue. A total of 12 solid teratomas were generated from these 3 hESC lines. Eight weeks after injection, the formed teratomas were surgically harvested, and DNA was extracted from all teratomas and analyzed by the high-resolution Affymetrix SNP 6.0 array. (Table 1).
Cells at the same passage as the injected ones were cultured in vitro up to 8 weeks, until teratomas were extracted, to compare their developmental adaptation capacity with the in vivo developing cells.
We assessed the genomic variation by in vitro and in vivo growth comparing mutation coverage (in kb) of the whole genome:
In in-vitro growth, no significant arise of new mutations could be observed. In in-vivo growth, comparing teratomas with cell cultures until teratoma extraction, we observed an increase of mutation coverage of about 30% (median) Table 1. See supplementary data of CNVs of all 3 hESC and relative teratomas (Fig. S1, S2, S3; Supplementary Data available online at
Discussion
hESCs and hiPSCs offer an immense potential as a source of cells for regenerative medicine, but the possible ability of these cells to produce tumors in vivo presents a major impediment for the achievement of this prospect in clinical reality. We have used the high-resolution Affymetrix SNP 6.0 arrays to detect chromosome changes during the differentiation process in vivo after transplantation of the cells in SCID mice and harvesting them later for SNP and FISH analyzes.
Our results on the frequent mutations in vitro confirmed our earlier findings obtained using SNP analyses [23, 24]. The observation of larger chromosomal changes in vivo within teratomas than in in-vitro cultured cells was more unexpected. The teratomas were composed of mature tissues, and did not display any teratocarcinoma features. As visualized by SNP arrays, there were clearly more new mutations in vivo than in vitro during the same period. In vitro, only one new change was seen for each 3 lines. In vivo, in teratomas, there were 67 new variations covering a total of 17.3 Mb in the line HS364. FISH analysis revealed that this was probably due to X mosaicism for XY and XX cells present in the teratoma. In a teratoma from the line HS368, 8 new mutations were seen in the SNP array, and the interphase FISH analysis revealed that 74% of the cells had a normal XY content, whereas 21% showed one X centromere signal and 3 SHOX signals. Changes were seen in all 3 lines, smaller ones using the SNP array. Some gains could be verified by interphase FISH. Our findings confirm that there may be differences between lines how prone they are for mutations, as suggested earlier by Catalina et al. and Närvä et al. [18, 24].
The increase of mutations up to 30% in the teratomas suggests that while the adaptation to the in vitro environment is already achieved, and the rate of the new mutations is small, when injected in the mice, the stem cells readapt to the new environmental conditions.
The 8-week time in vivo was still relatively short, and thus it remains speculative if more changes, and particularly malignant ones, might be formed during longer periods. Such studies are highly indicated to assess the safety of clinical cell transplantations.
Even though containing genomic changes, the tissues seen morphologically in the teratomas did not have any malignant features. They were not invasive or metastatic, and they did not have particularly frequent mitoses. The teratocarcinoma-like hESC line that we reported earlier [22, 23] (Feki et al., Hovatta et al.) was clearly invasive from the very beginning, and it had a chromosomal constitution typical of malignant tumors. Whether a teratoma formed by pluripotent stem cells with mature tissue components turns to a malignant tumor during a longer time is not known. It is not acceptable to leave fast-growing, large teratomas to mice for longer periods. How in vitro predifferentiated cells maintain their genetic stability after transplantation remains to be studied.
On the other hand, only differentiated cells are used in regenerative medicine. Differentiated cells do not have any more teratoma-forming capacity. The results of this study underline the importance of removing all the pluripotent stem cells from the cell population aimed at transplantation. Only some remaining pluripotent cells might be a safety risk. There are different options in removing the pluripotent cells, the most promising on being the use of a panel of 3 to 4 antigens against pluripotency factors as described by Tang et al. [29]
Footnotes
Acknowledgments
We would like to thank Hilda Lunden-Miguel for helping us in the culturing and maintenance of these 3 hESC lines and the Swedish Research Council for funding.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.