
Abstract
Autophagy plays an important role in homeostasis, development, and disease, functioning both as a survival and cell death pathway. However, despite its importance in cell physiology, there is little information about the role of autophagy in stem cells and, in particular, on its implication in their survival and/or cell death. We describe here that in vitro, human mesenchymal stem cells (hMSCs) exhibited a high level of constitutive autophagy. Inhibitors of autophagy such as Bafilomycin A1 (Baf-A1) inhibited the proteolytic degradation associated with autophagy in these cells. In addition, we show that a knockdown in the expression of Bcl-xL is accompanied by a loss of autophagic proteolytic ability. Indeed, Bcl-xL seems to exert a tight control on autophagy regulation, since its reintroduction by a protein construct PTD-Bcl-xL resulted in the reacquisition of autophagy. We show that the suppression of autophagy through the knockdown of Bcl-xL influenced hMSC survival and differentiation. This study expands our knowledge on the control exerted by Bcl-xL on autophagy and illustrates the important role of autophagy in the maintenance and differentiation of adult hMSCs.
Introduction
H
Macroautophagy (hereafter referred to as autophagy) is an evolutionary-conserved catabolic program controlled by the ATG family of genes [7] to recycle proteins and organelles. During autophagy, regions of the cytoplasm become sequestered in double-membrane vesicles known as autophagosomes. These autophagosomes undergo a stepwise maturation process that includes fusion with acidified endosomal and/or lysosomal vesicles, resulting in the delivery of the cytoplasmic contents to the lysosomal compartment. Upon fusion, the contents of the autophagosomes are degraded, and the resulting degradation products are either reused to maintain basal macromolecular synthesis or oxidized by the mitochondria to maintain bioenergetics [8,9]. Recently, emphasis has been placed on understanding the physiological role of autophagy in homeostasis and survival. In mammals, autophagy exists at a basal level and controls homeostatic functions. Recent studies have extended the cytoprotective role of autophagy in the maintenance of cell viability by showing that ATG genes are necessary for survival under different settings in mammals [10,11]. These studies elegantly pinpoint that autophagy is critical for maintenance of bioenergetics and cell viability in vitro, and also plays an essential role in survival of the whole organism in vivo.
Beside the physiological role in tissue homeostasis, autophagy is also paradoxically associated with cell death. This arose from the observation that autophagy is commonly observed in dying cells when massive elimination is required in organs [12]. The existence of autophagic cell death rather than cell death with autophagy [13] has been an important point, because autophagy and apoptosis are often activated together in response to stress [14,15], although have very distinct morphologies [16]. In mammals, the BCL-2 family of proteins plays a dual role in the control of apoptosis and autophagy [17]. Apoptosis can be defined as suicidal cell death with a particular morphology that includes chromatin condensation and is under the control of the BCL-2 family of proteins. The BCL-2 family can be divided into antiapoptotic proteins (e.g., Bcl-2 and Bcl-xL) that possess 4 Bcl-2 homology (BH) domains (BH1–BH4), proapoptotic multidomain proteins (e.g., Bax and Bak) that possess 3 BH domains (BH1–BH3), and the proapoptotic BH3-only proteins (e.g., Bad, Noxa, Puma) [18].
Recent reports suggest that autophagy also plays a role during development [19]. Autophagy has a very distinct role in the maturation of various hematopoietic lineages, including erythroid and lymphoid lineages, [20] and recently, it was shown that autophagy is necessary for the maintenance of adult hematopoietic stem cell, and inhibition of autophagy results in a dysregulated myeloid proliferation [21]. Here, we report that basal autophagy is present in hMSCs, and that this autophagy is dependent on Bcl-xL.
Materials and Methods
Unless stated otherwise, all chemicals were from Sigma-Aldrich, and all cell culture materials were obtained from Gibco (Life Technologies).
MSC isolation and culture
Bone marrow samples were obtained from healthy donors from the Department of Orthopedics at Centre Hospitalier Universitaire de Nantes. The bone marrow cells were isolated by density-gradient centrifugation (Ficoll). The cells collected at the interface were cultured in an alpha-MEM modified with ribonucleosides and deoxyribonucleosides supplemented with 20% fetal calf serum, with 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (complete media) in an atmosphere of 5% CO2 and 95% humidity. MSC cultures were used between passage 2 and 10 passage to avoid changes in proliferation and differentiation rates [22]. Cultures were kept at subconfluent levels (about 75% confluency) and passaged every 5–7 days. Differentiation was induced in hMSCs by culturing the cells for 48 h in a complete medium containing 20 ng/mL human recombinant (h) bFGF (100-18B; PeproTech) and 20 ng/mL hrEGF (100-15; PeproTech). The cells were then cultured in a complete medium containing 10 ng/mL hrBDNF (B-3795; Sigma) to induce differentiation along the neuronal pathway. Osteogenic differentiation: Osteogenic differentiation was induced in vitro by culturing in the Poietics™ hMSC Osteogenic Differentiation (Lonza) for 21 days. Differentiation was determined by staining of mineralized nodules by alizarin red staining (2% alizarin red S; pH 4.2 in H2O) for 2 min.
After 3 weeks of differentiation, the cells were cultured in the Dulbecco's modified Eagle medium (DMEM) with 4.5 g/L glucose supplemented with 10% fetal calf serum, with 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (DMEM-10) in an atmosphere of 5% CO2 and 95% humidity.
For lentiviral infection, cells were cultured with lentiviral particles (Sigma) at a multiplicity of infection=15 in a complete medium 48 h before analysis. The lentiviral particles used for Bcl-xL were TRCN0000033499, TRCN0000033500, TRCN0000033501, TRCN0000033502, and TRCN0000033503 (in bold are those used in our study). Alternatively, a HIV-1 lentivirus-based vector was used to introduce shRNAs into the cells. ShRNAs were cloned under the control of the U6 promoter between Bam-H1 and Hind-III sites in pSilencer 2.1 (Ambion), according to the manufacturer's protocol. PCR-amplified fragments were further subcloned between Xba-I and Xho-I sites of the FG12 lentivector.
Bcl-xL was reintroduced into the cells using a Permeable Transmembrane Domain protein construct, PTD-Bcl-xL (PTD sequence=YGRKKRRQRRR). The cells were cultured in the presence of 20 μg/mL PTD-Bcl-xL for 16 h at 37°C. For transfection of the cells with a plasmid, hMSCs (106) were nucleofected with 2 μg plasmid and pEGFP-C1 or the construct MAP-LC3 insert (pLC3) using the Amaxa human MSC nucleofector kit according to the manufacturer's instructions (Lonza). Twenty-four hours after transfection, the cells were used in experiments. Cell viability and numbers were determined by Trypan blue exclusion using the Countess™ automatic cell counter (Life Technologies).
Fluorescence-activated cell-sorting analysis
Bone marrow MSCs were analyzed for cell surface antigen expression by fluorescence-activated cell sorting (FACS). Briefly, 2×105 cells were incubated in phosphate-buffered saline (PBS) containing 2 mM ethylenediaminetetraacetic acid and 0.5% bovine serum albumin (BSA) for 30 min at 4°C with CD11c-PE (BD-Pharmingen), CD41-PE (Immunotech), CD34-FITC (Immunotech), CD44-FITC (AbD Serotec), CD90-PC5 (Beckman Coulter), CD105-FITC (AbD Serotec), and CD117-APC (Beckman Coulter). Identical IgG isotypes (BD Bioscience) were used as negative controls. Cells were washed twice in PBS before analyzed on FACScalibur (BD Biosciences) using Cell Quest Pro software. A minimum of 10,000 events were acquired for each condition. The threshold was adjusted in the forward and the scatter dot plot to exclude cell debris.
Autophagy assays
Labeling of autophagosomes: Autophagy was induced by amino acid and serum starvation as follows: cells were washed 3 times with PBS and incubated for 6 to 9 h in the Hank's Buffered Salt Solution (HBSS) buffered with 2.2 g/L NaHCO3 and supplemented with 0.1% BSA. Autophagic vacuoles were stained with monodansylpentane (MDH) as described by Niemann et al. [23]. Briefly, cells were grown on gelatin-coated glass coverslips to ∼60% confluency, then transferred into HBSS for 6 h, washed with PBS, and incubated for 30 min with 200 μM MDH at 37°C in the dark, then washed with PBS, and mounted with a mowiol polymerizing agent for an immediate observation under UV (λex=359 nm) on a Leica DMLB microscope. Digital pictures were acquired with a Leica DC 300-F camera. Alternatively, the autophagosomes were quantified by the presence of green fluorescent protein (GFP)-labeled MAP-LC3.
Degradation of long-lived proteins: Degradation of radioactive L-[14C]valine-labeled long-lived proteins was measured as follows: cells were incubated for 24 h in a complete medium with 0,1 μCi L-[14C]valine to label total proteins. Radioactivity was then prechased for 1 h in the complete medium in the presence of an excess of L-valine (10 mM) to remove the contribution of short-lived protein degradation. Finally, cells were incubated for 6 to 9 h in either the complete medium or the HBSS in the presence or in the absence of Bafilomycin A1 (Baf-A1) and an excess of L-valine. Supernatants were collected and free amino acids precipitated in 80% trichloroacetic acid (TCA), whereas proteins in the adherent cells were precipitated in 10% TCA. The radioactivity was quantified in the scintillation liquid analyzer Tri-Carb 2100TR (Packard). Proteolysis is expressed as the percentage of free radioactivity released into the supernatant relative to the total radioactivity.
Lysosome labeling
Cells were cultured on gelatin-coated glass coverslips until ∼60% confluency and then incubated in for 30 min with 50 nM LysoTracker Red DND-99 (Invitrogen) at 37°C in the dark. The cells were then washed with PBS and mounted with mowiol for immediate observation under a Leica DMLB microscope. Digital pictures were acquired with a Leica DC 300-F camera.
Video microscopy
Time-lapse video-microscopy experiments were performed using a Zeiss Axiovert 200-M inverted microscope (Carl Zeiss) and the AxioVision 4.8 program. Dishes were placed inside the Incubator XL-3, on a heating insert M06 (37°C) topped with a CO2-cover HM connected to a CO2 controller that maintained the environmental CO2 concentration at 5% for the duration of filming. Digital pictures were acquired and saved every 10 min over 48 h using an AxioCam MR digital camera. The series of photographs were displayed as continuous time-lapse movies for analyses.
Western blots
Total proteins were extracted in 1% NP-40, 0.5% sodium-deoxycholate, and 0.1% sodium dodecyl sulfate (SDS) supplemented with a protease inhibitor cocktail from Roche Diagnostics. Protein concentration was determined using the Bradford assay (Bio-Rad). Protein extracts were separated on SDS-polyacrylamide gel electrophoresis, transferred onto polyvinylidene fluoride membrane (Millipore), and revealed with electrochemiluminescence (Roche Diagnostics). Primary antibodies were used at 1/1000 dilution: mouse monoclonal anti-actin (Chemicon, Millipore), rabbit polyclonal anti-Beclin-1 (NB500-249; Novus Biologicals), and rabbit monoclonal anti-Bcl-x (Epitomics); rabbit antisera directed against human LC3 (L7543; Sigma) was used at 1/250 dilution. Horseradish peroxidase-conjugated secondary antibodies were from Bio-Rad. Quantification was performed with ImageJ software.
Results
Human MSCs exhibit strong basal and inducible autophagy
We have recently shown that undifferentiated hMSCs are extremely resistant to apoptosis [22], and as such could be an interesting tool to study alternate cell death/survival programs such as autophagy.
Human bone marrow mesenchymal stem cells (hMSCs) were isolated from bone marrow aspirates after adhesion to plastic tissue culture Petri dishes in a complete medium as described previously [22]. To limit errors, which could occur due to the presence of senescent or differentiated cells observed in later passages [24], we used cells between passages 2 and 10. The FACS analysis of the hMSCs showed the following classical mesenchymal phenotypes: CD45+, CD90+, and CD105+, and no classical hematopoietic markers such as CD34-, HLA DR-, CD11c-, CD14-, and CD117- [22]. Note that for all the experiments performed, at least 3 different bone marrow aspirates were used to validate the data obtained in this study.
Analysis of autophagy in hMSCs was detected after staining the autophagosomes in vitro with MDH [23] and the microtubule-associated protein light chain 3B (LC3B), an autophagy-controlling ubiquitin-like protein that binds to autophagosomes. The data in Fig. 1A and B show that there is an important basal autophagy in hMSCs, as untreated cells already exhibit high amount of MDH or GFP-LC3-labeled vesicles. In addition, autophagy was upregulated by either rapamycin (an inducer of autophagy) or amino acid starvation (cultured in HBSS) and then detected by staining the autophagosomes with MDH (Fig. 1A). A number of stained vacuoles were observed in the untreated hMSCs. After treatment with rapamycin or starvation, the number of labeled vacuoles increased dramatically. Similar observations were observed in hMSCs transfected with a GFP-LC3 plasmid (Fig. 1B). We observed that the number of GFP-labeled vesicles increased in cells cultured in HBSS or treated with 100 nM rapamycin. This basal autophagy was present in all cultures regardless of the number of cells present and the number of passages (data not shown). Quantification of autophagy cells cultured in complete media or in HBSS showed no statistical difference in the mean number of autophagosome vesicles (AV) present in the cells (n=200; P=0.2382); however, the mean volume of the AV was statistically significant (P=0.00003; Fig. 1C). Concomitantly, labeling of the lysosomes using a lysotracker showed no co-localization between AV and the lysosomes (Supplementary Fig. S1; Supplementary Data are available online at

Autophagy in hMSCs.
During autophagy, LC3 is cleaved into LC3-I, which is then conjugated with phosphatidylethanolamine and identified as LC3-II, localized in the membranes of autophagosomes [25]. Monitoring LC3-II is widely used to assay the autophagic activity in cells. However, LC3-II can be degraded in the autophagosome due to its localization in the autophagosomes. In addition, an accumulation of LC3-II has been observed when the process of autophagosome–lysosome fusion or downstream protein degradation is impaired [26], to exclude this possibility that the accumulation of LC3-II was also determined in the presence of the inhibitor of vacuolar H(+)-ATPase Bafilomycin A1 (Baf-A1), which inhibits lysosome acidification and prevents lysosomal degradation. As shown in Fig. 1D, Baf-A1 caused the accumulation of LC3-II and not LC3-I, suggesting that the autophagic flux was intact in these cells. Next, we analyzed the effect of Beclin-1, a major actor in autophagy, knockdown in these cells under starvation. Western blot analysis of Beclin-1 knockdown in hMSCs indicated an approximate 80% decrease in Beclin-1 expression (Supplementary Fig. S2). As can be seen in Fig. 1E, the knockdown of Beclin-1 completely abrogated the formation of autophagic vesicles when compared to cells infected with shscr.
Further, to validate the presence of autophagy in these cells, we analyzed the degradation of L-[14C]valine-labeled long-lived proteins after 6 h of starvation (Fig. 2A). Starvation as assessed by culturing the cells in HBSS caused a 2-fold increase in proteolysis, which was almost completely inhibited when the cells were cultured in HBSS and treated with 0.1 μM Baf-A1.
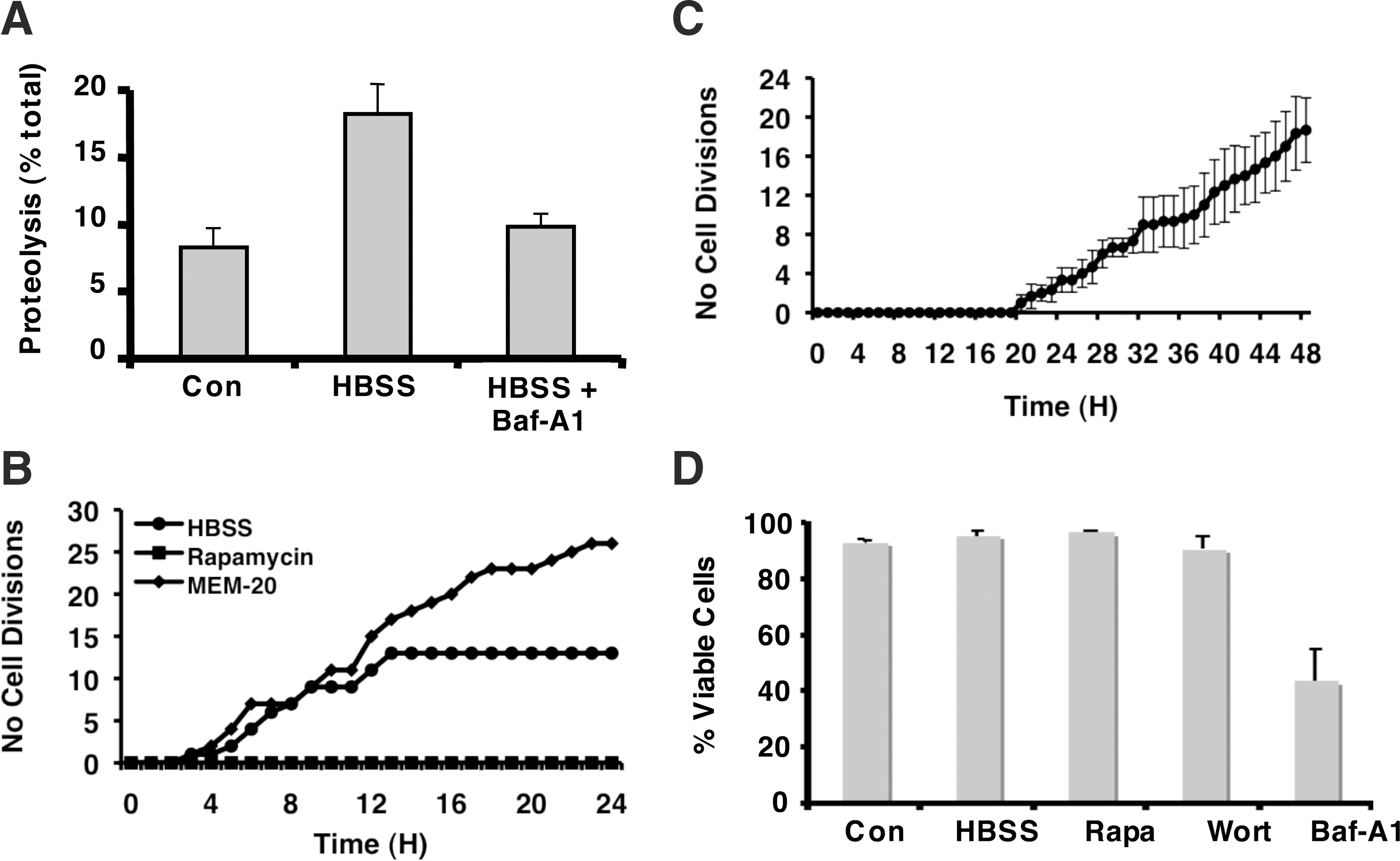
Measure of autophagy in hMSCs.
To analyze these cells under starvation, the proliferation of hMSCs was assessed by estimating the number of cell divisions using time-lapse microscopy under normal conditions, under starvation, or during a rapamycin treatment. As shown in Fig. 2B, cells cultured in the continual presence of serum continued to proliferate, whereas cell divisions were observed in the cultures under starvation during the first 15 h, and then the cells became quiescent, and no further cell divisions were observed. However, this did not lead to any significant cell death (Supplementary Fig. S3) even after 7 days (data not shown).
To validate the quiescent state of the cells after starvation, hMSCs were cultured in HBSS for 72 h and then cultured in complete media; the number of cell divisions was determined by video microscopy. As demonstrated in Fig. 2C, the cells started to proliferate after about 20 h in complete media. Autophagy is controlled by the PI3K-signaling cascade and is negatively regulated by mTOR [27]. It has been shown that stimulation of class I PI3K inhibits autophagy at the sequestration step, while class III PI3K is necessary to activate autophagic sequestration [28]. As shown in Fig. 2D, wortmannin (an inhibitor of PI3Ks), which inhibits autophagic sequestration, or rapamycin (an activator of autophagy) did not induce significant cell death. Similar results were obtained with 3-methyladenine, another inhibitor of autophagic sequestration (data not shown). However, Baf-A1, a V-ATPase inhibitor that inhibits the formation of autolysosomes (Supplementary Fig. S4), had a significant effect on cell death (Fig. 2D). Thus, these data show that wortmannin and 3-methyladenine, which inhibit both class I and class III PI3K, have no effect on cell viability, but have opposite effects on the regulation of autophagy, whereas in contrast, Baf-A1, which inhibits the terminal step in the autophagic process, has a more potent effect on viability.
Role of Bcl-xL in autophagy in hMSCs
We have recently shown that Bcl-xL was a key molecular regulator of autophagy in the HCT116 cell line [29]. Since hMSCs expressed Bcl-xL, but not Bcl-2 [22], we performed Bcl-xL knockdown experiments in these cells and analyzed its effect on autophagy. For this, we used 5 different shBcl-xL sequences delivered by lentiviral infection. The results in Supplementary Fig. S5 showed that shBcl-xL500 and shBcl-xL501 gave the maximum knockdown of Bcl-xL, whereas shBcl-xL499 had no effect on the level of expression of Bcl-xL. For the rest of the experiments presented here, we have used shBcl-xL501 to knockdown Bcl-xL and shBcl-xL499 as the control. In the first set of experiments, hMSCs infected with shBcl-xL499 or shBcl-xL501 were cultured in complete media or in HBSS for 6 h, and then autophagy was determined by MDH staining. The data in Fig. 3A illustrate that the cells infected with shBcl-xL499 were capable of autophagy, whereas the cells infected with viral particles containing shBcl-xL501 were incapable of autophagy even under starvation.
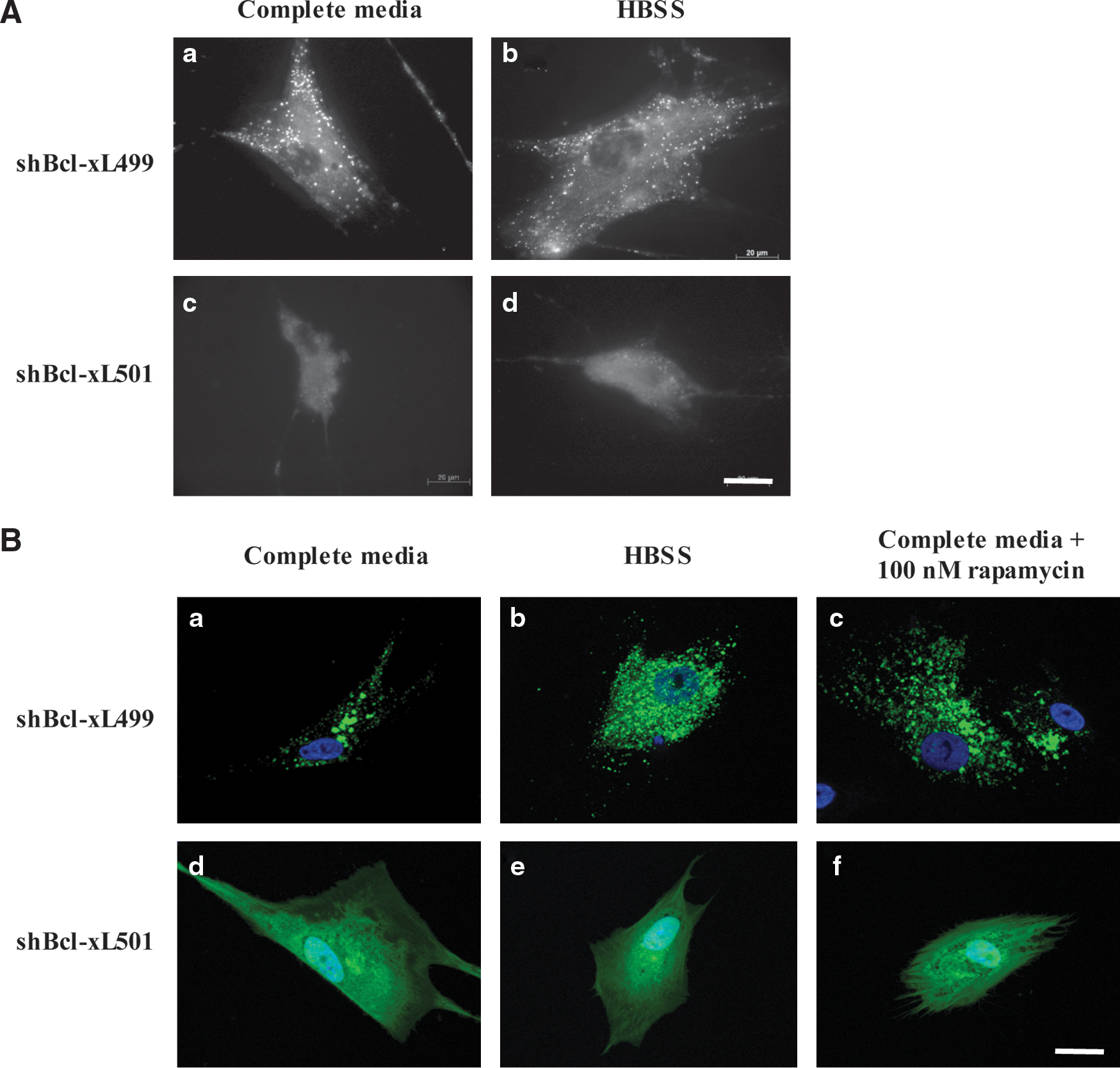
Bcl-xL stimulates survival autophagy.
To validate these results, cells infected with the different viral particles were transfected with GFP-LC3. These cells were then cultured in either complete media or HBSS, or were treated with rapamycin. The results in Fig. 3B show that GFP staining remained diffuse in cells infected with shBcl-xL501 under all conditions, whereas in cells infected with shBcl-xL499, a punctate GFP staining was observed under control conditions, and this staining increased under starvation and in the presence of rapamycin, suggesting the presence of autophagosomes in these cells. The results showed that knockdown of Bcl-xL markedly inhibited the autophagic response in hMSCs.
Human MSCs were subjected to a double infection using shBcl-xL499 and GFP or shBcl-xL501 and GFP. We used the double infection, because we were not able to select after infection, and this could introduce false positives. Forty-eight hours postinfection, the cells were cultured in the presence of a PTD-Bcl-xL construct (PTD=permeable transmembrane domain). The PTD peptide coupled to Bcl-xL permitted the internalization of Bcl-xL into the cells [30]. After 16 h of incubation in the presence of PTD-Bcl-xL, the cells were cultured in a complete medium or in HBSS, and then the presence of autophagic vesicles was determined after MDH staining. The data in Fig. 4A show that the reintroduction of Bcl-xL into cells, in which the expression of Bcl-xL was altered, resulted in the reacquisition of autophagic vesicles in these cells. These results confirm that Bcl-xL is necessary for autophagy in hMSCs. Note that infection of hMSCs with the shBcl-xL501 or shBcl-xL499 plus GFP gave similar results to those obtained in the absence of the GFP plasmid (Supplementary Fig. S6). In addition, the number of AV observed after the reintroduction of Bcl-xL into cells that had a maximal knockdown of Bcl-xL (shBcl-xL501) was not different from that detected in cells infected with shBcl-xL499, suggesting that there was no additive effect.

Reintroduction of Bcl-xL into Bcl-xL knockdown hMSCs.
As stated above, hMSCs do not express Bcl-2, and we asked whether the effect of Bcl-xL on autophagy could be replaced by Bcl-2. Human MSCs that had previously been infected by shBcl-xL501 were transfected with pCMV or pBcl-2 and then incubated in a complete medium or in HBSS. The results in Fig. 4B show that the cells transfected with pCMV contained no autophagic vesicles, and even under starvation, no autophagic vesicles were present. However, after transfected with a plasmid containing Bcl-2, these cells were autophagy competent and the number of autophagic vesicles augmented under starvation. As expected, the number of autophagic vesicles was much lower in these cells as compared to control hMSCs expressing Bcl-xL (data not shown).
Autophagy in differentiation-induced hMSCs
We have noted that hMSCs are resistant to cell death induced by a variety of drugs, unless they are engaged into a differentiation process [22]. In continuum, we analyzed autophagy in hMSCs and in hMSCs induced to differentiate into osteoblasts (see Materials and Methods). Differentiation of hMSCs into osteoblasts was determined after red alizarin staining of the extracellular matrix (Fig. 5A).
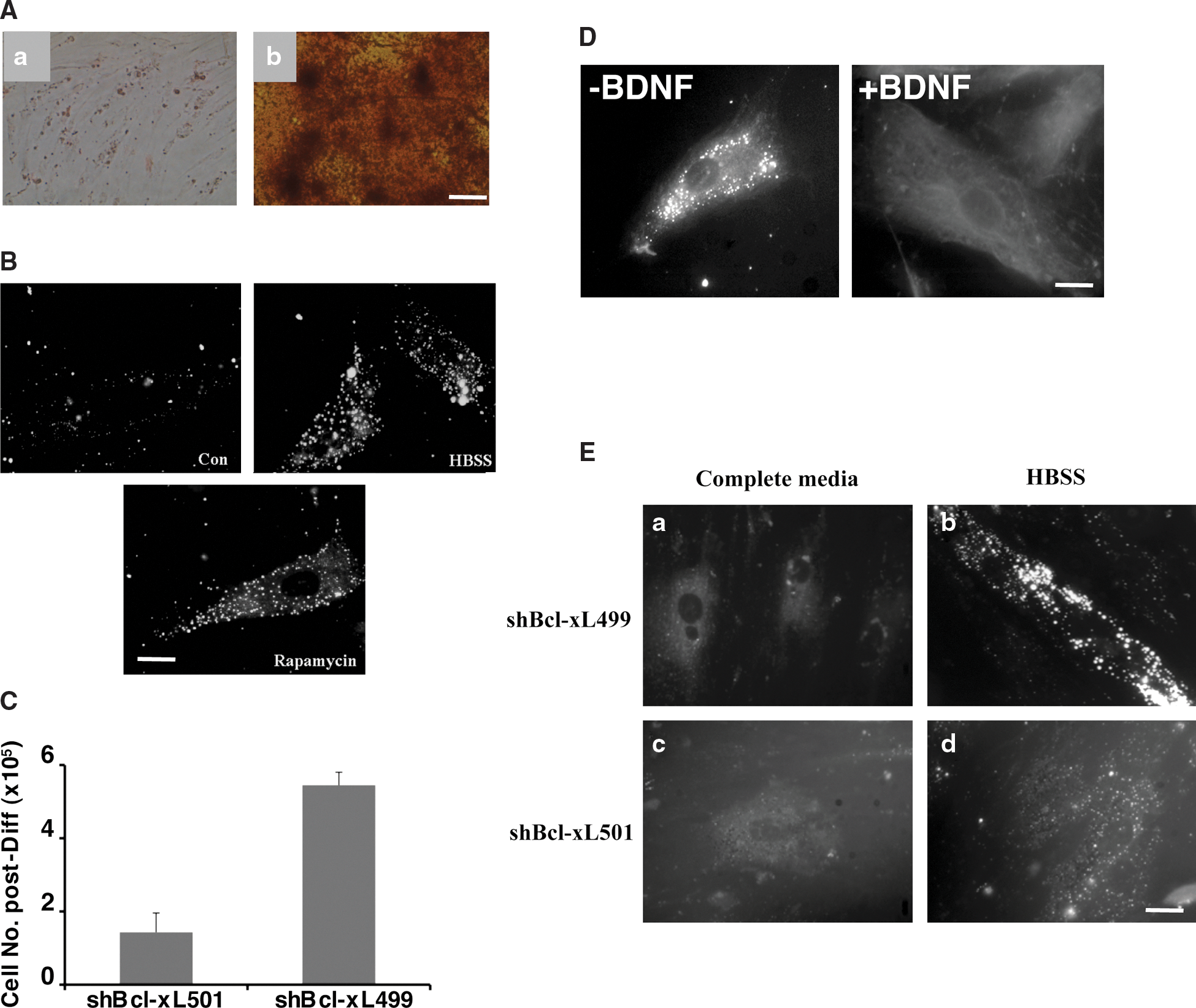
Autophagy in differentiated cells.
As shown in Fig. 5B, no basal autophagy was detected in differentiated osteoblasts; however, it was possible to induce autophagy in these cells after a treatment with rapamycin or under conditions of starvation. To validate the role of Bcl-xL in autophagy, hMSCs were infected with shBcl-xL499 and shBcl-xL501 and then induced to differentiate along the osteogenic pathway. As expected, we observed that very few cells survived the differentiation process, and the number of cells obtained after the 3 weeks of osteogenic differentiation was 3.8 times less in shBcl-xL501-infected cultures when compared to shBcl-xL499-infected cultures (Fig. 5C); this could be due to the survival function of Bcl-xL during either apoptosis or autophagy. However, since similar results were obtained with hMSCs infected with shBeclin-1, this autophagy is likely to be involved in survival (data not shown).
In addition, autophagy was induced by starvation in undifferentiated hMSCs and in hMSCs induced to transdifferentiate along the neuronal pathway. This transdifferentiation was induced in hMSCs along the neuronal pathway by preincubation for 48 h with 20 ng/mL bFGF and 20 ng/mL EGF, followed by incubation in the presence of 10 ng/mL BDNF for 24 h. As depicted in Fig. 5D, undifferentiated cells had a basal autophagy that was absent once differentiation was initiated in these cells. Note that similar results were obtained with hMSCs induced to differentiate into adipocytes or chondrocytes (data not shown).
During the differentiation of hMSCs into osteoblasts, there is an expression of Bcl-2 in these cells. To determine if the expression of Bcl-xL was necessary for autophagy in osteoblasts, these cells were infected with shBcl-xL499 or shBcl-xL501 and were cultured in a complete medium or HBSS for 24 h and then stained with MDH. As shown in Fig. 5E, the knockdown of Bcl-xL in osteoblasts reduced the number and the size of the autophagosomes present under conditions of starvation, suggesting that Bcl-2 can partially supplement for Bcl-xL in osteoblasts.
Discussion
The role of autophagy has been particularly studied during tissue remodeling during development and as a stress response in parallel with apoptosis [27]. It has been suggested that autophagy is necessary in the maintenance of adult hematopoietic stem cells, thereby protecting these cells against malignant transformation [21]. These authors have shown that normal quiescent hematopoietic stem cells are maintained in hypoxic niches, providing the optimal microenvironment to sustain their functions and life-long integrity. Hematopoietic stem cells and MSCs share the same bone marrow niche; in fact, MSCs express high levels of hematopoietic stem cell maintenance factors [31].
Recent works have established in embryonic [32] and in adult cardiac and hematopoietic stem cells [33, 34] that autophagy is required for self-renewal and differentiation. A possible role for autophagy in cellular reprogramming has been discussed by Vessoni et al. [35].
In this report, we show that basal autophagy is abundant in hMSCs (Figs. 1 and 2) and can be augmented under stress (starvation) or after inhibition of mTOR. However, after differentiation of the hMSCs into osteoblasts or transdifferentiation of hMSCs into neurons (Fig. 5), basal autophagy is completely inhibited. The absence of a basal autophagy does not imply that these differentiated cells are not capable of autophagy, since autophagy can be induced either by starvation or by inhibition of mTOR. We have recently described a similar proautophagic function of Bcl-xL in HCT-116 colorectal cells [29].
We also show that a knockdown of Bcl-xL completely blocked autophagy in these cells. In contrast, Zeng et al. [36] reported that in nonstarved glioblastoma cells, neither Bcl-2 nor Bcl-xL was the normal endogenous binding entity for Beclin 1, suggesting that depending on the settings, these proteins are not obligate partners. Opposite to autophagic cell death, survival autophagy was stimulated by Bcl-2 and Bcl-xL. Bcl-2 and Bcl-xL also seemed committed in the regulation of autophagosome formation, but apparently through nonredundant mechanisms. Such observations are in keeping with the data published that suggested a possible functional difference between Bcl-2 and Bcl-xL in the stimulation of autophagy. [29] We found that Bcl-xL controls the autophagosome formation (Figs. 3 and 4). The mechanism, either direct or indirect, by which Bcl-xL activates autophagy, is not known at the moment. However, studies performed with Bax(-/-) Bak(-/-) cells suggest that one of the physiological roles of these proteins in absence of apoptotic insults may be the suppression of autophagy [37]. Bcl-xL could thus stimulate autophagy by counteracting this inhibition. We reach here in nontransformed cells the very same conclusion that Bcl-xL is involved in cytoprotective autophagy. The molecular mechanism by which Bcl-xL acts on this particular form of autophagy remains to be elucidated.
Of note, lymphocytes derived from Bcl-xL(-/-) mice could differentiate into mature lymphocytes, although the total number of mature cells was less; similar results were observed with other bone marrow cellular components [38]. In this study, we found similar results with hMSC differentiation into several lineages (Fig. 5).
To conclude, we found that both Bcl-2 and Bcl-xL stimulate survival autophagy and help the formation of autophagosomes through nonredundant mechanisms, with Bcl-xL acting on the whole independently of Beclin 1. This work is the first to establish the requirement for autophagy in the maintenance and differentiation of hMSCs and to point out a novel and essential role of Bcl-xL in the control of stem cells. We have shown in a previous study that Bcl-xL knockdown decreased the survival of undifferentiated hMSCs (22). However, Bcl-xL is the key protein in the sensitivity to apoptosis in undifferentiated cells, but its function is bolstered by Bcl-2 in differentiated cells (22). From the latter study and our present work, one could postulate that survival of hMSCs could be controlled by high autophagy and low apoptotic sensitivity in undifferentiated cells and by high apoptotic sensitivity and low autophagy in differentiated hMSCs.
Footnotes
Acknowledgments
We thank Gwenola Bougras for technical assistance, Philippe Hulin from the Cellular and Tissular Imaging Core Facility (MicroPICell), Université de Nantes, for his aid with all the microscopic analyses, and Paul Pilet from the Plateau Technique Microimagerie, Université de Nantes, for his assistance with the electron microscopy. We also thank Dr. Claire Pecqueur for critical reading of the article and fruitful discussions and Dr. Christelle Guegan for the PTD-Bcl-xL protein. A special program from Equipe Labellisée la Ligue Contre le Cancer supported this study.
Author Disclosure Statement
The authors indicate no potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.