
Abstract
Transplantation of cardiac progenitor cells (CPCs) is currently in early clinical testing as a potential therapeutic strategy. Superoxide is increased in the ischemic myocardium and poor survival of cells is one of the major limitations of cell transplantation therapy. Superoxide dismutase (SOD) levels were analyzed in c-kit-positive CPCs isolated from rat myocardium to identify their roles in protection against oxidative stress-induced apoptosis in vitro. CPCs were subjected to oxidative stress using xanthine/xanthine oxidase (XXO) and little apoptosis was detected. CPCs contained significantly higher levels of SOD1 and SOD2 as compared with adult cardiac cell types, both at the protein and activity levels. Both SOD1 and SOD2 were increased by XXO at the mRNA and protein level, suggesting compensatory adaptation. Only knockdown of SOD2 and not SOD1 with siRNA sensitized the cells to XXO-apoptosis, despite only accounting for 10% of total SOD levels. Finally, we found XXO activated Akt within 10 min, and this regulated both SOD2 gene expression and protection against apoptosis. Rat CPCs are resistant to superoxide-induced cell death, primarily through higher levels of SOD2 compared to adult cardiac-derived cells. Exposure to superoxide increases expression of SOD2 in an Akt-dependent manner and regulates CPC survival during oxidative stress.
Introduction
E
Oxidative stress is increased in the ischemic myocardium and indirect evidence suggests the vulnerability of CPCs to oxidative stress [4]. Irrespective of the cell types used, poor survival and engraftment of cells are 2 of the major limitations of cell transplantation therapy. For example, survival of CPCs and mesenchymal stem cells (MSCs) are less than 10% within 4 days of transplantation within the ischemic myocardium [5, 6]. The survival of cells, such as skeletal myoblasts and cardiomyoblasts, are improved if antioxidants, such as superoxide dismutase (SOD) and Tempol (SOD mimetic), are delivered to the myocardium during cell transplantation [7, 8]. Further, enhanced endogenous expression of antioxidants, including SOD2, protects endothelial progenitors during oxidative stress [9]. In addition to the effect of reactive oxygen species (ROS) and antioxidants on survival, they also regulate other important properties of stem cells, such as their self-renewal and senescence [10, 11].
Although CPCs are now in phase I clinical trials (CADUCEUS, clinical trials identifier NCT00893360; and SCIPIO, clinical trials identifier NCT00474461), many of the basic properties, such as their antioxidant levels and their response to physiological stresses, such as oxidative stress remain unknown. Recent studies demonstrate that these cells contain heterogeneous populations that are more prone to senescence and cell death. While some populations are still being discovered, alterations in expression of the insulin-like growth factor receptor (IGFR) and angiotensin II types 1 and 2 receptors (AT1R and AT2R) lead to CPC dysfunction [12, 13]. As dysregulation of both of these pathways can lead to oxidative stress, the purpose of this study was to examine the antioxidant capacity of CPCs to better understand the adaptations of CPCs under pro-oxidant conditions.
Methods
Isolation of CPCs and cardiomyocytes
Endogenous cardiac resident progenitor cells were isolated from rat myocardium as previously described [14] with slight modifications. Healthy adult Sprague-Dawley rats (Charles River Labs) were euthanized; hearts were excised and washed with sterile Hank's balanced salt solution (HBSS) and their ventricles were minced to small pieces. The extracellular matrices in the minced hearts were digested for 30 min at 37°C using 50 mg of collagenase type-2 dissolved in 50 mL of sterile HBSS. The digested tissue suspension was passed through a 70-μm cell strainer and centrifuged. The cell pellet was resuspended with 2 mL of anti c-kit (Santa Cruz H-300)-coated magnetic beads for 2 h at 37°C. C-kit-positive fractions of the cells bound to the beads were separated using a magnetic particle concentrator and washed 3 times with sterile HBSS containing bovine serum albumin to remove nonspecifically bound cells. Finally, the bead-bound cells were grown in culture media and expanded.
Cardiomyocytes were isolated from 1–2 days old Sprague-Dawley rat pups (Charles River Labs) as previously described [15]. Cardiac fibroblasts (FBs) were isolated from adult Sprague-Dawley rats by collagenase digestion of the heart, passing the cells through a 70-μm cell strainer and centrifugation. Following resuspension, cells were cultured for 1 h at 37°C and adherent cells were kept, while the supernatant was discarded.
Cell culture
C-kit-positive cells from the rat myocardium were cultured in growth media that has Ham's F-12 media (Cellgro) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Hyclone), l-glutamine, penicillin/streptomycin (Invitrogen), 10 ng/mL leukemia inhibitory factor (Sigma), and 10 ng/mL basic fibroblast growth factor (Sigma). Before commencing the experiments, the cells were grown to 80% confluence, serum starved in starvation media containing Ham's F-12 media supplemented with l-glutamine, insulin/transferrin/selenium (ITS) serum supplement (Cellgro), and penicillin/streptomycin. Single-cell cloning was performed to test clonability of the cells. Briefly, c-kit-positive cells were counted and subjected to serial dilution until there was no more than 1 cell per mL of growth media. The diluted cell suspension was distributed to the wells of 24-well plates such that only 12-wells of the 24-wells received a cell. Cells growing from a single colony were subsequently expanded and tested for CPC markers using flow cytometry.
Cardiomyocytes and cardiac FBs were maintained in Dulbecco's modified Eagle's medium (DMEM) (Cellgro) supplemented with 10% FBS (Hyclone), l-glutamine, and penicillin/streptomycin (Invitrogen). The cells were serum starved overnight before experiments in DMEM supplemented with l-glutamine and penicillin/streptomycin.
Xanthine/Xanthine oxidase treatment
Superoxide was induced in the medium using xanthine/xanthine oxidase (XXO) system. Briefly, Ham's F12 supplemented with l-glutamine, ITS serum supplement (Cellgro), and penicillin/streptomycin was used as the treatment media for the cells. For XXO treatment, 1 mM xanthine and 10 mU/mL Xanthine oxidase were added, along with 10 U/mL catalase to have increased flux of superoxide in the XXO system. The flux of ROS generated was measured using the Amplex red assay kit (Invitrogen) as per the manufacturer's protocol after addition of exogenous SOD to convert all superoxide radicals to hydrogen peroxide. For groups receiving oxidative stress more than 24 h, the treatment media was subjected XXO addition once every 24 h to maintain the free radical levels. For time-matched control experiments, each experiment had various indicated termination points with each time point having its own respective control and XXO-treated cells.
SOD activity assay
Total SOD activity was measured using the SOD assay kit (Dojindo Molecular Technologies) using manufacturer's protocol. This assay is based on inhibition of color forming reaction between superoxide and water soluble tetrazolium (WST) in the presence of active SOD. Briefly, proteins were extracted by lysing the cells overnight at 4°C using the cell lysis buffer (1 mM EDTA, 150 mM NaCl, 10 mM KH2PO4, 10 mMTris-HCl, 1% NP-40) containing cocktails of protease and phosphatase inhibitors (Sigma) and stored at −20°C until further analysis. During the day of analysis, the cell lysates and WST were incubated in a 96-well plate and the absorbance at 450 nm was measured kinetically at 37°C for 30 min. SOD2 activity was calculated using the same protocol in the presence of 4 mM potassium cyanide, which inhibits the activities of SOD1 and SOD3. Since the amount of extracellular SOD inside the cells is minimal [16], the difference between the activities of total SOD and SOD2 is estimated as the activity of SOD1. All the activities were reported after normalizing to the protein content in the samples. The protein content was measured using the micro BCA protein assay kit (Thermo Scientific) according to manufacturer's protocol.
SOD knockdown experiments
Lipofectamine RNAiMax (Invitrogen) was used to transfect the cells with specified siRNAs as per manufacturer's protocol. Various duplexes of siRNAs for SOD1, SOD2 gene silencing and nonspecific scrambled siRNA (3′-5′–CGUUAAUCGCGUAUAAUACGCGUAT; 5′-3′–AUACGCGUAUUAUACGCGAUUAACGAC) were obtained from IDT. About 10 nM siRNAs were reverse transfected to CPCs using RNAiMax, and the duplexes were tested for knockdown after 24 h of transfection using real-time (RT)-polymerase chain reaction (PCR). The duplex giving the best gene silencing (SOD1: 5′-3′ - GGAAAUGAAGAAAGUACAAAGACTG; 3′-5′: CACCUUUACUUCUUUCAUGUUUCUGAC), SOD2: 5′-3′ - AGAAUGUUAGCCAAAGAUACAUAGT; 3′-5′ - CCUCUUACAAUCGGUUUCUAUGUAUCA) was chosen for further experiments. CPCs were transfected with chosen siRNAs and allowed to grow for 48 h before beginning XXO treatment.
Apoptosis detection
Sub-G1 cell fractions containing fragmented DNAs were stained with propidium iodide (PI) as follows. Briefly, the cells were fixed in cold (−20°C) 70% ethanol, washed with phosphate-buffered saline (PBS), and centrifuged at 250 g. DNA was extracted with 0.2 M Na2HPO4 containing 0.004% Triton (Sigma) followed by staining for 1 h at 37°C with 1 mL PBS containing 20 μg propidium iodide (Sigma) and 200 μg DNAse-free RNAse (Sigma). The stained cells were analyzed by flow cytometry; single cells were gated and the percentage of sub-G1 fraction was quantified. Additionally, in independent experiments, cells were fixed with 4% paraformaldehyde and apoptosis was determined using TUNEL assay per manufacturer's instructions.
Western blotting and quantitative real-time PCR
Equal amounts of denatured proteins from cell lysate were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis. The separated proteins were blotted onto a polyvinylidene fluoride (PVDF) membrane using the wet transfer procedure. Protein-blotted PVDF membranes were blocked with 4% milk in tris-buffered saline containing 0.1% Tween and probed with antibodies for SOD1, SOD2, GAPDH, or beta-actin (Santa Cruz Biotechnology). The membranes were incubated with horse radish peroxidase-bound secondary antibodies and subjected to chemiluminescence reaction and the signals were developed and quantified on Kodak Image station 4000 MM PRO and Carestream Molecular Imaging software.
For qPCR, RNAs were extracted from CPCs using Trizol® reagent (Invitrogen) according to manufacturer's protocol. c-DNAs were synthesized by reverse transcribing the mRNA using Superscript reverse transcriptase (Invitrogen) along with oligo dT and random hexamers as primers. Real-time polymerase chain reactions were run on StepOnePlus™ Real-Time PCR System (Applied Biosystems) with SYBR Green PCR master mix (Applied Biosystems) using primers for, SOD1, SOD2, catalase, and GPX1. All the reaction products were normalized to expression levels of 18 s across samples.
Statistics
All statistical analyses were performed using Graphpad Prism 5.0 software as described in the figure legends. All data are expressed as mean±SEM. P-values of less than 0.05 are considered significant.
Results
Isolation of c-kit-positive CPCs from adult rat myocardium
Heart cells were separated from the 8–10 weeks old Sprague-Dawley rat myocardium after digesting the extracellular matrix. The C-kit-positive cell population was isolated from the homogenous cell mixture using immunomagnetic separation by incubating with anti-c-kit antibodies. The cells bound to the magnetic beads were cultured for about 3 weeks and the colonies were subsequently expanded. Immunocytochemistry followed by flow cytometric analysis show that the isolated cells were >90% c-kit positive (Supplementary Fig. S1; Supplementary Data are available online at
CPCs are resistant to oxidative stress-induced apoptosis
In our preliminary experiments, CPCs were subjected to XXO or lumazine/xanthine oxidase-induced oxidative stress. Increasing concentrations of xanthine (200 μmole/L to 2.5 mM) were added to CPC treatment media with or without 10 mU/mL xanthine oxidase. Following 24 h of oxidative stress, DNA of CPCs were stained with PI and the sub-G1 DNA fractions in the cells were quantified using flow cytometry. At all substrate concentrations of xanthine, there was no difference between the sub-G1 fractions in the control and XXO-treated cells (Supplementary Fig. S2). Replacing xanthine with lumazine as a substrate to increase the flux of superoxide generation had no effect on sub-G1 fractions (Supplementary Fig. S2, beige bar). In our subsequent studies, treatment media containing 1 mmole/L xanthine, 10 U/mL catalase, and 10 mU/mL xanthine oxidase was used to generate the oxidative stress. In those conditions, dihydroethidium-based high-performance liquid chromatography analysis showed the presence of superoxide in the media even 15 h after XXO addition (Supplementary Fig. S3A). In addition, preliminary studies with amplex red assay show that around 75 μmole/L of superoxide flux was generated over 15 h using this substrate/enzyme concentration (Supplementary Fig. S3B).
Using the above treatment conditions, we compared oxidative stress-induced apoptosis in CPCs to that of a more mature cardiac cell type, neonatal cardiomyocytes, and analyzed the data using 2-way analysis of variance (ANOVA). Similar to preliminary observations, CPCs had no significant increase in the sub-G1 fraction following 24 h of XXO treatment compared to time-matched controls. In stark contrast, myocytes subjected to same treatment conditions had significantly more cells in the sub-G1 fraction compared to the control cells (P<0.05; Fig. 1A). To confirm whether this was truly apoptosis, cell death was also quantified using TUNEL assay in independent experiments conducted under the same treatment conditions. While 2-way ANOVA demonstrated no significant differences in basal cell death between the groups, TUNEL-positive cells were only significantly increased in myocytes treated with XXO (P<0.05; Fig. 1B, C). Taken together, these data demonstrate that CPCs are more resistant to oxidative stress-induced cell death than more mature cardiac cells.
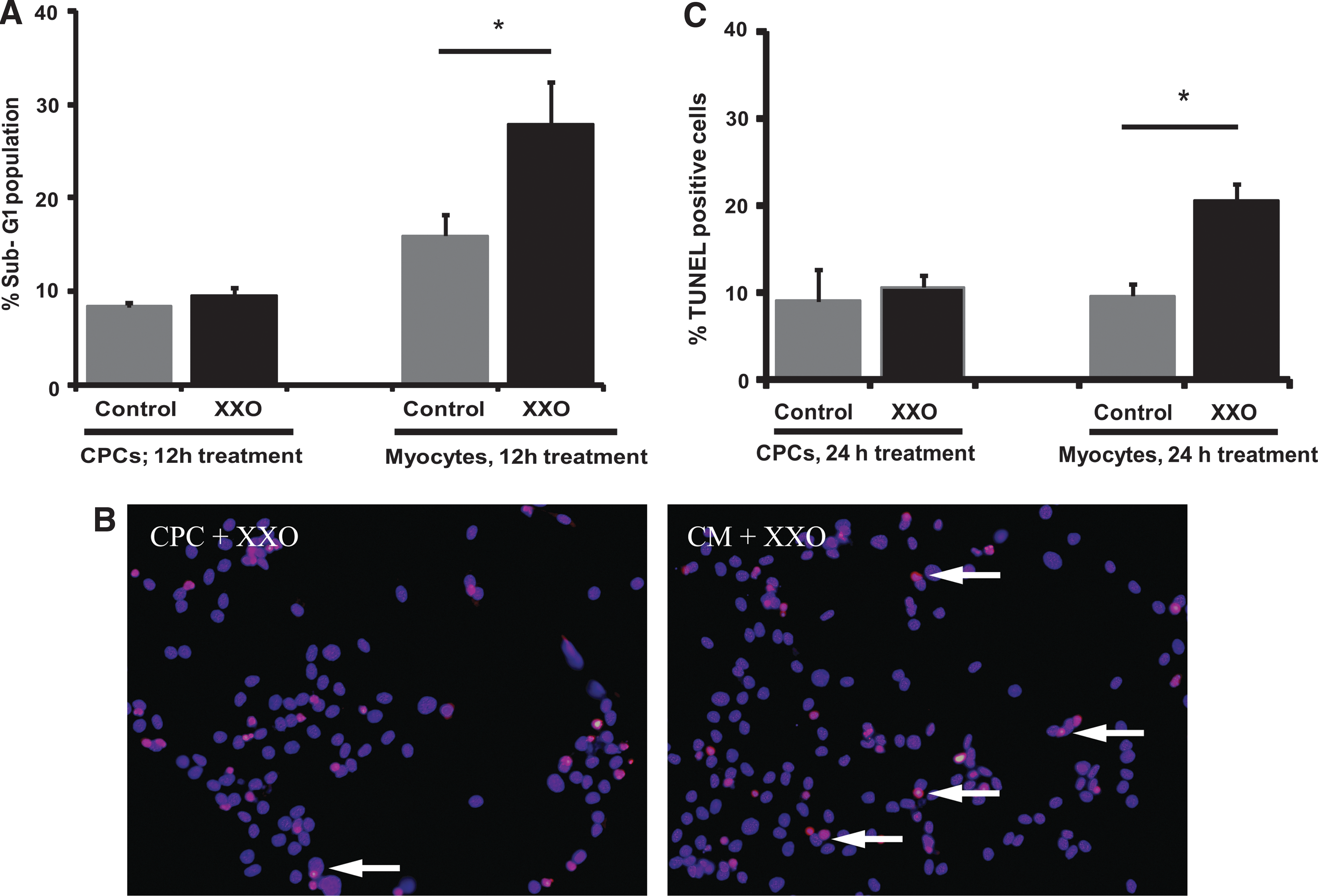
CPCs are more resistant to apoptosis following XXO treatment compared to myocytes.
CPCs have higher basal SOD levels compared to cardiomyocytes and FBs
To determine whether resistance to XXO-induced apoptosis was due to higher scavenging capacity, SOD activities were determined in CPCs and compared with 2 adult cell types, primary cardiomyocytes and cardiac FBs. SOD1 activity in CPCs was 3.76±0.40 U/mg protein, which was significantly higher than both FBs and neonatal cardiomyocytes (P<0.01; Fig. 2A). Similarly, basal activity levels of SOD2 followed the same trend in these 3 cell types. CPCs had 0.29±0.03 U/mg protein of SOD2 activity, significantly more than FBs (0.03±0.01 U/mg protein; P<0.01) and neonatal cardiomyocytes (0.21±0.05 U/mg protein; P<0.05) (Fig. 2A). In all cell types tested, only about 10% of total SOD activity was due to SOD2, while 90% or greater of total activity was due to SOD1 (Fig. 2B). In addition to the activity levels, the basal protein levels of both SOD1 and SOD2 were analyzed semiquantitatively using Western blotting. As shown in Fig. 2C, CPCs contained significantly higher protein levels of both SOD1 and SOD2 as compared to myocytes (P<0.01 and P<0.05, respectively). In the same fashion, CPCs also had significantly more SOD1 and SOD2 protein (P<0.001 and P<0.01, respectively), as compared to FBs (Fig. 2D). These data demonstrate a significantly higher superoxide scavenging capacity in CPCs compared with adult cardiac cell types.
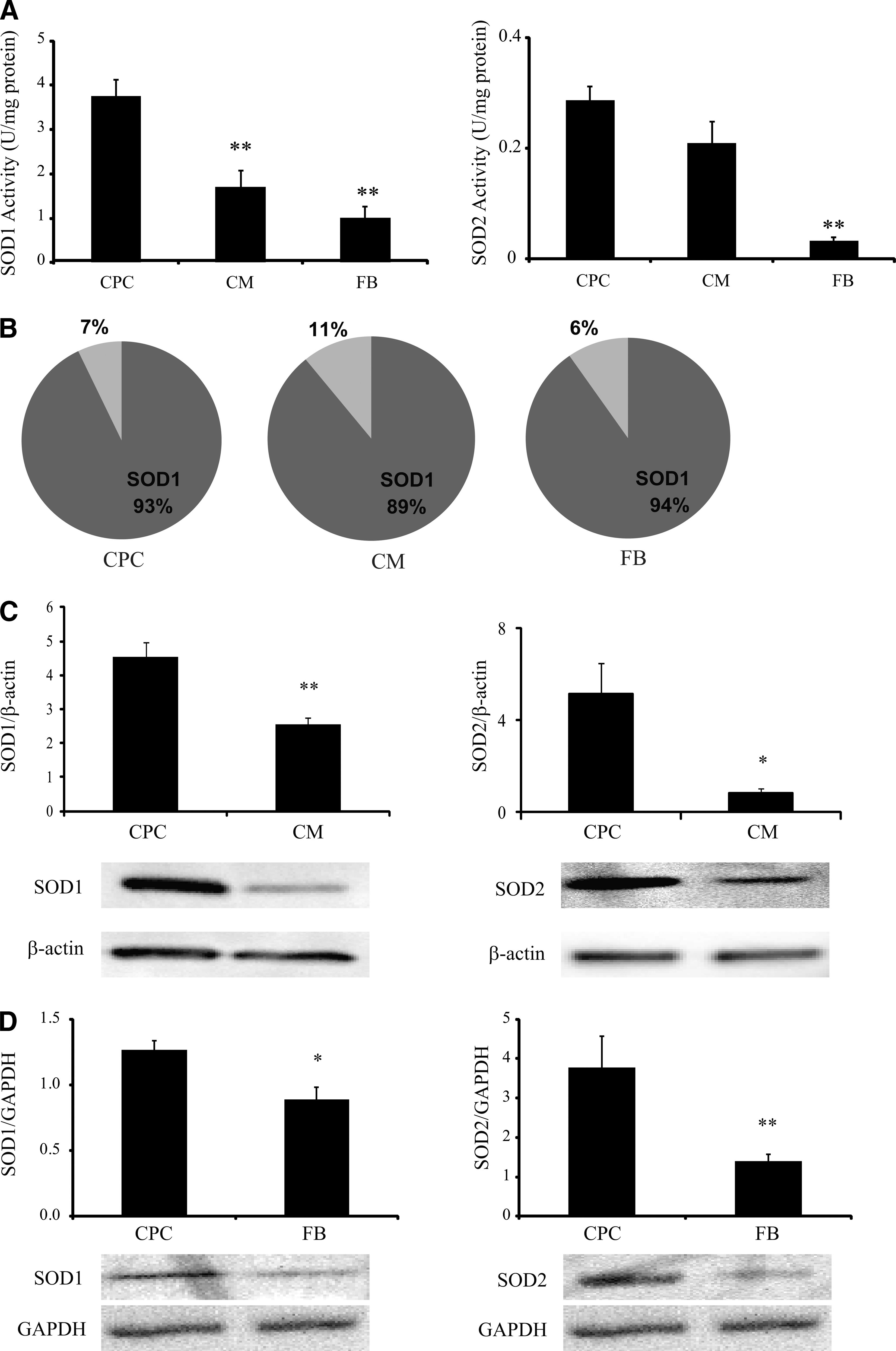
CPCs have higher basal SOD activity and protein levels compared to myocytes and fibroblasts.
XXO treatment increases the expression of SOD mRNAs
To determine whether CPCs could also compensate against XXO-induced injury by increasing SODs, qPCR was conducted for both sod1 and sod2 gene expression. After 3 h of XXO addition, the expression of sod1 mRNA significantly increased (P<0.01) by 1.95-fold before returning back to control levels (Fig. 3A). Similarly, after 6 h of XXO treatment, sod2 mRNA expression was significantly (P<0.05) increased by 1.89-fold compared to control (Fig. 3B) before returning to control levels. These data demonstrate that CPCs increase expression of SODs following oxidative injury.

XXO treatment increases SOD1 and SOD2 expression in CPCs.
SOD activity increases in CPCs after XXO treatment
To investigate whether XXO-induced changes in mRNA led to increased protein expression and activity, Western and activity analysis were performed. As Fig. 4A demonstrates, there was no increase in protein levels at early time points, though levels of SOD1 increased at 48 h and SOD2 increased at both 24 h and 48 h. CPCs were also treated with XXO for up to 48 h, and both SOD1 and SOD2 activity were determined and compared with time-matched controls. Two-way ANOVA analysis of the data showed that basal SOD1 activity in control cells dropped by 38.6%±6.1% within 48 h. However, this drop was not statistically significant (Fig. 4B). In contrast, following 48 h of XXO treatment in CPCs, SOD1 activity increased significantly (P<0.001) by 1.42±0.13-fold compared to its time-matched control (Fig. 4B). Basal SOD2 activity remained similar to control levels during the treatment, (Fig. 4C) and similar to SOD1 results, SOD2 activity also increased significantly (P<0.01) by 1.98±0.27-fold with XXO treatment compared to the time-matched controls (Fig. 4C). A slight, but not statistically significant decrease was seen early (3–6 h) in both SOD1 and SOD2 in XXO-treated cells.

XXO treatment increases SOD1 and SOD2 protein and activity levels in CPCs.
SOD2 protects CPCs from apoptosis following XXO treatment
To determine which SOD isoform conferred the most protection, CPCs were subjected to siRNA-induced gene silencing experiments. Following siRNA screening, 2 siRNAs that gave selective and efficient silencing of SOD1 and SOD2 expression were selected. After 24 h of transfection with siSOD1, expression of SOD1 mRNA was reduced by 95%, as compared to transfection-alone control (Supplementary Fig. 4A) with no effect on SOD2 mRNA levels. In a similar manner, transfection of CPCs with siSOD2 reduced the expression SOD2 mRNA levels by 95% compared to transfection-alone control, while the levels of SOD1 mRNA were unaffected (Supplementary Fig. 4B). Western blotting for SOD1 and SOD2 confirmed that gene knockdown translated to protein knockdown. Following 48 h of siRNA transfections, TUNEL assays were conducted in the presence and absence of XXO to investigate the oxidative stress-induced apoptosis in CPCs. As shown in Fig. 5, there was no significant change in basal or XXO-stimulated TUNEL-positive cells. Interestingly, XXO treatment induced a significant (P<0.05) 2.99-fold increase in the percentage of TUNEL-positive cells following siSOD2 treatment with no effect on basal cell death (Fig. 5A, B). Further, these data were confirmed by PI measurement of cells in sub-G1 (Fig. 5C). In either assay, there was no effect of SOD1 knockdown on basal or XXO-induced apoptosis. These data demonstrate that knockdown of only SOD2 sensitizes CPCs to XXO-induced apoptosis.

Silencing of SOD2 and not SOD1 sensitizes cells to XXO-induced apoptosis.
Regulation of SOD2 and apoptosis by Akt
As Akt plays an important role in cell survival, we first examined whether XXO exposure could induce phosphorylation of Akt. As the representative blot and grouped data in Fig. 6A demonstrate, Akt was rapidly phosphorylated (2.32±0.44-fold; P<0.01) in 10 min in response to XXO. This remained significantly increased at 20 min before returning to baseline by 30 min. To determine whether Akt phosphorylation regulated gene expression, we used both dominant negative Akt adenovirus (Ad-DNAkt) and the inhibitor of Akt phosphorylation LY-294002 (PI-3 kinase inhibitor; LY 10 μmole/L). CPCs were treated with 50 MOI of either Ad-DNAkt ([17] a generous gift of Dr. Kathy Griendling) or an adenovirus encoding lacZ (Ad-lacZ) for 5 h, and media was replaced overnight. The following day, cells were treated with XXO for 10 min and levels of both phospho- and total Akt were determined by Western blotting. Figure 6B shows a representative Western blot demonstrating XXO-induced phosphorylation of Akt in the presence of Ad-lacZ that is completely inhibited by Ad-DNAkt. As Fig. 6C shows, cells treated with Ad-lacZ, and then exposed to XXO for 6 h demonstrated increased sod2 expression compared to control CPCs (2.51±0.53-fold; P<0.05). This increase was significantly decreased in Ad-DNAkt-treated cells (1.52±0.31-fold P<0.05 versus Ad-lacZ+XXO). Similar results were seen with pretreatment of CPCs with LY for 30 min, which completely abrogated the increase in XXO-induced sod2 gene expression. While sod2 expression was increased by 1.95±0.19-fold by 6 h of XXO treatment (P<0.05), there was a significant reduction in LY-treated cells (Fig. 6D; 1.31±0.03-fold; P<0.05).
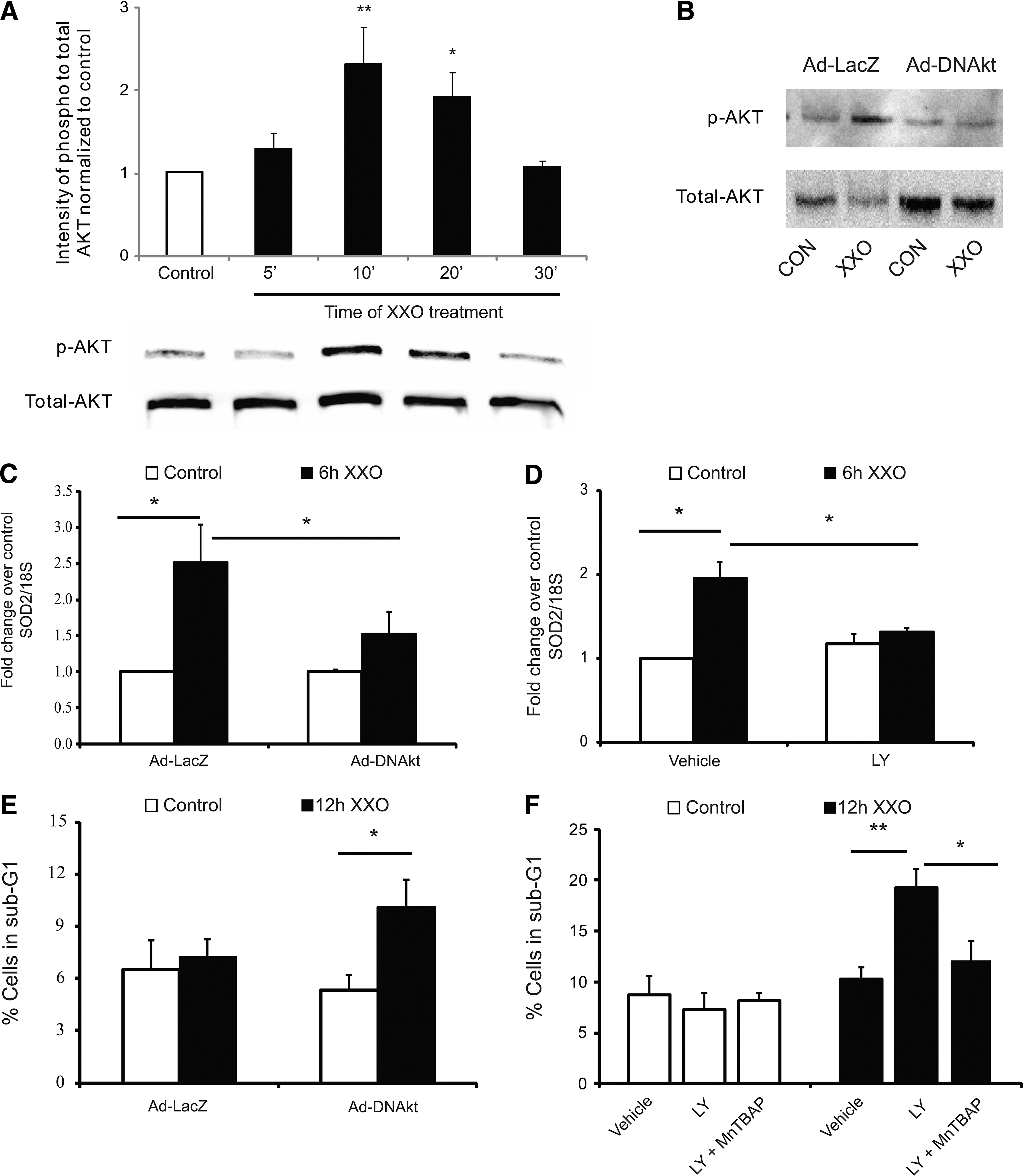
Akt activation regulates SOD2 gene expression and XXO-induced apoptosis.
To determine whether XXO-regulation of Akt phosphorylation controlled apoptosis, we pretreated cells with Ad-lacZ or Ad-DNAkt before 12 h of XXO. As the grouped data in Fig. 6E demonstrate, treatment of cells with DN-Akt sensitized the cells to apoptosis as measured by PI staining (P<0.05 vs. control cells). To further confirm this, we treated cells for 30 min with LY before addition of XXO or XXO plus the SOD2 mimetic MnTBAP (50 μM) for 12 h. Similar to our initial data, there was no change in apoptosis in CPCs treated with XXO in the absence of LY (Fig. 6F). In contrast, in LY-treated cells, XXO significantly increased the percentage of sub-G1 cells from 10.2±1.0 to 19.2±1.9 (P<0.01). Treatment of CPCs with XXO in the presence of the SOD2 mimetic MnTBAP completely inhibited the effect of LY, demonstrating a critical role for XXO-induced Akt activation leading to increased SOD2 expression (P<0.05 vs. XXO+LY).
Discussion
Various cell surface markers are used in the isolation of cardiac stem and progenitor cells [18]. The population identified by the presence of the surface marker c-kit has robust cardiovascular differentiation ability [14] and is currently in early clinical trials. Therefore, we isolated c-kit-positive cells expressed in the rat myocardium using the immunomagnetic sorting method. The current isolation yielded a population that is greater than 90% c-kit positive and this percentage varies from 60% to 90% from passages 4 to 15. Although, the exact reason for variation in the percentage of c-kit expression is not known, c-kit receptors are known to undergo ligand-induced dimerization and internalization [19]. C-kit expression is essential for the promotion and regulation of cardiac stem cell differentiation [20]; however, lower percentage (20%) of c-kit expression does not reduce its cardiogenic potential [1]. It is important to note that c-kit expression is not exclusive to cardiac stem cells and is also found in many other progenitors, including hematopoietic lineage, as well as mature cell lines, such as mast cells, astrocytes, sweat glands, and breast glandular epithelial cells [21]. Therefore, the c-kit-positive cells were tested for the presence of other markers, such as Sca-1, MDR-1, and CD34 that are reported to exist in cardiac stem cells and also tested for its clonability [18]. Our analysis showed that the isolated cells were indeed clonogenic and were positive for cardiac stem cell markers Sca-1, MDR-1, and CD34 and negative for the hematopoietic marker CD45. Additionally, the cells used in this study were positive for the expression of early cardiac transcription factors, such as Nkx-2.5 and GATA-4. Therefore, they were termed as CPCs instead of cardiac stem cells that lack the expression of these transcription factors [14].
XXO is a physiologically relevant system to generate reactive oxygen species–particularly superoxide and hydrogen peroxide [22]. Our preliminary experiments confirmed the generation of free radicals in the treatment media following XXO addition. Further, XXO is known to induce apoptosis in many cell types [23, 24]. Therefore, in our studies, XXO was used to investigate the effects of oxidative stress on CPC apoptosis. Following PI staining of DNA, increased fractions of fragmented DNA generated due to cell death can be observed behind the diploid peak of normal, nonfragmented DNA. Quantification of this hypodiploid or sub-G1 fraction gives a simple estimate of what are believed to be apoptotic cells [25]. While previous reports show apoptosis with 100 μM xanthine and 10 mU/mL XXO in HUVECs [23], our PI staining studies in CPCs showed that there was no difference in the sub-G1 fraction between XXO-treated cells and control cells even at higher xanthine concentrations. This interesting observation prompted us to compare the oxidative stress-induced apoptosis in CPCs and neonatal cardiomyocytes. Independent apoptosis quantification using PI staining and TUNEL assay showed increased apoptosis in myocytes following XXO treatment with no effect of XXO in CPCs. While there were slight variations in basal cell death, these were not considered significant and could be due to the fact that cardiomyocytes are terminally differentiated cells.
SODs are one of the major endogenous antioxidant enzymes providing first line protection against oxidative stress. For example, higher SOD2 and catalase levels in circulating endothelial progenitors protect them from oxidative stress-induced apoptosis compared to related cell types [9]. In addition, enhanced expression of antioxidant genes is considered a stemness trait in many progenitor cells [26]. Therefore, the basal SOD protein and activity levels of CPCs were compared with other adult cardiac cell types. Interestingly, CPCs had significantly higher protein levels of both SOD1 and SOD2 enzymes compared to FBs and myocytes, suggesting this as a potential mechanism by which CPCs could be protected from oxidative stress unlike cardiomyocytes. SOD activity assays confirmed Western analysis, though SOD2 levels were not significantly lower in cardiomyocytes compared to CPCs, despite the large difference in protein levels. As SOD2 activity is dependent on other cofactors in addition to protein levels, these need to be examined in more detail in future studies.
Oxidative stress is known to increase the expression of antioxidant enzymes in many other cell types, such as epithelial cells and FBs [27 –29]. Therefore, the effect of oxidative stress on the antioxidant enzyme gene expression in CPCs was investigated. SOD2 is known to be regulated by external factors, while SOD1 is often thought to be constitutively expressed in various cell types [30]. RT-PCR analysis of CPC mRNAs showed that XXO treatment induced almost 2-fold higher expressions of sod1, sod2, and gpx1 mRNAs in 3–6 h, perhaps suggesting a compensatory mechanism. SOD proteins are known to undergo proteasome-induced degradation [31]. Further, their levels drop within 2 h of oxidative stress [32]. Consistent with published results, SOD activities measured in CPCs show that the cells lose about 25% of total SOD activity within 6 h of oxidative stress [33]. However, after 48 h of oxidative stress, CPCs had not only regained activity, but had significantly higher total SOD activity levels compared to control cells. Additionally, 2-way ANOVA conducted on CPCs with time-matched controls demonstrate significantly higher activity of SOD1 and SOD2 following 48 h of XXO treatment, suggesting that the increased SOD mRNA expression led to changes in protein levels. While there appears to be some lag between the new mRNA expression (3–6 h) and new protein expression (24–48 h) and activity levels (48 h), this may be due to both the highly regulated destruction and generation of the protein. Whereas published studies show a decrease in protein levels following generation of hydrogen peroxide (SOD product), our studies demonstrate no decrease in protein levels, perhaps due to the compensatory increase in expression. Thus, in our system, there appears to be a constant balance being struck between degradation and generation that result in a net increase following 24–48 h.
To determine if enhanced SOD levels were responsible for the resistance of CPCs to oxidative stress-induced apoptosis, siRNA-based gene silencing experiments were conducted to determine the contribution of SOD1 and SOD2 to this phenotype. While decreased SOD1 had no significant effect on protection against XXO-induced apoptosis, our results showed that by decreasing the expression of SOD2, CPCs had significantly higher apoptosis following XXO treatment. This result is interesting given that total SOD2 activity is only 10% of total activity in CPCs. Similar observations exist in the literature that show that SOD2, but not SOD1, is critical to protect the cells against oxidative stress-induced apoptosis [34]. SOD2 is known to maintain mitochondrial membrane integrity, which when disrupted leads to cytochrome release and begins the chain of events leading to apoptosis [35]. The result also implicates the importance of maintaining the mitochondrial homeostasis and oxidative stress level in CPCs.
In addition to cell survival, senescence of endogenous CPCs is another important issue facing regenerative therapies [36], and SOD2 deficiency is implicated in senescence and apoptosis in mouse models [37]. Recent studies have shown that the human myocardium has a young and senescent population of CPCs marked by the presence or absence, respectively, of insulin-like growth factor-1 receptors [12]. Further, senescent CPCs are more prone to oxidative damage and apoptosis [4]. Previous studies have shown that addition of IGF-1 or activation of the downstream Akt signaling reduces the apoptosis following serum deprivation-induced oxidative stress [38, 39]. Our unpublished data in select human samples demonstrate potential differences in SOD2 in subsets of human CPCs and studies are ongoing to determine the role of SOD2 in human CPCs.
Akt signaling is known to control the expression of SOD1 and SOD2 [32, 40]. Further, following oxidative stress, Akt is known to be activated within 10 min [41], and evidence from our current study demonstrated Akt activation within 10 min of XXO treatment in CPCs. Additionally, our experiments demonstrated an increase in XXO-induced apoptosis following inhibition of Akt with either a dominant negative approach or chemical inhibition with LY-29004. One potential explanation may be that increased SOD2 leads to rapid dismutation of superoxide into hydrogen peroxide, which has been shown to stimulate Akt activation [42]. While it is entirely possible that Akt may be inducing survival independent of SOD2 gene regulation, addition of the SOD2 mimetic MnTBAP completely rescued the phenotype of Akt inhibition, suggesting a potential link. While it is unclear how Akt is regulating SOD2 expression in these cells, it was previously shown to occur through PI-3 kinase/Akt regulation of forkhead transcription factors in endothelial cells [43]. This will be examined in future CPC studies to determine whether similar mechanisms are seen.
In summary, our data demonstrate that CPCs contain higher levels of SODs compared to adult cell types and are able to increase levels further during oxidative insult. These higher basal levels, in particular that of SOD2, are responsible for the resistance to XXO-induced apoptosis in rat CPCs. Induction of SOD2 gene expression is regulated by Akt, and this confers resistance to XXO-induced apoptosis. Future studies will need to be performed to determine the importance of this axis in human CPCs with clinical trials underway.
Footnotes
Acknowledgment
This work was supported by HL094527 and HL089120 to MED.
Author Disclosure Statement
The authors have nothing to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.