
Abstract
Pancreatic progenitor (PP) cells are tissue-committed cells, which can differentiate into all kinds of pancreatic cells. They are potential candidates for regeneration of pancreatic tissue. However, it is unfeasible to acquire PP cells from pancreatic tissues and expand them in vitro. Generation of PP cells from adipose tissue-derived mesenchymal stem cells (AD-MSCs) would provide an unlimited source of PP cells. Here we developed a 2-step stepwise protocol, which induced AD-MSCs to generate FOXA2- or SOX17-positive definitive endoderm (DE) (5 days) and pancreatic and duodenal homeobox gene 1 (PDX1)-positive PP cells (4–6 days). By mimicking the developmental progress in embryonic development, we optimized the timing and combination of cytokines to activate the key signaling pathways during pancreatic development. We found that activating the Nodal/Activin signal with Activin A could induce differentiation of AD-MSCs toward DE, which could be further promoted by the Wnt signaling pathway activator Wnt3a. Besides, transient T (BRACHYURY)+ mesendodermal cells were observed during formation of DE from AD-MSCs. Subsequently, the Wnt signaling pathway inhibitor Dkk1 along with retinoic acid/FGF2 (60 ng/mL) further induced AD-MSC-derived DE cells to differentiate into PDX1-positive PP cells. The derived PP cells were capable to form pancreatic endocrine or exocrine cells. In conclusion, we established a stepwise protocol that could derive DE and PP cells from AD-MSCs. It might provide an unlimited source of autologous PP cells for pancreatic diseases.
Introduction
U
The pancreatic and duodenal homeobox gene 1 (PDX1) has a critical role in the early embryonic development of the pancreas [3]. PDX1-positive cells could give rise to both pancreatic endocrine and exocrine cells, which has been verified by lineage-tracing experiments [4]. Therefore, PDX1-positive cells are considered to be pancreatic progenitor (PP) cells, which are tissue-committed cells and can differentiate into all cells of pancreatic lineages [5,6]. It might be a potential candidate of seed cells for tissue engineering in pancreas reconstruction or for replacement therapy in pancreatic diseases. However, it is unfeasible to acquire PP cells from pancreatic tissues and expand them in vitro [6].
Several protocols have been set up to direct embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) to differentiate into PP cells, but the induction efficiency is still low [7 –10]. In addition, the ethical/legal issues, safety concerns, and risks of teratoma formation related to the starting materials also limit their usage in translational medicine [6]. Studies have demonstrated that mesenchymal stem cells (MSCs), which can be isolated from many fetal and adult tissues, possess multilineage differentiation potential [11,12]. More importantly, unlike ESCs or iPSCs, which are prone to form teratomas in vivo, no teratomas were observed after transplantation of human MSCs in immunodeficient recipients [13]. Therefore, MSCs represent a good cell source for regenerative medicine. Recently, MSCs derived from adipose tissue, bone marrow, umbilical cord blood, and pancreas have been shown to generate insulin-secreting cells [13 –15]. However, these studies did not describe if MSCs possess the capacity to be differentiated into definitive endoderm (DE) and PP cells in vitro. In addition, efficient protocols for differentiation of pancreatic exocrine cells from adult stem cells have not been developed yet.
During pancreatic development, epiblast cells first ingress in the primitive streak (PS) to form mesendoderm and DE, and then DE is regionalized and forms the posterior foregut and develops sequentially into prospective pancreatic endoderm, pancreatic endoderm, PPs, endocrine and exocrine progenitors, and islets, acinar and duatal cells. According to pancreatic development, formation of DE and PP cells is a prerequisite for the induction of cells to differentiate toward mature pancreatic cells [5,16,17].
In this study, we developed a 2-step protocol to generate DE and PP cells from adipose tissue-derived MSCs (AD-MSCs) by mimicking multiple signals that occur during pancreatic development (Fig. 1) [18]. In the first step, AD-MSCs were treated with Activin A and Wnt3a to derive DE. In the second step, DE derived from AD-MSCs was induced to generate PP cells using a cocktail of retinoic acid (RA), Dkk1, FGF2, and other cytokines. These PP cells expressed PDX1 and could differentiate into pancreatic, exocrine, and endocrine cells. To our knowledge, we are the first to show that adult human MSCs could be used to generate PPs efficiently.
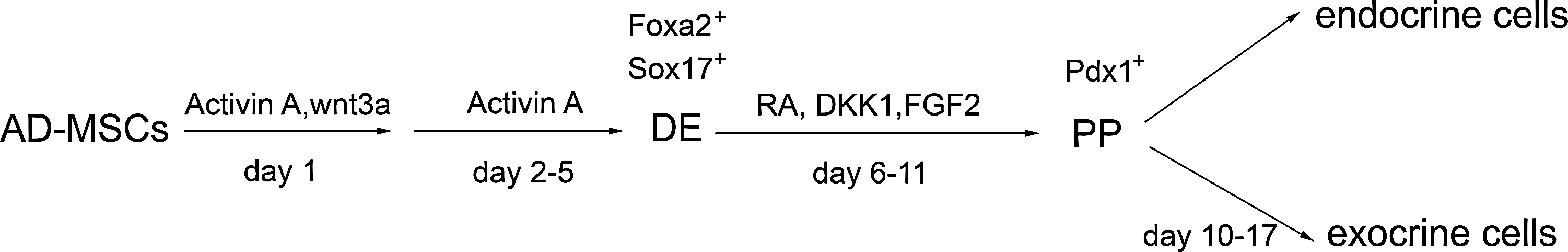
Differentiation protocol for generation of DE and PP cells from AD-MSCs. Exposure of AD-MSCs in BM supplemented with 5 ng/mL Activin A and 50 ng/mL Wnt3a from day 0 to day 1, and then in BM supplemented with 5 ng/mL Activin A from day 2 to day 5 resulted in the formation of DE cells (FOXA2+ or SOX17+). AD-MSC-derived DE cells differentiated into PP cells (PDX1+) after being cultured in PPM containing RA, DKK1, FGF2, and other cytokines for 4–6 days. AD-MSC-derived PP cells were further induced into endocrine and exocrine cells after being cultured in specific medium for 8 days. AD-MSCs, adipose tissue-derived mesenchymal stem cells; DE, definitive endoderm; PP, pancreatic progenitor; BM, basic medium; PPM, pancreatic progenitor medium; RA, retinoic acid; PDX, pancreatic and duodenal homeobox gene.
Materials and Methods
Isolation of human AD-MSCs
Human adipose tissue was obtained from patients undergoing tumescent liposuction, according to procedures approved by the Ethics Committee at the Chinese Academy of Medical Sciences and Peking Union Medical College. The isolation procedure was described in our previous study [19]. After isolation, resuspended cells were plated in a 75-cm2 culture flask at a density of 2×106 cells in 12 mL of a regular growth medium and incubated at 37°C in a humidified environment containing 5% CO2. A regular growth medium contained a 58% Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12 medium (DF-12; Gibco), a 40% medium complete with trace elements-201 (Gibco), 2% fetal bovine serum (FBS; Gibco), 1×10−9 M insulin–transferrin–selenium (Gibco), 1×10−9 M dexamethasone, 1×10−4 M ascorbic acid 2-phosphate, 20 ng/mL interleukin-6, 10 ng/mL epidermal growth factor, 10 ng/mL platelet-derived growth factor BB (all purchased from Sigma-Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco).
Differentiation conditions
The basic medium (BM) was the DMEM (Gibco) supplemented with 4.5 g/L
AD-MSCs of passage 2 were seeded (5×105 cells/well) in the regular growth medium in 6-well plates (Nunclon). After adherence, the culture medium was switched to the BM with different concentrations of Activin A (0, 0.1, 1, 5, 10, and 100 ng/mL) or 10 μM SB431542 for 5 days. The effect of Activin A concentration on generation of DE form AD-MSCs was evaluated by measuring the expression of DE-specific genes. Based on the preliminary results, 5 ng/mL Activin A was used in the following experiments (Fig. 1). Cells were divided into 3 groups. In the first group, AD-MSCs were cultured in the BM with 5 ng/mL Activin A for 5 days. In the second group, AD-MSCs were cultured in the BM with 5 ng/mL Activin A and 50 ng/mL Wnt3a for 1 day, and then in the BM with 5 ng/mL Activin A for 4 additional days. In the third group, AD-MSCs were cultured in the BM with 5 ng/mL Activin A and 2 μM Wortmannin for 5 days. The effect of Wnt3a or Wortmannin on DE generation was evaluated by measuring the expression of DE-specific genes. Based on the preliminary results, 5 ng/mL Activin A and 50 ng/mL Wnt3a were used in the following experiments. For PP cells differentiation, day 5 AD-MSCs in the second group were cultured in the PPM for 4–6 days (Fig. 1).
For pancreatic endocrine cell differentiation, day 9 AD-MSCs (PP cells) were cultured in the pancreatic endocrine cells specification medium (the BM supplemented with 10 ng/mL Exendin-4, 10 ng/mL Activin A, 10 mM nicotinamide, 2% B-27, and 1% N2) for 8 days. For pancreatic exocrine cells differentiation, day 9 AD-MSCs (PP cells) were cultured in the pancreatic exocrine cells specification medium (the BM supplemented with 10 ng/mL Exendin-4, 15 ng/mL FGF7, 10 mM nicotinamide, 2% B-27, and 1% N2) for 8 days (Fig. 1). The medium was changed every other day. Wnt3a and Dkk1 were purchased from R&D Systems, Inc. EGF, FGF2, RA, Exendin-4, nicotinamide, SB431542, and Wortmannin were purchased from Sigma-Aldrich. FGF7 and Activin A were purchased from Peprotech. Glutamax-I, NEAA, B-27, and N2 were purchased from Gibco.
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA was extracted using TRIZOL (Invitrogen) according to the manufacturer's instruction. Two micrograms of total RNA from each sample was reverse transcribed using M-MLV (Takara) in a final volume of 30 μL. The polymerase chain reaction (PCR) amplification was carried out using the IQ5 System (Bio-Rad) with SYBR Green Mastermix (Takara). All quantitative real-time PCR (qRT-PCR) results were carried out in duplicate and normalized to GAPDH and β-actin. The primer sequences used for qRT-PCR are listed in the Supplementary Table S1 (Supplementary Data are available online at
siRNA and transfection
siRNAs were used to selectively silence the expression of the T gene in differentiated AD-MSCs. Three T (Brachyury) siRNAs (CCUCGAAUCCACAUAGUGATT, GCAAAUCCUCAUCCUCAGUTT, and CUCCAACCUAUUCUGACAATT) and negative control (UUCUCCGAACGUGUCACGUTT) were synthesized by Shanghai GenePharma Co., Ltd. siRNAs were transfected into cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction.
Immunofluorescence assays
The cultured cells were fixed for 10 min at 4°C in ice cold methanol, washed several times in phosphate-buffered saline (PBS), and then permeabilized in PBS+0.1% Triton X-100 for 10 min on ice, and then blocked for 30 min in PBS+0.5% Tween-20 containing 1% bovine serum albumin (BSA). The primary antibodies were incubated overnight at 4°C, washed with PBS, and then incubated with the secondary antibodies at room temperature for 1 h. The incubated cells were washed in PBS, and Hoechst 33342 (Sigma-Aldrich) was used to visualize nuclei. The sources of antibodies and dilutions used are summarized in the Supplementary Table S2.
Flow cytometry assays
The cultured cells were analyzed for cell antigen expression by fluorescence-activated cell sorting (FACS). Briefly, 1×105 cells were fixed in 4% paraformaldehyde, and then permeabilized with PBS+0.1% Triton X-100 for 10 min. Cells were incubated at 4°C for 1 h with the following primary antibodies: FOXA2, T (BRACHYURY), SOX1, and PDX1 or isotype antibodies, which served as negative controls to set the quadrants in each graph. Then, cells were incubated at room temperature for 30 min with the following secondary antibodies: goat anti-mouse-FITC, goat anti-rabbit-FITC, or rabbit anti-goat-FITC accordingly.
Western blot analysis
Cells were suspended in a radio immunoprecipitation assay (RIPA) lysis buffer (Beyotime Institute of Biotechnology). Samples (40 μg of total protein/well) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and the protein bands on the gel were transferred onto polyvinylidene fluoride membranes. The membranes were processed with the primary antibody followed by a horseradish peroxidase-conjugated secondary antibody (Supplementary Table S2). GAPDH was used to verify that similar amounts of protein were loaded in all lanes.
Total insulin content and release assays
For a glucose-stimulated insulin release assay, about 6×105 differentiated cells were used. After preincubation with the KRBH buffer (120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.1 mM MgCl2, 25 mM NaHCO3, 10 mM HEPES buffer, and 0.1% BSA, pH 7.4) at 37°C for 3–6 h, cells were incubated with the KRBH buffer containing 5.5 mM glucose at 37°C for 1 h. The supernatants were collected and the same cells were further incubated under 22 mM glucose for an additional 1 h. The supernatants were collected and analyzed using the Human C-peptide ELISA kit (Millipore Corporation). The total intracellular C-peptide was extracted by incubating differentiated cells or AD-MSCs overnight in acid ethanol (18 mL 10M HCl, 1L 70% ethanol) at 4°C. The total C-peptide content was measured after the cell pellet was sonicated in 200 μL acid/ethanol. The conversion factor for human C-peptide is 1 μg/L corresponds to 331 pM.
Statistical analysis
Each experiment was performed at least 3 times. For qRT-PCR data, C-peptide release and the total C-peptide content, the statistical significance was calculated by the Student–Newman–Keuls test or the Student's t-test. SPSS 13.0 software was used for analysis. The statistical significance was set at P<0.05 (indicated by “*”).
Results
Optimal concentration of Activin A to induce AD-MSCs to differentiate toward DE
Many cytokines and signal pathways have been suggested to govern the pancreatic development in a sequential fashion. For example, a high level of Nodal signaling was reported to promote DE formation [8,9,20 –23]. Since Activin A could replace the Nodal/Crypto signal [17] and generate DE cells from ESCs by activating the Nodal/Activin signaling pathway, we plan to evaluate if Activin A plays a same role in promoting DE formation from AD-MSCs and find the optimal concentration. AD-MSCs were treated with Activin A at different concentrations (0, 0.1, 1, 5, 10, and 100 ng/mL) for 5 days. We found that the expression of DE-specific genes were markedly upregulated on day 5 in a dose-dependent manner. Maximal expression of DE-related genes (Foxa2 and Sox17) [9,24] was observed after 5 ng/mL Activin A treatment. Meanwhile, expression of mesendoderm-related genes [Eomes, T (Brachyury), Mixl1, and Gsc] [9,24] was also upregulated in the presence of 5 ng/mL Activin A. The expression of Sox7, which is expressed in primitive endoderm and extraembryonic endoderm (ExEn), but not in DE [25], was not upregulated in the presence of Activin A. This suggests that the expression of Foxa2 and Sox17 is not involved in the differentiation of ExEn. Mesoderm-related genes (Tbx6 and Kdr) [24] were not changed in the presence of Activin A. Interestingly, ectoderm-related genes (Sox1 and Pax6) [24,26] were also upregulated, suggesting that AD-MSCs may differentiate into ectodermal cells after Activin A treatment. Besides, the expression of MET-related genes (E-cad and N-cad) [24] were upregulated in a dose-dependent manner (Fig. 2A). Results of FOXA2, SOX17, T (BRACHYURY), EOMES, GSC, and SOX1 from western blot confirmed these findings (Fig. 2B). In the immunofluorescence assay, we randomly selected 3 fields and counted the number of positive and negative cells, and then calculated the percentage. The average proportions of FOXA2+, SOX17+, T (BRACHYURY)+, and SOX1+ cells were about 80%, 80%, 30%, and 70%, respectively, in the presence of 5 ng/mL Activin A on day 5 (Fig. 2C). Inhibition of Nodal/Activin signaling by SB431542 led to decreased expression of a 3 germ layer-related gene (Foxa2, Sox17, T (Brachyury), Gsc, Eomes, Mixl, Kdr, Sox1, and Pax6) and loss of FOXA2+, SOX17+, T (BRACHYURY) +, and SOX1+ cell populations on day 5 (Supplementary Fig. S1). These results showed that activation of Nodal/Activin signaling by Activin A have an important role in the generation of DE cells (FOXA2- or SOX17-positive) from AD-MSCs, and 5 ng/mL Activin A may be the optimal concentration during this induction process.

Effects of different concentrations of Activin A on generation of DE cells from AD-MSCs.
WNT signal pathway activator Wnt3a promotes generation of DE cells from AD-MSCs
To increase the proportion of DE cells and decrease the proportion of mesoderm and ectoderm cells from AD-MSCs, the effects of cytokines related to multiple endogenous signaling pathways were investigated in the inducing system. Activation of Wnt signaling and/or inhibition of PI3K signaling have been demonstrated to have a synergistic effect with Nodal signaling in the induction of DE from ESCs [27]. We compared the efficiency of DE differentiation from AD-MSCs in the presence of Activin A (aA), Activin A and an activator of Wnt signaling Wnt3a (aAW), or Activin A and an inhibitor of PI3 kinase Wortmannin (aAWo). After 5 days' culture, qRT-PCR showed that the expression of DE-related genes (Foxa2 and Sox17) was most prominent in aAW culture (Fig. 3A). On the contrary, the expression of mesoderm-related genes Kdr and Tbx6 was the lowest in aAW culture (Fig. 3A). The results of FOXA2, SOX17, T (BRACHYURY), and SOX1 were further confirmed by western blot (Fig. 3B). FACS demonstrated that aAW did selectively induce more FOXA2-positive cells (DE cells) and T (BRACHYURY) -positive cells (mesendoderm cells) than both aA and aAWo (Fig. 3C, D), but less SOX1-positive cells (ectoderm cells) (Fig. 3C, D). It is worth mentioning that the western results (Fig. 3B) and the FACS results (Fig. 3C, D) for T (BRACHYURY) do not completely agree with the qRT-PCR results for T (Brachyury) (Fig. 3A), especially in aAWo culture. Unlike the DE marker Foxa2 and Sox17, we did find that the basic expression level and induced expression level of the T gene varied greatly among different batches of AD-MSC samples. Whether post-transcriptional processing and regulation take place in this process deserves further experiments.

Canonical Wnt signaling activator Wnt3a promotes DE generation from AD-MSCs when combined with Activin A. Passage 3 AD-MSCs were divided into 3 groups. In the first group (aA), AD-MSCs were treated with 5 ng/mL Activin A for 5 days. In the second group (aAW), AD-MSCs were treated with 5 ng/mL Activin A and 50 ng/mL Wnt3a for 1 day, and then with 5 ng/mL Activin A for 4 days. In the third group (aAWo), AD-MSCs were treated with 5 ng/mL Activin A and 2 μM Wortmannin for 5 days.
These results are similar to the previous reports that demonstrated Wnt3a and Activin A are synergistic in inducing DE cells from ESCs and iPSCs, while the PI3K signal pathway is not involved in this process. Thus, 5 ng/mL Activin A and 50 ng/mL Wnt3a were chosen to induce the generation of DE cells from AD-MSCs in the following experiments.
Transient T (Brachyury)+ cells appear during the formation of DE cells
Next, we delineated the dynamic change in the generation of the DE cell from AD-MSCs. In the epiblast of mice, Nodal is one of the first markers of PS formation, and its rapid upregulation in differentiating ESCs suggests that the cells are initiating the transition toward a PS-like cell population [9]. When AD-MSCs were treated with Activin A and Wnt3a for 5 days, the Nodal expression peaked on day 3 and decreased thereafter (Fig. 4A). During this process, a strong expression of the mesendoderm-related marker T (Brachyury) was noticed on day 2. Expression of DE-related genes (Foxa2 and Sox17) was upregulated on day 1 and persisted to day 5. Gsc, expressed in mesendoderm and Tbx6, expressed in mesoderm were downregulated on day 1 and upregulated on day 2, but then decreased on day 5. Expression of another mesoderm marker Kdr was downregulated on day 1, upregulated on day 4 (and higher than basic levels), but then was decreased to basic levels below on day 5. Expression of the ExEn marker Sox7 was upregulated on day 2, and then decreased to the basic level on day 5 (Fig. 4A), suggesting that subtle differences may exist in the induction process to DE cells between AD-MSCs and ESCs. The sequential gene expression dynamics recapitulated those in gastrulation, mesendoderm [5,9], and DE development [28,29]. These data indicated the transient presence of a T (BRACHYURY) + mesendoderm cell population during AD-MSC differentiation.

Kinetics of mRNA and protein expression of key genes during DE differentiation in the presence of 50 ng/mL Wnt3a and 5 ng/mL Activin A.
We then observed the dynamic changes of FOXA2, SOX17, T (BRACHYURY), and SOX1 by western blot. When expression of the T (BRACHYURY) protein reached its peak, expression of FOXA2 and SOX17 protein was only marginally detectable (at day 2 or 3 of differentiation). The highest expression of FOXA2 and SOX17 was observed around day 5 when T (BRACHYURY) expression decreased to a low level (Fig. 4B). Besides, SOX1 protein expression reached its peak at day 5 of differentiation (Fig. 4B). In agreement with western blot analysis, immunofluorescence experiments showed that FOXA2- or SOX17-positive cells began to appear at day 1 or 0, respectively. By day 5, the proportion of FOXA2- or SOX17-positive cells reached maximal. (Fig. 4C) We also observed that T (BRACHYURY)-positive cells (66.7%) at day 3 of differentiation also expressed FOXA2 (22%). (Fig. 4C) Their positive rates were evaluated by counting positive cell ratios of randomly selected fields. We speculated that T (BRACHYURY)-positive cells might have a role in the formation of FOXA2-positive cells. Besides, the proportion of GSC-positive cells reached maximal at day 1, and then decreased at day 5 (Fig. 4C).
To further investigate the role of T (BRACHYURY) in generation of DE from AD-MSCs, we used siRNA to downregulate T (Brachyury) expression. qRT-PCR revealed that the expression of DE-related genes (Foxa2 and Sox17), mesendoderm-related genes (Mixl, Gsc and Eomes), and mesoderm-related gene (Kdr) all decreased at day 5, but the expression of ectoderm-related genes (Sox1 and Pax6) were not changed significantly (Fig. 5A). These results were confirmed by western blot and immunofluorescence staining (Fig. 5B, C). Collectively, these results demonstrated that the transient formation of T (BRACHYURY)-positive cells (mesendoderm stage cells) is very important to generate DE cells from AD-MSCs.
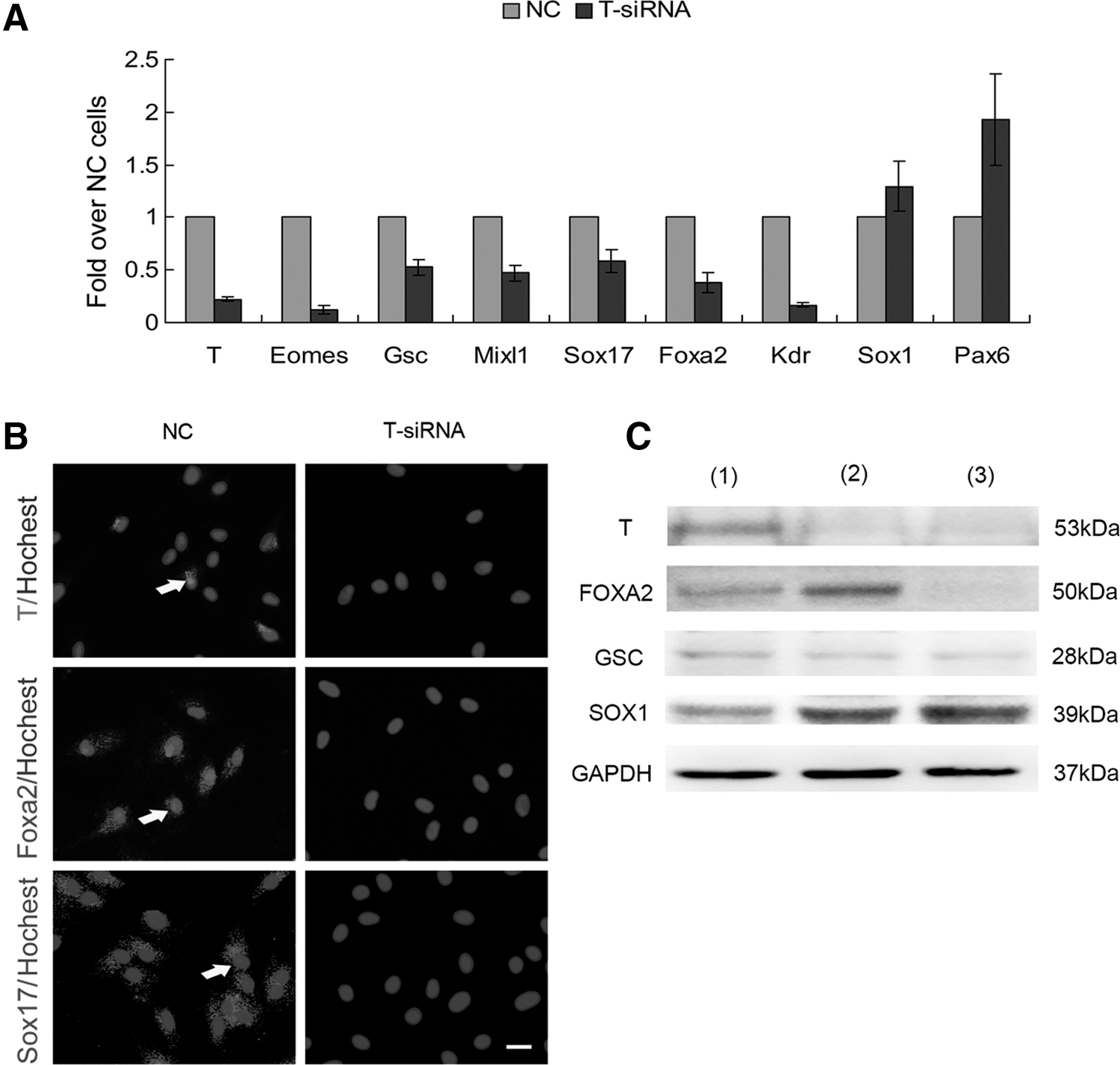
Downregulation of T (Brachyury) mRNA and protein expression by siRNA.
Differentiation of DE cells toward PP cells
After deriving DE cells from AD-MSCs, we tried to induce DE cells into PDX1-positive PP cells. For this purpose, DE cells were cultured in the PPM containing RA, Dkk1 (the Wnt signaling pathway inhibitor), FGF2 (60 ng/mL), and other cytokines for 6 days. These factors are involved in the pancreatic endoderm cell development [22]. Expression of pancreatic endoderm and progenitor cell markers, such as Pdx1, P48, Sox9, Hnf6, Hnf1b, Nkx6.1, and Nkx2.2, were upregulated (Fig. 6A). Western blot and immunofluorescence staining showed that PDX1 was detectable at day 5 and peaked at day 9 (Fig. 6B, C). FACS showed that the proportions of PDX1- and FOXA2-positive cells were as high as 93.1%±1.21% and 99.6%±0.21% at day 9, respectively (Fig. 6D, E). The majority of PDX1-positive cells also coexpressed FOXA2 (Fig. 6B).

Characterization of AD-MSC-derived PP cells by stepwise differentiation.
Collectively, RA, WNT inhibitor (Dkk1), and FGF2 are conducive to the transition of DE cells into PP cells. This is consistent with previous studies revealing that activation of the RA signal and FGF signal and inhibition of the Wnt signal are important for pancreatic development [30 –32].
High differentiation potential of PP cells derived from AD-MSCs
To investigate the differentiation potential of AD-MSC

Differentiation potential of AD-MSC-derived PP cells. PP cells can be differentiated into endocrine and exocrine cells by stepwise differentiation for 8 days.
In addition, after a 8-day culture in the pancreatic exocrine induction medium, expression of exocrine cell-related genes, including Amylase (AMY), Elastase 1, Carboxypeiptidase A (CPA), and chymotrypsinogen B (CTRB) [33], were upregulated by qRT-PCR (Fig. 7B). Immunofluorescence showed that Amylase-positive cells were present at day 17 and reached about 90% (Fig. 7F). Based on these results, we concluded that PP cells derived from AD-MSCs have the potential to differentiate into pancreatic endocrine and exocrine lineages.
Discussion
In the present study, we showed for the first time that stage-specific DE cells and PP cells could be generated from adult human AD-MSCs with a novel 2-step differentiation protocol by sequentially activating multiple signaling pathways via a combination of Activin A/Wnt3a and RA/Dkk1/FGF2. These DE and PP cells express specific markers and have the capacity to differentiate into both endocrine and exocrine cells.
During embryonic development, formation of DE cells is a necessary step for differentiation toward PP cells [34]. Based on this principle, we first generated DE cells from AD-MSCs. We found that Nodal/Activin signaling played a key role in the generation of DE from AD-MSCs as in ESCs [7,8]. However, the DE-related gene and protein expression is highest in the presence of 5 ng/mL Activin A, rather than 100 ng/mL Activin A for ESCs or iPSCs [7 –10]. The reason for the difference is unknown. In addition to inducing expression of DE-related genes, Activin A also induced expression of mesendoderm- and ectoderm-related genes, but had little effect on mesoderm-related genes. These suggest that the differentiation potential of AD-MSCs might have been changed after Activin A treatment.
To increase the efficiency in generating DE cells from AD-MSCs, we simultaneously used the canonical Wnt signaling pathway activator Wnt3a and found there was a synergistic effect between Wnt3a and Activin A, which is in consistent with the previous studies of ESC differentiation [31]. However, using the Wnt signaling activator Wnt3a alone was not sufficient to generate DE cells (data not shown) from AD-MSCs. Interestingly, during differentiation of AD-MSCs toward DE cells, the concomitant use of Wnt3a and Activin A significantly decreased the proportion of ectoderm cells. Hori et al. and Zhang et al. reported that inhibition of PI3K signaling results in an additional improvement of DE cell induction from ESCs by exit from self-renewal and entry into a specific lineage program [7,35]. However, we did not find that the exogenous addition of the inhibitor of PI3K signaling had any detectable synergistic effect on DE induction from AD-MSCs, which suggests that PI3K signaling may be not necessary for AD-MSCs to differentiate toward DE cells.
During embryonic development, T (BRACHYURY)-positive mesendoderm cells have been suggested to be the origin of DE cells. During the induction of AD-MSCs toward DE cells, we did observe a transient expression of T (Brachyury). Suppression of T (Brachyury) decreased formation of DE cells, indicating that T (BRACHYURY)-positive cells may represent mesendodermal progenitors that subsequently acquire a DE fate as judged by rapid upregulation of Foxa2 or Sox17 and decreased expression of T (Brachyury). How T (Brachyury) influences the differentiation of AD-MSCs toward DE deserves to be studied in the future.
We showed a combination of RA, intermediate concentration of FGF2 and Dkk1 may generate PDX1-positive PP cells from AD-MSC
Previous studies showed that Wnt signaling should be enhanced for epiblast cells to differentiate toward mesendoderm. However, in differentiation from DE to pancreatic endoderm, Wnt signaling should be inhibited [45 –47]. Although Wnt suppression is important in the formation of pancreatic endoderm, few studies have taken the advantage of Wnt inhibitors in differentiation protocols to generate PP cells in vitro. We blocked Wnt signaling in DE cells with Dkk1, which exerts its action by binding to a component of the Wnt receptor and preventing its activation. As expected, Dkk1 combined with an intermediate concentration of FGF2, and RA significantly induced differentiation of PP cells.
Chandran et al. have reported [48] that AD-MSCs could be differentiated into islet-like cells, which were confirmed in our works. Our protocol is different from theirs. We adopt a stepwise protocol that first generates DE cells from AD-MSCs, and then PP cells from DE cells. The advantage of our protocol is that both DE cells and PP cells can be generated efficiently. In addition, the intermediate product, DE cells, may be used to generate both pancreas and liver cells, even lung and gut cells. Most importantly, the final product is PP cells, but not islet-like cells, which may be used to generate both endocrine cells and exocrine cells. These PP cells might be used as new seed cells for transplantation or induced into mature pancreatic endocrine and exocrine cells to treat pancreatic disease. The proliferation potential of AD-MSC-derived PP cells, as well as their safety and efficiency after transplantation will be evaluated in the future.
Footnotes
Acknowledgments
This study was supported by grants from the ‘‘863 Projects'’ of the Ministry of Science and Technology of People's Republic of China (no. 2011AA020100), the National Natural Science Foundation of China (no. 30700321), the National Key Scientific Program of China (no. 2011CB964901), and the Program for Cheung Kong Scholars and Innovative Research Team in University-PCSIRT (no. IRT0909).
Author Disclosure Statement
All authors have no conflict of interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.