
Abstract
Human induced pluripotent stem cells (hiPSCs) exhibit pluripotency, proliferation capability, and gene expression similar to those of human embryonic stem cells (hESCs). hESCs readily form cartilaginous tissues in teratomas in vivo; despite extensive effort, however, to date no efficient method for inducing mature chondrocytes in vitro has been established. hiPSCs can also differentiate into cartilage in vivo by teratoma formation, but as with hESCs, no reliable system for in vitro chondrogenic differentiation of hiPSCs has yet been reported. Here, we examined the chondrogenic differentiation capability of hiPSCs using a multistep culture method consisting of embryoid body (EB) formation, cell outgrowth from EBs, monolayer culture of sprouted cells from EBs, and 3-dimensional pellet culture. In this culture process, the cell density of monolayer culture was critical for cell viability and subsequent differentiation capability. Monolayer-cultured cells exhibited fibroblast-like morphology and expressed markers for mesenchymal stem cells. After 2–3 weeks of pellet culture, cells in pellets exhibited a spherical morphology typical of chondrocytes and were surrounded by extracellular matrix that contained acidic proteoglycans. The expression of type II collagen and aggrecan in pellets progressively increased. Histological analysis revealed that over 70% of hiPSC-derived pellets successfully underwent chondrogenic differentiation. Using the same culture method, hESCs showed similar histological changes and gene expression, but differentiated slightly faster and more efficiently than hiPSCs. Our study demonstrates that hiPSCs can be efficiently differentiated into the chondrogenic lineage in vitro via generation of mesenchymal progenitor cells, using a simplified, multistep culture method.
Introduction
Human induced pluripotent stem cells (hiPSCs) have been generated from somatic cells by introducing Oct3/4 and Sox2 along with either Klf4 and c-Myc or Nanog and Lin28, using retroviruses or lentiviruses [11 –13]. hiPSCs exhibit pluripotency, proliferation capability, and gene expression similar to those of human embryonic stem cells (hESCs), but do not present the same ethical problems. Immune rejection can be avoided by the establishment of hiPSC banks from donors with various human leukocyte antigen (HLA) types. Moreover, to reduce the risk of tumorigenicity, new methods for generating iPSCs without viral vectors have been developed [14 –16]. Therefore, hiPSCs are viewed as a promising new tool for regenerative medicine, disease pathogenesis studies, and drug screening. hESCs readily form cartilaginous tissues in teratomas in vivo, but the proportion of chondrocytes arising from spontaneous differentiation via embryoid body (EB) formation in vitro is very low. Three-dimensional (3D) pellet culture and micromass culture have been used successfully for in vitro chondrogenic differentiation of MSCs and mature chondrocytes [17 –19]. These culture systems allow cell–cell interactions analogous to those that occur in precartilage condensation during embryonic development, and can induce terminal differentiation of mesenchymal progenitor cells into hypertrophic chondrocytes. Besides pellet culture and micromass culture, many attempts have been made to induce chondrocytes from ESCs in vitro, including coculture with irradiated articular chondrocytes [20] or limb bud progenitor cells from a developing embryo [21], culture with a conditioned medium [22], genetic manipulation (e.g., Sox9 expression) [23], and use of biomaterials [24]. However, both coculture with other animal cells and culture with a conditioned medium lack reproducibility and carry the risk of pathogen transmission. Genetic manipulation using virus-based vectors poses risks for clinical applications. Thus, in spite of the many efforts, an efficient culture method for inducing mature chondrocytes from hESCs has not yet been established.
hiPSCs have been reported to generate cartilaginous tissue in teratoma in vivo [11 –13], but limited data exists at present regarding the in vitro chondrogenic differentiation of hiPSCs. When dissociated EB cells generated from fetal neural stem cell-derived hiPSCs were grown with a chondrogenic medium on agarose-coated wells, they expressed chondrogenic differentiation markers detectable by immunofluorescence staining [25]. However, immunofluorescence did not clearly reveal morphological characteristics of chondrocytes; a large and spherical shape with clear cytoplasm surrounded by an extracellular matrix [25]. Thus, a reproducible method for in vitro chondrogenic differentiation of hiPSCs must be established. Since hiPSC-derived chondrocytes can be used to study the pathogenesis of genetic disorders, such as skeletal dysplasia, and to develop drug screening systems for cartilage diseases, it would be desirable to develop in vitro differentiation methods that mimic physiological differentiation processes and do not require genetic manipulation.
In the current study, we examined the chondrogenic differentiation capability of hiPSCs by combining a pellet culture system, the most reliable method reported to date, with spontaneous differentiation via EB formation, which mimics early embryonic development by forming the 3 germ layers (mesoderm, ectoderm, and endoderm). We also compare and discuss the chondrogenic cells induced from hiPSCs and hESCs using our culture protocol.
Materials and Methods
Cell culture
hiPSC line 201B7 (B7) was previously generated by introducing 4 reprogramming factors (Oct3/4, Sox2, Klf4, and c-Myc) into dermal fibroblasts from the facial dermis of a 36-year-old Caucasian woman [11]. The hESC line H9 was obtained from the WiCell Research Institute (Madison, WI,
Chondrogenic differentiation
For EB formation, hiPSC and hESC colonies were harvested by treating with 1 mg/mL collagenase type IV, and then plated onto suspension culture dishes, where they were allowed to aggregate in a maintenance medium without bFGF. After 7 days as a suspension culture, EBs were transferred to gelatin-coated dishes and cultivated for 1 week in DMEM containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. EBs and cells sprouted from EBs were harvested and dissociated with 0.25% trypsin/EDTA (Life Technologies, Inc., Grand Island, NY,
Flow cytometry
Cells were incubated with saturating levels of antibodies for 1 h at 4°C. The following fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated antibodies recognizing human antigens were used: TRA-1-60 (No. 560380), CD44 (No. 348052), CD90 (No. 555596), CD106 (No. 555647), CD146 (No. 550315), CD166 (No. 559263), CD34 (No. 348057), and CD45 (No. 347463) (BD Biosciences); CD73 (No. 12-0739) and CD105 (No. 12-1057) (eBioscience, Inc., San Diego, CA,
Real-time reverse transcription–polymerase chain reaction
Total RNA was extracted from cell pellets using the RNeasy® Mini Kit (Qiagen, Chatsworth, CA,
Histological analysis
Cell pellets were fixed in 2% paraformaldehyde, dehydrated, and embedded in paraffin. The sections were cut at a thickness of 4 μm and stained with Alcian blue and toluidine blue, as previously described [28]. For immunohistochemical analysis, a labeled streptavidin–biotin staining kit (LSAB2 System-HRP) (Dako, San Antonio, TX,
Statistical analysis
Real-time RT-PCR experiments were performed independently at least 3 times and gave highly similar results. Data are presented as the mean±SD. Statistical analysis was performed using one-factor analysis of variance. Differences were considered statistically significant when P<0.05.
Results
Mesenchymal differentiation of hiPSCs via EB formation
We used a multistep culture method combining spontaneous differentiation via EB formation, cell outgrowth from EBs on gelatin-coated dishes, monolayer culture after cell dissociation into single cells, and 3D pellet culture (Fig. 1). Chondrogenic progenitor cells are derived from MSCs, which originate from the mesoderm and neural crest. Therefore, we first tried to promote differentiation of undifferentiated hiPSCs into the mesenchymal lineage. hiPSCs contained in EBs retained pluripotency in vitro; we used immunofluorescence analysis to confirm the expression of markers for mesoderm (α-smooth muscle actin), ectoderm (β3-tubulin), and endoderm (forkhead box A2; FOXA2) on cultured EBs (Supplementary Fig. S1; Supplementary Data are available online at
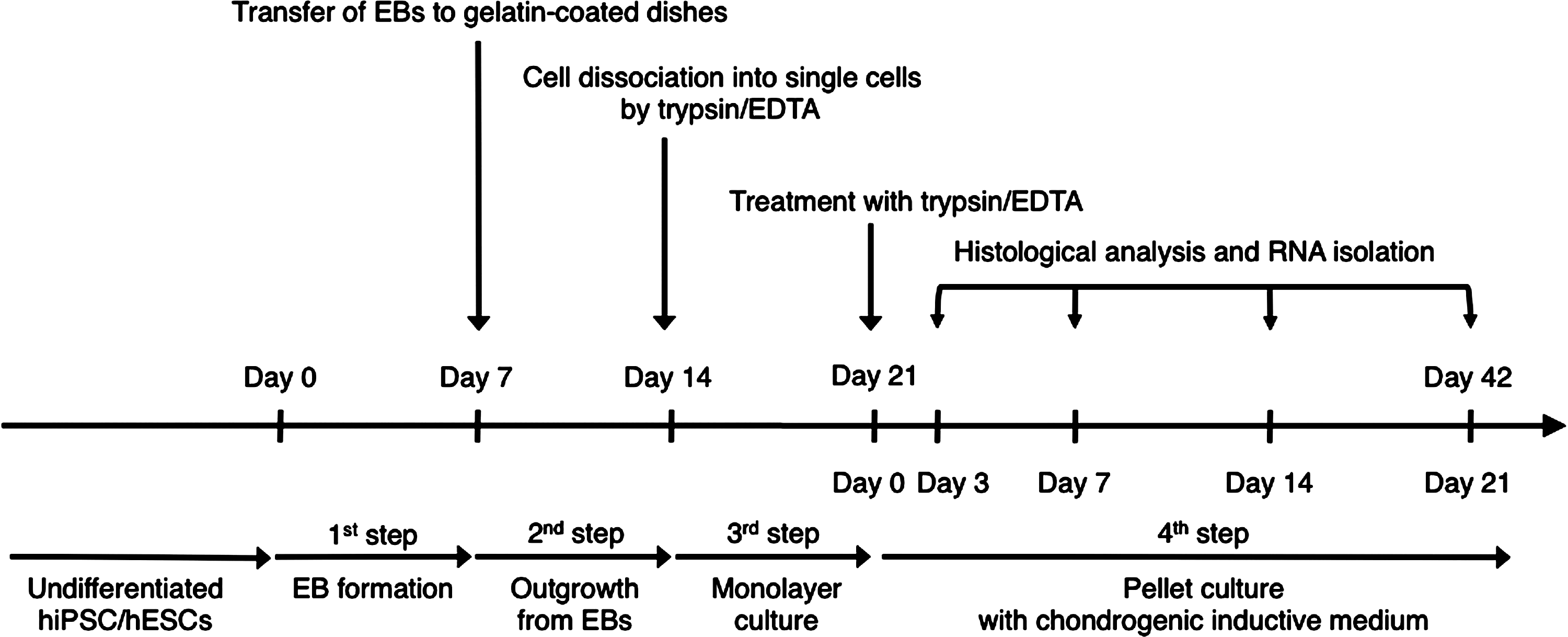
Schematic diagram of the culture protocol for chondrogenic differentiation of human induced pluripotent stem cells (hiPSCs) and human embryonic stem cells (hESCs). The differentiation culture protocol consists of 4 steps: (1) Embryoid body (EB) formation in suspension culture dishes; (2) Cell outgrowth from EBs on gelatin-coated dishes; (3) Monolayer culture under mesenchymal stem cell (MSC) growth conditions after dissociation into single cells by trypsin/EDTA; and (4) Three-dimensional pellet culture in the chondrogenic induction medium. During pellet culture, histological and gene expression analyses were performed at days 3, 7, 14, and 21.
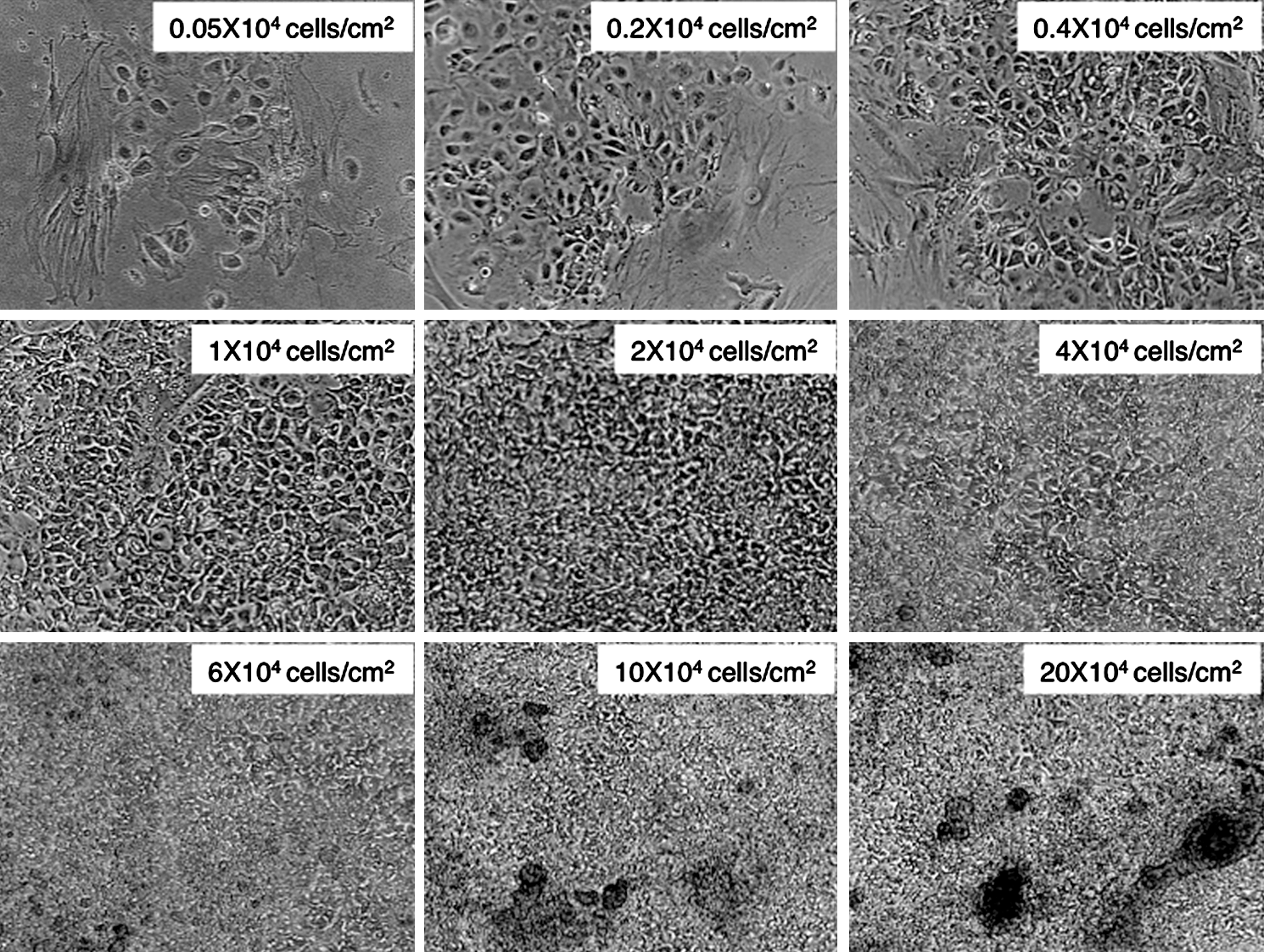
Ideal cell density of hiPSCs for monolayer culture. Phase-contrast micrograph of monolayer cultured hiPSCs at 40× magnification 7 days after plating onto adhesive culture dishes. To determine the ideal cell density, sprouted cells from EBs were dissociated by trypsin/EDTA and serially diluted to yield cell densities of 0.05–20×104 cells/cm2. A cell density at 1–2×104 cells/cm2 was determined to be ideal for cell proliferation without cell aggregation.
Next, before proceeding to pellet culture, we analyzed the expression of cell surface markers on hiPSCs in monolayer culture. Flow cytometric analysis demonstrated that the majority of hiPSCs expressed CD90 (Thy-1), CD44, and CD166 (activated leukocyte cell adhesion molecule) (Fig. 3). A small fraction of hiPSCs was positive for CD146 (MUC18), CD73, CD105 (endoglin), and CD140a (platelet-derived growth factor receptor α [PDGFRα]) (Fig. 3). CD73, CD90, CD105, CD44, CD166, and CD146 are known to be markers for MSCs [29,30]. PDGFRα is a marker for paraxial mesodermal cells, from which cartilage tissues originate, especially in mouse development [31]. TRA-1-60, an undifferentiated cell marker both for hiPSCs and hESCs, was not detected in monolayer-cultured hiPSCs (Fig. 3), suggesting that the multistep culture process promoted differentiation of hiPSCs. CD34 and CD45, hematopoietic markers that are not expressed in MSCs [29], were also not detected in monolayer-cultured hiPSCs (Fig. 3). However, we did not detect the expression of the MSC markers Stro-1 and CD106. Stro-1 has been established as a monoclonal IgM derived from mice immunized with human CD34-positive bone marrow cells [32]. Recently, it was reported that Stro-1 is a 75-kDa endothelial antigen, and that its expression on MSCs might result from induction of MSCs to endothelial lineage [33]. Monolayer-cultured hiPSCs in our method may commit to differentiating into mesenchymal lineage cells, which do not contain endothelial antigens. Furthermore, few reports have demonstrated the expression of Stro-1 on MSCs derived from hiPSCs and hESCs, although many attempts have been made to induce MSCs from hiPSCs and hESCs [34]. In a recent report describing one-step derivation of MSC-like cells from hiPSCs, MSC-like cells did not express Stro-1, although the cells clearly expressed CD73, CD90, CD105, CD144, and CD166 [35]. A Nestin(+)/CD271(−)/Stro-1(−) cell population from hESCs was recently reported to be mesenchymal-like precursors [36]. MSCs induced from hiPSCs or hESCs in vitro may exhibit different characteristics, including surface marker expression, from those of MSCs isolated primarily from bone marrow and other organs. CD106 is also known as vascular cell adhesion molecule-1 [37]. Several groups have reported changes in the expression of cell surface markers, including CD106, following prolonged cultivation [37,38]. Treatment with hyaluronan, a major glycosaminoglycan ligand of CD44, has also been reported to upregulate the expression of CD106 in MSCs [39]. Thus, it is possible that our culture method, which consists of 3 weeks of culture under 3 different conditions, attenuates the expression of CD106. Nonetheless, expression of CD90, CD44, CD166, CD146, CD73, CD105, and CD140a and the lack of CD34 and CD45 suggest that at least a fraction of hiPSCs in monolayer culture differentiated along the mesenchymal lineage. Indeed, osteogenic differentiation of monolayer-cultured hiPSCs was confirmed using a previously described induction medium [7] (data not shown).
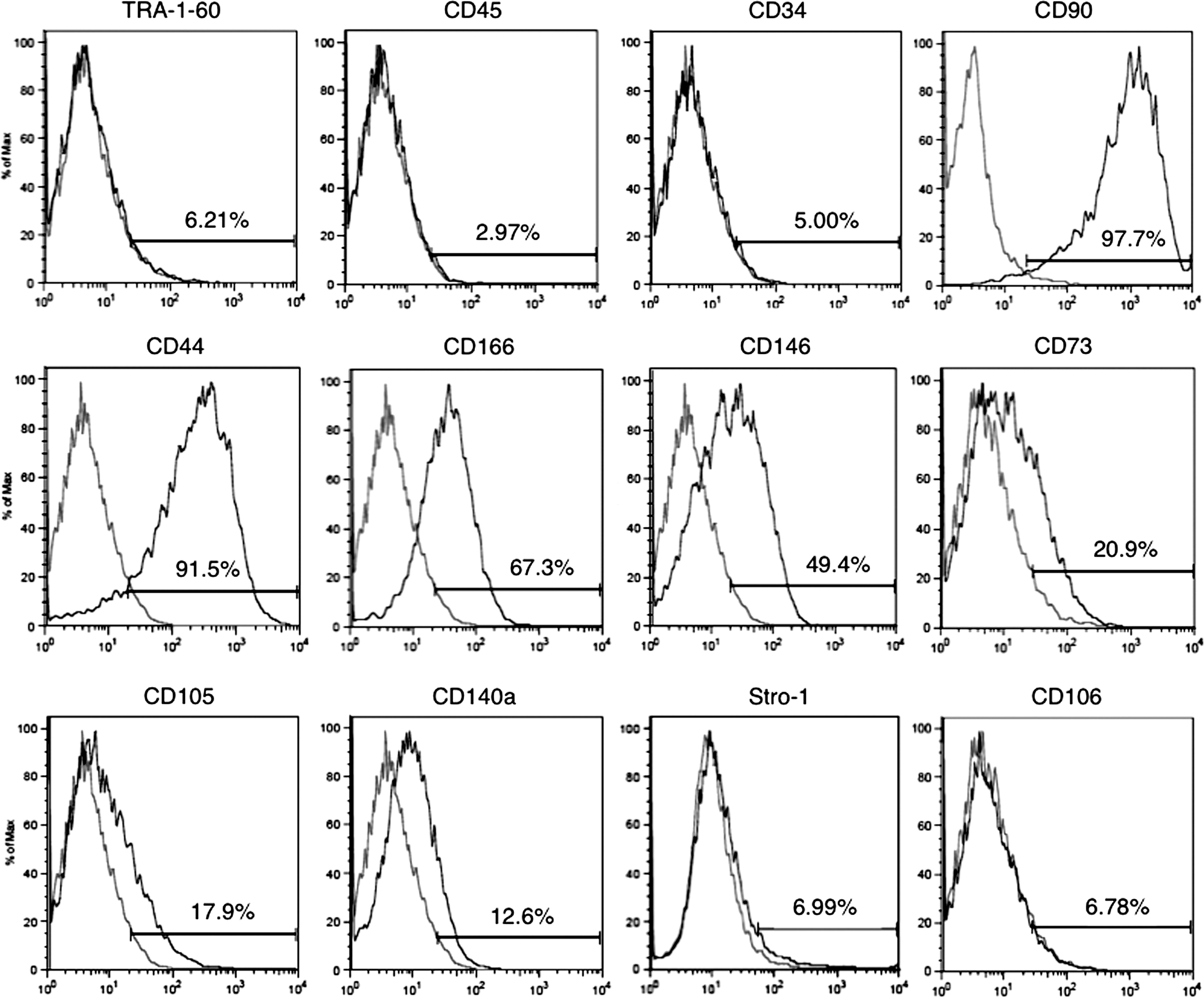
Expression of MSC markers by monolayer cultured hiPSCs. Cells were cultivated in monolayer culture under MSC growth conditions for 7 days, and then dissociated by trypsin/EDTA for pellet culture. Flow cytometric analysis of cell surface marker expression on dissociated hiPSCs revealed that the major fraction of hiPSCs expressed CD90, CD44, and CD166, and that a fraction of hiPSCs expressed CD146, CD73, CD105, and CD140a. hiPSCs were negative for TRA-1-60, a marker of undifferentiated cells, and for hematopoietic markers (CD34 and CD45). Gray lines show staining with the relevant isotype-matched control antibodies.
Chondrogenic differentiation of hiPSCs by pellet culture
The original in vitro chondrogenic differentiation method was initially established using rabbit and human bone marrow-derived mesenchymal progenitor cells [17,18]. The high-density 3D microenvironment is thought to facilitate cell–cell and cell–matrix interactions and to mimic in vivo limb development, in which mesenchymal condensation occurs before chondrogenic induction. In a pellet or micromass culture system, cells change their morphology, express chondrogenic differentiation markers, and produce an extracellular matrix that contains acidic proteoglycans, which stain positive for Alcian blue and toluidine blue. In a previous report, micromass culture using hESCs dissociated from EBs exhibited chondrogenesis superior to that of a two-dimensional (2D) EB direct-plating outgrowth system [40].
In the present study, we dissociated EBs and selected only the cells that proliferated in monolayer culture in the MSC growth medium. After 3 days of pellet culture, little Alcian blue staining was observed, and hiPSCs inside the pellet exhibited fibroblast-like morphology (Fig. 4A). At day 7 of induction, pellets showed partial Alcian blue staining and contained some large spherical cells with typical chondrocyte-like morphology (Fig. 4A). At day 14 of induction, spherical cells and interstitium, which was clearly stained with Alcian blue, were evident throughout the pellet (Fig. 4A). At day 21 of induction, the entire pellet was intensely stained with Alcian blue, suggesting maturation of cartilaginous extracellular matrix (Fig. 4A).
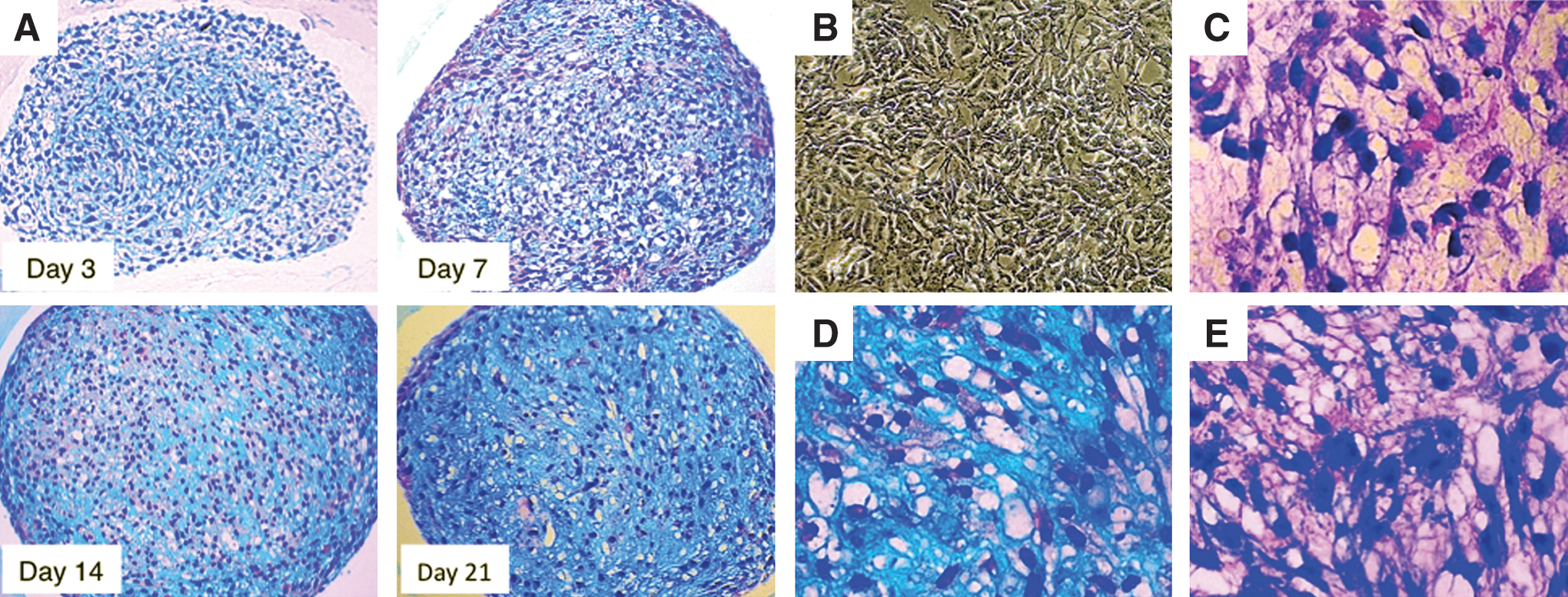
Histological analysis of chondrogenic differentiation from hiPSCs (B7). Histological analysis of paraffin-embedded cell pellets stained with Alcian blue/hematoxylin/eosin (HE) revealed progressive chondrogenic differentiation of hiPSCs. The positive area and intensity of Alcian blue staining, which indicated the existence of acidic proteoglycans, increased progressively
Before chondrogenic induction by pellet culture, hiPSCs in monolayer culture were small and fibroblast-like (Fig. 4B). However, at day 21 of induction, hiPSCs located inside the pellet exhibited large spherical morphology (Fig. 4C) and were surrounded by Alcian blue–positive (Fig. 4D) and toluidine blue–positive (Fig. 4E) acidic proteoglycans. At day 28 of induction, many pellets exhibited internal necrosis (data not shown). Thus, in our method, the differentiation of hiPSCs appeared to reach a plateau at day 21 of induction. The cell morphology and positive staining for Alcian blue and toluidine blue indicated that hiPSCs differentiated into the chondrogenic lineage. To confirm the chondrogenic differentiation capability of hiPSCs, we examined 2 more hiPSC lines. One line (iPS-TIG107) was generated from TIG-107 fibroblasts derived from the skin of an 81-year-old Japanese woman by introducing 4 reprogramming factors using retroviruses. Another line, 253G1 (G1), was derived from the same donor as B7, but generated by introducing 3 factors (Oct 3/4, Sox2, and Klf4, but not c-Myc) using retroviruses [41]. These 2 iPSC lines (iPS-TIG107 and G1) exhibited chondrogenic differentiation capability and time course similar to those of B7 (Supplementary Fig. S4 and S5), although the level of matrix synthesis appeared to be smaller in G1-derived pellets.
Comparative analysis of chondrogenic differentiation capability of hiPSCs and hESCs
We performed the procedure described above with hESCs (H9) and compared their chondrogenic differentiation capability to that of hiPSCs. During 21 days of induction, hESCs demonstrated a sequence of histological changes similar to that observed with hiPSCs (Fig. 5A). At day 3 of induction, some areas of hESC-derived pellets were faintly stained with Alcian blue, and some hESCs already exhibited chondrocyte-like spherical morphology. At day 7 of induction, the whole hESC-derived pellet was stained with Alcian blue. In comparison, hiPSC-derived pellets showed only partial staining at this time. At day 14 of induction, the whole pellet was more strongly stained with Alcian blue. At day 21 of induction, the appearance of cells with chondrocyte-like morphology and Alcian blue staining of the interstitium were more apparent, although the difference between days 14 and 21 was not significant. Thus, compared to hiPSCs, hESCs exhibited slightly faster progression of chondrogenic induction. Before chondrogenic induction by pellet culture, the morphology of monolayer-cultured hESCs was also fibroblast-like, similar to that of hiPSCs (Fig. 5B).
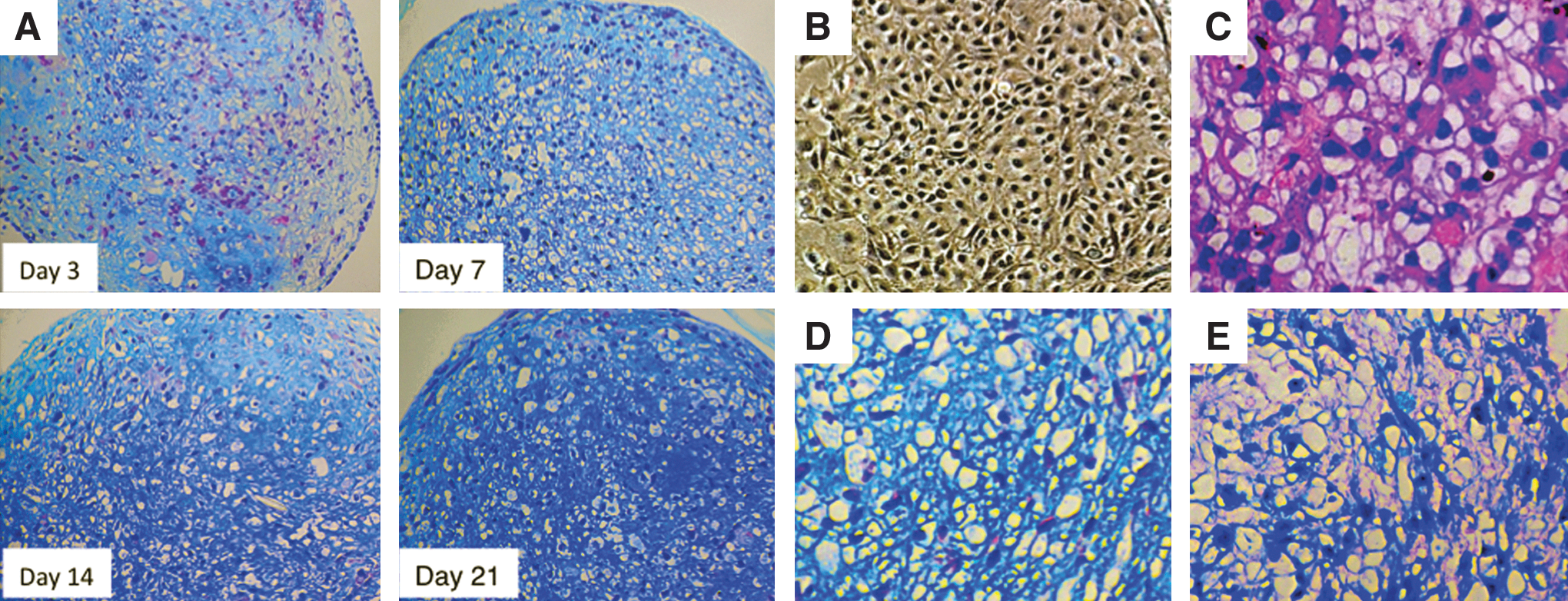
Histological analysis of chondrogenic differentiation from hESCs (H9). Histological analysis of hESC-derived pellets stained with Alcian blue/HE indicated progressive chondrogenic differentiation similar to that observed with hiPSCs. The positive area and intensity of Alcian blue staining increased progressively
hESCs began showing morphological changes at day 3, and matured progressively thereafter (Fig. 5C). At day 21, the hESC-derived pellets showed intense Alcian blue staining (Fig. 5D) and toluidine blue metachromasy (Fig. 5E), as was the case for hiPSCs. At day 28 of induction, many hESC-derived pellets also exhibited internal necrosis (data not shown). About 85% of hESC-derived pellets (11 successful pellets/13 experiments), compared to 73% of hiPSC-derived pellets (8 successful pellets/11 experiments), exhibited successful chondrogenic differentiation. We also applied this culture protocol to another hESC line, KhES-1 [42], and found that it exhibited similar chondrogenic differentiation (Supplementary Fig. S6). These data demonstrate that in our multistep culture method, both hiPSCs and hESCs differentiate into the chondrogenic lineage, although hESCs exhibited slightly faster progression and increased efficiency of chondrogenic differentiation compared to hiPSCs.
Time course of gene expression of chondrogenic differentiation markers
We analyzed the time course of chondrogenic differentiation marker expression by real-time RT-PCR. The chondrogenic transcription factor Sox9 is required for precartilage condensation and directly regulates the transcription of type II collagen and aggrecan [43 –45]. Type II collagen is an early chondrogenic differentiation marker, and aggrecan is a major sulfated proteoglycan of the cartilage matrix and a highly specific marker of differentiated chondrocytes [45]. SOX9 expression was already present in hiPSCs at day 0 of monolayer culture (Fig. 6A), temporarily decreased at day 3 before the elevation of expression of COL2A1 and ACAN, and gradually increased thereafter. COL2A1 exhibited low expression at day 0, but was gradually upregulated at day 3 and 7, and was over 60-fold upregulated by day 14 (Fig. 6B). The expression of ACAN was also quite low initially (at days 0 and 3), but was dramatically upregulated at day 7 and increased further thereafter (Fig. 6C). The expression of COL10A1, a specific marker for hypertrophic chondrocytes, was not detected at any time during the culture period (data not shown). hESCs showed similar trends in SOX9, COL2A1, and ACAN expression, but the increase in COL2A1 levels was more modest, and the increase of ACAN was larger in hESCs than in hiPSCs (Fig. 6D–F). COL10A1 expression was not detected in hESCs at any time during the culture period (data not shown).
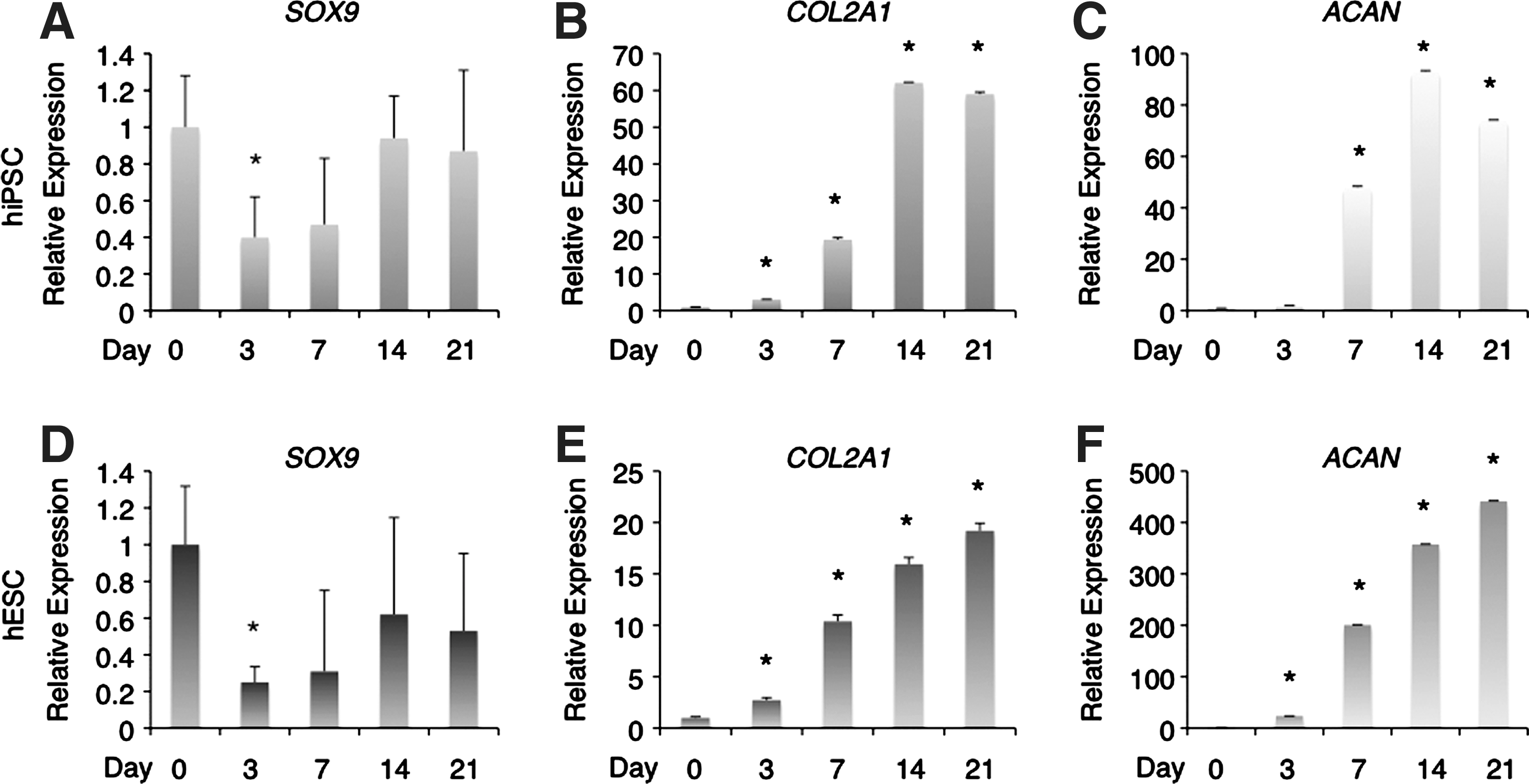
Marker gene expression during chondrogenic differentiation of hiPSCs and hESCs. The expression of chondrogenic differentiation markers was analyzed by real-time reverse transcription–polymerase chain reaction. Both hiPSCs and hESCs in monolayer culture exhibited high expression of SOX9
Immunohistochemical analysis of chondrogenic differentiation markers
After confirming the expression of chondrogenic differentiation markers in both hiPSC- and hESC-derived culture pellets at the mRNA level, we next examined the expression of these markers by immunohistochemistry. Sections of hiPSC-derived pellets stained with hematoxylin/eosin (HE) exhibited spherical cell morphology characteristic of chondrocytes (Fig. 7A). Although we tried several different antibodies, we detected no type II collagen signal in hiPSC-derived pellets at day 21 of induction (Fig. 7B). Weak aggrecan expression was detected in hiPSC-derived pellets at day 21 of induction (Fig. 7C). Staining of NHAC-kn-derived pellets with HE revealed large spherical cell morphology and rich interstitium, suggesting greater maturation in NHAC-kn-derived pellets than in hiPSC-derived pellets (Fig. 7E). Unlike hiPSC-derived pellets, NHAC-kn-derived pellets exhibited strong positive staining both for type II collagen and aggrecan (Fig. 7F, G). Human BMMSC (hBMMSC)-derived pellets were cultivated in DMEM/F12, which contained only 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, for 21 days as negative control. hBMMSC-derived pellets did not exhibit cell morphological change (Supplementary Fig. S7A) or the expression of chondrogenic markers; Type II collagen (Supplementary Fig. S7B) and aggrecan (Supplementary Fig. S7C). Positive signals for human-specific mitochondria in the hiPSC-, NHAC-kn-, and hBMMSC-derived specimens (Fig. 7D, H, Supplementary Fig. S7D) suggested that lack of staining for chondrogenic markers was not due to a general loss of antigenicity during specimen processing. In the specimens from hESC-derived pellets, type II collagen was not detected, and aggrecan was weakly detected, as with the hiPSC-derived pellets (data not shown). Taken together, these data suggested that hiPSCs and hESCs differentiated into chondrogenic lineage cells with similar characteristics, but neither hiPSCs nor hESCs yielded fully mature chondrocytes.
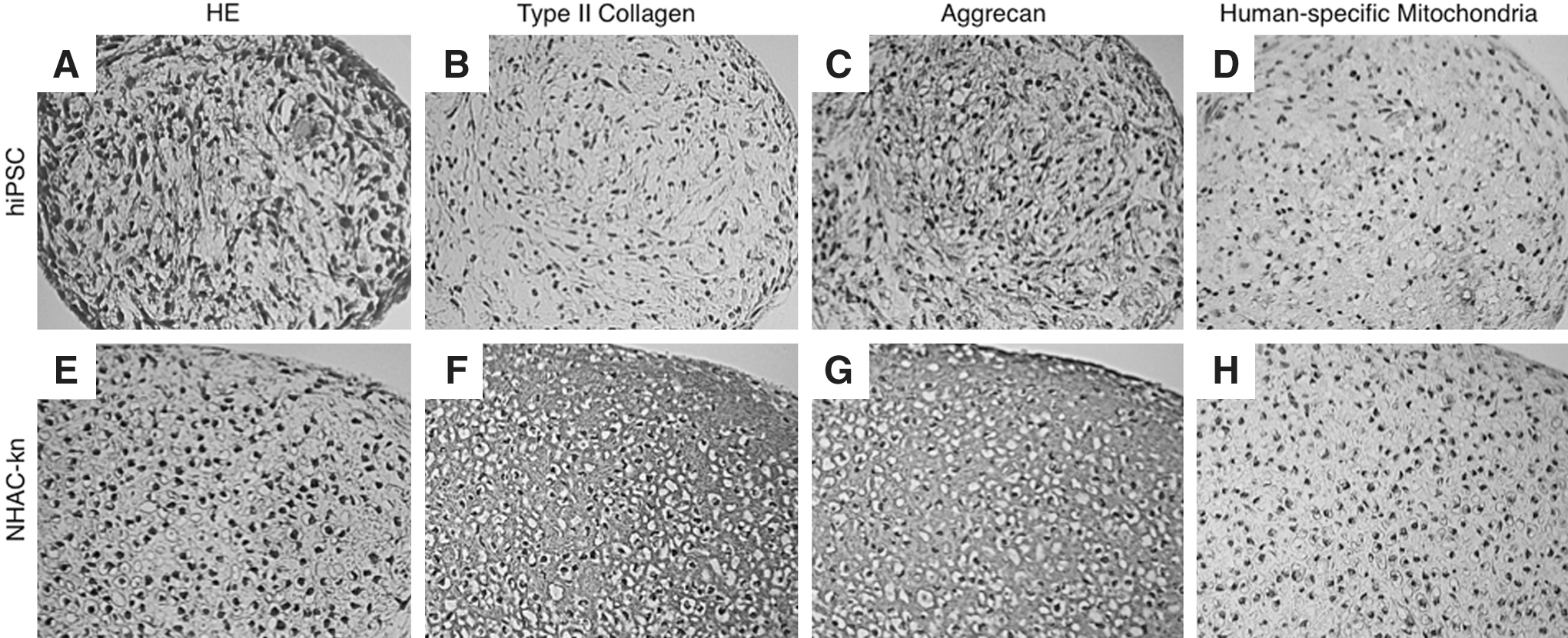
Immunohistochemical analysis of chondrogenic differentiation markers in pellets derived from hiPSCs. Sections from paraffin-embedded hiPSC pellets cultivated for 21 days in the chondrogenic induction medium were stained with HE
Discussion
In this study, we developed a simplified, reproducible method for differentiating hiPSCs into the chondrogenic lineage in vitro. Our method does not involve genetic manipulation, making it suitable for studying the mechanism of idiopathic genetic diseases, such as skeletal dysplasia, and for evaluating drug effects on chondrocytes. Our protocol consists of 4 steps: (1) EB formation, which involves spontaneous differentiation and mimics the formation of the 3 germ layers in vitro; (2) Cell outgrowth from EBs to increase cell number; (3) Monolayer culture to remove undifferentiated cells and select cells that can adapt to MSC growth conditions; and (4) 3D pellet culture, which is important for cell–cell and cell–matrix interactions. Our pellet culture medium was supplemented with TGF-β3 and dexamethasone, since these factors were previously demonstrated to produce the highest chondrogenic potential in 3D culture reported to date [46]. In our protocol, differentiation into the mesenchymal lineage during EB formation and monolayer culture was critical. In addition, cell density was a crucial factor for monolayer culture. The ideal cell density for monolayer culture in our method was the same as that previously reported for induction of chondrocytes from hESCs in an arginine-glycine-aspartate–modified hydrogel combined with EB formation and monolayer culture [47], suggesting that the optimal culture condition for chondrogenic differentiation of hiPSCs is very close to that of hESCs.
Other attempts to induce MSCs from ESCs and iPSCs have also been reported. For instance, MSCs or MSC-like cells have been derived from hESCs by either transfection of a human telomerase reverse transcriptase (hTERT) gene [48] or by coculture with mouse OP9 cells [34]. However, the use of exogenous genetic material or mouse cells in those protocols introduces, respectively, the risk of tumorigenicity or contamination by animal pathogens. To overcome these problems, hESCs have been plated directly on gelatin-coated dishes without EB formation and cultured with bFGF and PDGF-AB [49]. In that study, after 1 week, CD105-positive and CD24-negative cells were sorted as MSCs. Those cells expressed MSC markers, such as CD44, CD49a, CD105, and CD166. However, expression of type II collagen mRNA decreased progressively after chondrogenic induction, even though the immunostaining for type II collagen was positive. Recently, mesenchymal progenitor cells were induced from murine iPSCs using a method similar to ours [50]. In that method, murine iPSC-derived EBs were transferred to culture dishes and cultivated in DMEM supplemented with 10% FBS and 10−6 M all-trans retinoic acid. Sprouted cells from EBs were dissociated with trypsin/EDTA, seeded onto gelatin-coated dishes, and cultivated in the MesenPRO RS™ Medium (Invitrogen) supplemented with bFGF. The resulting induced mesenchymal progenitor cells expressed markers for MSCs. Thus, a culture protocol consisting of EB formation, cell outgrowth from EBs, and monolayer culture after dissociation into single cells appears to be suitable for mesenchymal differentiation from iPSCs and ESCs. This method is simple, reproducible, and results in good cell viability, although some details still need to be refined. In the present study, to reduce the risk of cell damage, we did not use cell sorting. However, purification of mesenchymal progenitor cells by cell sorting may improve the efficiency of chondrogenic differentiation, and should be combined with our approach in future studies.
The expression of sequential markers of chondrogenic differentiation in vitro is inconsistent among studies. Sox9 belongs to the Sry-related high-mobility-group box (Sox) transcription factors and directly regulates the expression of several chondrogenic markers, including type II collagen and aggrecan [43 –45]. Sox9 was identified as a causative gene of campomelic dysplasia, with heterozygous patients exhibiting disproportionately short stature, bowing of the limbs, low ears, depressed nasal bridge, talipes equinovarus, long philtrum, and micrognathis [51,52]. Heterozygous Sox9 mouse mutants exhibited the same skeletal abnormalities as campomelic dysplasia patients, and histological analysis revealed smaller and delayed chondrogenic mesenchymal condensation and enlargement of the hypertrophic zone in association with premature mineralization [53]. During mouse embryogenesis, Sox9 is expressed in all chondroprogenitors and chondrocytes except for hypertrophic chondrocytes. Thus, Sox9 is required for the commitment of undifferentiated MSC to the chondrogenic lineage, for mesenchymal condensation, and for inhibiting hypertrophic conversion of proliferating chondrocytes. Gong et al. reported that micromass culture using hESCs (H9) resulted in chondrogenic differentiation, with Sox9 expression elevated at the early stage and strikingly downregulated at the intermediate stage of inductive culture [54]. In their report, no type X collagen expression was detected even at late stages of induction, which the authors deemed favorable, because chondrocytes in articular cartilage do not normally undergo hypertrophic maturation. Meanwhile, a report by Toh et al., in which hESCs (line H1 and H9) were differentiated into hypertrophic chondrocytes following micromass culture of EB-derived cells, demonstrated the expression of type X collagen, which was detected by both PCR and immunohistochemistry [40]. However, in another report Toh et al. induced chondrogenic differentiation of hESCs (H9) by a combination of micromass culture of EB-derived cells and pellet culture after cell dissociation; they demonstrated that Sox9 expression increased continuously for 21 days in micromass culture, whereas no type X collagen expression was detected even after pellet culture for another 4 weeks [55]. In a recent study, chondrocytes could be induced from hESCs in 2D culture by a multistep process that applied different cell density and different combinations of growth factors to each stage [56]. In that study, differentiated cells expressed Sox9, CD44, and type II collagen, but did not express type X collagen. In our experiments, the expression of SOX9 was already elevated at the start of pellet culture, decreased promptly, and increased again at a later stage. Because Sox9 inhibits hypertrophic conversion of proliferating chondrocytes [53], increased expression of SOX9 at a late stage might explain why the expression of type X collagen was not detected at any time during the culture period. Moreover, negative expression of type X collagen is not necessarily unfavorable, because hyaline cartilage, which constitutes articular cartilage, does not express type X collagen [54].
The cause of the discrepancy between gene expression and protein expression in cell pellets needs to be elucidated. Although RT-PCR revealed that expression of type II collagen and aggrecan mRNAs was obviously increased, immunohistochemistry detected only weak staining for aggrecan and no staining for type II collagen. The reason for negative immunostaining of type II collagen is not known. However, a discrepancy between gene expression and protein expression of type II collagen has been reported previously, suggesting that mRNAs containing AU-rich elements are destabilized when translated [49]. There is a possibility that translation might be destabilized in our culture conditions. Another possibility is that cells in pellets are still immature chondrocytes, and do not produce enough collagen to be detected by immunohistochemistry. To achieve in vitro chondrogenic differentiation, 3D pellet culture in the presence of TGF-β superfamily members seems to be essential. While pellet culture is definitely an effective tool, it is not without its limitations. Another critical factor is oxygen tension. The physiological environments of both articular cartilage and bone marrow exist within a range of 1%–7% oxygen. Previous studies have shown that hypoxia promotes chondrogenic differentiation of MSCs [57,58] and that hypoxia increases the expansion potential of MSCs [59,60]. Additionally, Sox9 is upregulated by hypoxia, resulting in increased expression of type II collagen and aggrecan in human articular chondrocytes. Thus, it is possible that hypoxic culture conditions would promote chondrogenic differentiation of hiPSCs in combination with our protocol. In addition, the combination of TGF-β in the induction medium with various growth factors, such as growth and differentiation factor 5 [61], cell sorting by MSC markers, and subjecting cultures to cyclic hydrostatic pressure [62] might also improve maturation of chondrocytes in our method, resulting in enhanced expression of type II collagen and aggrecan protein.
In our method, the ability of hESCs to differentiate into the chondrogenic lineage was similar to that of hiPSCs, although hESCs exhibited slightly faster progression and greater efficiency of chondrogenic differentiation compared to hiPSCs. However, a slight difference in the progression and efficiency of chondrogenic differentiation might arise from clonal variation, since different ESC and iPSC lines can vary in their characteristics [63,64]. In our study, G1-derived pellets seemed to produce less matrix than B7 and iPS-TIG107 lines. It is difficult for our method to control evenly the differentiation time course and the characteristics of induced chondrocytes from different cell lines.
In conclusion, we have developed a simplified and reproducible culture method that allows hiPSCs and hESCs to be differentiated into the chondrogenic lineage via generation of mesenchymal progenitor cells. Although the inductive culture method still needs improvement, our study provides an important foundation for cartilaginous tissue engineering, which will benefit regenerative medicine, pathogenesis studies of idiopathic skeletal dysplasia, and the development of drug screening systems for cartilaginous diseases.
Footnotes
Acknowledgments
We thank J. Toguchida and A. Nasu, Center for iPS Cell Research and Application, Kyoto University, and H. Yao, Department of Transfusion Medicine and Cell Therapy, Kyoto University, for technical advice. This work was supported by the project for realization of regenerative medicine and Grant-in-Aid for Scientific Research (22591009) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grant-in-Aid for Scientific Research from the Ministry of Health, Labor, and Welfare of Japan, and Novo Nordisk Study Award for Growth and Development 2011.
Author Disclosure Statement
The authors indicate no potential conflict of interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.