
Abstract
Inhibition mechanisms of protein kinase B (Pkb)/Akt and its consequences on related cell signaling were investigated in human umbilical cord blood stem cells (hUCBSCs) exposed to monocrotophos (MCP, an organophosphate pesticide). In silico data reveal that MCP interacts with kinase and c-terminal regulatory domains of Akt1, resulting into a total docking score of 5.2748 and also forms H-bond between its N-H and Thr-291 residue of Akt1, in addition to possessing several hydrophobic interactions. The main cause of Akt inhibition is considered to be the strong hydrogen bond between N-H and Thr-291, and hydrophobic interactions at Glu-234, and Asp-292 in the vicinity, which is usually occupied by the ribose of ATP, and interaction with residue Phe-161, thus leading to a significant conformational change in that particular portion of the protein. In silico data on Akt inhibition were confirmed by examining the downregulation of phosphorylated (Thr308/Ser493) Akt1 in MCP-exposed hUCBSCs. MCP-mediated altered levels of pAkt downstream targets viz., downregulated pGSK3β (Ser9), unchanged GSK3αβ, and upregulated levels of Bad, P53, and caspase-9 further confirm the inhibition of pAkt. The cellular fate of such pAkt inhibition was confirmed by increased terminal deoxynucleotide transferase dUTP nick-end labeling positive cells, reduced mitochondrial membrane potential, and the activation of various MAPKs, proapoptotic markers-Bax, and caspases-9/3. Our data demonstrate that Akt1 plays a key role in MCP-induced apoptosis in hUCBSCs. We also identified that such cellular responses of human cord blood stem cells against MCP were due to strong binding and inhibition of kinase and AGC-Kinase-C terminal regulatory domains of Akt1.
Introduction
P
Phosphoinositide 3-kinase (PI3K) is the only enzyme that can phosphorylate PPI2 to PPI3, which eventually activates the cellular signaling in terms of cell proliferation and growth by means of activation of Akt1. In experimental conditions, the overexpression of a tumor suppressor (PTEN) was found to reverse this process by dephosphorylating PPI3 to PPI2 [4,11]. Akt also prevents the degradation of cyclin D1 by phosphorylating and inhibiting GSK3β, attenuates the cell cycle inhibitors such as p21waf1 and p27kip1 by phosphorylating these proteins, and also phosphorylates BAD, which triggers its release from Bcl-2/Bcl-xL proteins in the mitochondrial membrane as well as procaspase 9, and thus helps in preventing the apoptosis [4].
Multiple efforts have been made to discover small-molecule antagonists that may hamper the different functions of Akt1 domains or their interacting proteins, since Akt inhibition considered an effective therapy against different types of cancer [5,12 –16]. Some pesticides such as dieldrin and endosulfan induced ERβ-mediated activation of Akt phosphorylation in cortical neurons; however, in cerebellar granule cells, same dieldrin induced Akt Phosphorylation by multiple activation of ERα, ERβ, and G protein-coupled receptor 30. Thus, the extracts of these dieldrin- and endosulfan-treated cortical neurons induce the proliferative potential of cancerous MCF-7 cells [3]. Other reports indicate that the pesticide has role in inducing cell injuries by inhibiting pAkt that could result into hampered glucose metabolism in 3T3-L1 adipocytes [2]. Primarily, the Akt-mediated pathways have been explored in cancer to investigate the mechanisms of protection [5,13,15,16], but the mechanisms of Akt inhibition in noncancerous normal human stem cells are still to be explored. Thus, the present investigations were aimed to study the effect of the pesticide monocrotophos (MCP) on the activation of Akt/Pkb and their consequences on downstream signaling pathways in human umbilical cord blood stem cells (hUCBSCs). The reason for carrying out such types of studies in hUCBSCs is that because transplacental diffusion of pesticides and subsequent impaired hematopoiesis/and related disorders are well documented [17,18]; however, the underlying cell death mechanisms involved in hematopoietic stem cells are largely unknown. These cells are pluripotent in nature and give rise to all kind of hematopoietic and matrix cells in the organisms, and small injuries in these cells result into long-lasting consequences. The present investigation was carried out using an organophosphate pesticide, since the pesticide exposure in mice has already been reported to reduce the total number of bone marrow cells by arresting the hematopoietic and nonhematopoietic progenitor cells either in the G0/G1-phase or in the S/G2/M-phase, and subsequently impaired the hematological parameters. The results clearly indicated the significant long-lasting toxic effect of pesticides on the bone marrow microenvironment, which ultimately leads to the formation of a degenerative disease like aplastic anemia [19]. In the independent studies on infant acute and childhood leukemia, it has been reported that frequent exposure of insecticides during the gestation period and early stage of life poses significantly a higher risk of disease than later exposures. The hematotoxicological consequences through transplacental diffusion of pesticides were found to be greater than outdoor exposures of pesticides [20,21]. In the present investigations, MCP was selected as a model pesticide, as it is being used extensively worldwide for more than forty years, and its adverse outcomes have reported earlier by us [22,23].
Materials and Methods
In silico studies
Molecular modeling parameters and energy minimization
The molecular modeling of compounds viz., MCP and camptothecin, was performed with Protein kinase B (Pkb/Akt), a serine/threonine kinase, which plays a key role in the regulation of cell survival and proliferation, by using Sybyl X 1.3 molecular modeling and drug discovery software (
Molecular docking studies
To find out the possible bioactive conformations of different Akt inhibitors, the Sybyl X 1.3 interfaced with Surflex-Dock program [25] was operated to dock all the compounds into the active site of the Akt kianse (PDB code: 3CQU). The program automatically docks ligand into the binding pocket of an enzyme/receptor protein using a protomol-based algorithm and empirically produced scoring function. The protomol is a very important and necessary factor for docking algorithm and works as a computational representation of the proposed ligand that interacts into the binding site. Surflex-Dock's scoring function has several factors that play an important role in the ligand–receptor interaction, in terms of hydrophobic, polar, repulsive, entropic, and solvation, and it is a well-established and recognized method [26]. The most standard docking protocols have ligand flexibility into the docking process, while count the protein as a rigid structure [27]. Our molecular docking involves the following several steps: (1) the protein structure was imported into Surflex, and then hydrogens were added; (2) generation of protomol using a ligand-based strategy, and during this process, 2 parameters (first called protomol_bloat, which determines how far the site should extend from a potential ligand; and another called protomol_threshold, which determines deepness of the atomic probes that is used to define the protomol penetration into the protein) must be specified to form the appropriate binding pocket. Thus, in the current study, protomol_bloat was set to 0, and protomol_threshold was set to 0.50, when a reasonable binding pocket was obtained; and (3) all of the compounds were docked into the binding pocket, and 20 possible active docking conformations with different scores were obtained for each compound. During the docking process, all of the other parameters were assigned to their default values. Surflex-Dock total scores, which express the −log10 (Kd) units to represent binding affinities, were applied to estimate the ligand–receptor interactions of the newly designed molecules.
In vitro validation studies
Reagents and consumables
All the chemicals, reagents, and kits used in this study were purchased from Stem Cell Technologies and Sigma, unless otherwise stated. All cytokines and growth factors such as recombinant human basic fibroblast growth factor (rhbFGF), thrombopoietin (rhTPO), stem cell factor (rhSCF), and fetal liver tyrosine kinase 3 ligand (rhFLT-3 Ligand) were purchased from PeproTech. All the antibodies were procured from Chemicon International and Abcam. Culture wares and plastic wares were procured from Nunc and Corning Incorporated. Autoclaved Milli-Q water was used in all the experiments.
Ethics clearance for collection and transportation of human tissues
The entire study was carried out by following the protocols and procedures approved by the Institutional Human Ethics Committees of Indian Institute of Toxicology Research (IITR), Lucknow, India, and the CSM Medical University, Lucknow, India. The informed consent of parents was obtained before collecting blood from umbilical cord.
Isolation and purification of human umbilical cord blood hematopoietic stem cells
The entire study was carried out by following the protocols and procedures approved by the Institutional Human Ethics Committees of the IITR, Lucknow, India, and the CSM Medical University, Lucknow, India. The informed consent of parents was received before collection of the blood from umbilical cord. Mothers enrolled in the study were of age range 24.5±6.2 years. They fulfilled the entire inclusion criteria and were nonobese and free from malignancy or any other systemic disorder. Totally, 52 blood samples (∼40 mL/cord) were collected from the cord vein in a sterile container having an anticoagulant citrate dextrose buffer and immediately transported to the IITR, Lucknow, for further processing. Blood was diluted in the ratio of 1:1 with Dulbecco's phosphate-buffered saline (DPBS) without Ca2+ and Mg2+, pH 7.5 (Stem Cell Technologies). Subsequently, cord blood mononuclear cells were segregated through negative immunodepletion of CD3+, CD14+, CD19+, CD38+, and CD66b+ cells using a RossetteSep™ cord blood CD34 Pre-enrichment Cocktail (Stem Cell Technologies; Catalog No. 15631C), according to the instructions given by the manufacturer, followed by Ficollpaque™ (1.077 g/cm3; Stem Cell Technologies) density-gradient centrifugation (700 g for 30 min). CD34+ hematopoietic stem cells were isolated from mononuclear cells using the automated robotic and magnetic cell separator RoboSep™ (Stem Cell Technologies; Catalog No. 20000) and an EasySep™ human cord blood CD34-positive selection kit (Stem Cell Technologies; Catalog No. 18056) possessing monoclonal bispecific antibodies against the human antigen CD34 and iron-core dextran-coated nanoparticles as per instructions from the manufacturer.
Proliferation and bulk production of hematopoietic stem cells
Freshly isolated hematopoietic stem cells were cultured in a plastic 25-cm2 ultralow-attachment culture flask (Corning Incorporated) at a density of 1×105 cells/mL in 5 mL of a serum-containing medium, the myelocult medium (Stem Cell Technologies; Catalog No. H5150) supplemented with hydrocortisone (10−6 M) and other growth factors viz., rhbFGF (50 ng/mL), rhSCF (25 ng/mL), rhTPO (25 ng/mL), rhFLT-3 Ligand (10 ng/mL; PeproTech). Cells were maintained as suspension in a humidified atmosphere at 37°C and 5% CO2. Half of the medium was changed twice in a week. At each passage, cells were checked for the CD34+ marker to ascertain the purity of stem cells. Cells were subcultured at the confluence of 80%–90% in a myelocult medium supplemented with hydrocortisone and growth factor cocktails as earlier.
Purity analysis of stem cells
Enriched stem cells were analyzed for purity by quantifying the hematopoietic stem cell (CD34+) and primitive stem cell (Thy1+) markers. In brief, stem cells (1×105) were centrifuged at 200 g for 10 min and then stained with PE-Texas Red conjugated anti-CD34 and anti-Thy1 antibodies (Stem Cell Technologies) for 30 min at 4°C. The cells were then washed with DPBS and analyzed using flow cytometry (BD-FACS Canto) equipped with BD FACS Diva, version 6.1.2 software. The data are presented in percent population of stem cells.
Dose selection of MCP
Noncytotoxic doses of MCP used in the study were identified using standard endpoints of cytotoxicity, that is, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) and lactate dehydrogenase (LDH)-release assays.
MTT assay
The MTT assay was carried out as per the protocols described by us earlier [22]. In brief, stem cells (1×104 cells/well) were seeded in laminin- (25 μg/mL) precoated 96-well tissue culture plates (Corning Incorporated) and incubated under a high-humid environment in 5% CO2 for 24 h at 37°C. After that, the medium was replaced with a fresh medium containing different concentrations of MCP (10−3 to 10−8 M). The cells were reincubated for 12–96 h, and thereafter the MTT assay was done. Tetrazolium bromide salt (10 μL/well; 5 mg/mL of stock in PBS) was added 4 h before the completion of respective incubation periods. Then, the reaction mixture was carefully taken out, and 200 μL of culture-grade DMSO was added to each well by pipetting up and down several times until the content gets homogenized. After 10 min, the color was read at 550 nm using a Multiwell Microplate Reader (Synergy HT; Bio-Tek). Parallel sets without MCP were also run under identical conditions that served as basal controls.
LDH assay
LDH release is a method to measure the membrane integrity as a function of the amount of cytoplasmic LDH released into the medium. The LDH assay was carried out using the readymade commercially available LDH assay kit for in vitro cytotoxicity evaluation (TOX-7; Sigma). The assay was based on the measurement of the activity of LDH release from damaged cells. The resulting colored compound was measured after the deduction of background at 690 nm using a Multiwell Microplate Reader (Synergy HT; Bio-Tek) at 490 nm. The culture conditions and MCP exposure and incubation periods were similar to that in MTT assay. After the respective exposure period (12–96 h), cells were removed from the CO2 incubator and centrifuged at 250 g for 4 min. Then, the supernatant from each well was transferred to a fresh flat-bottom 96-well culture plates and processed for an enzymatic assay following the instructions and guidelines provided with the kit. A noncytotoxic dose (10−5 M) of MCP was used to study the alterations in the expression (mRNA and protein) of markers of apoptosis and cell death.
Oxidative stress
Reactive species generation
Estimation of MCP-induced reactive oxygen species (ROS) generation was carried out following the protocol described earlier by us [23]. In brief, cells (2×104 per well) were seeded in laminin- (25 μg/mL) precoated 96-well black-bottom culture plates and allowed to adhere for 24 h in 5% CO2–95% atmosphere at 37°C. Thereafter, the medium was aspirated, and cells were exposed to MCP (10−5 M), camptothecin (3 μg/mL), NAC (10 μM) and NAC+MCP (10 μM) for 6 h. After the exposure, cells were reincubated with 2′, 7′ dichlorodihydrofluorescein–diacetate (DCFH-DA) (20 μM) for 30 min at 37°C. Thereafter, the reaction mixture was replaced by PBS (200 μL per well). The plates were then kept on a rocker shaker platform for 10 min at room temperature in dark, and the fluorescence intensity was measured using a Multiwell Microplate Reader (Synergy HT; Bio-Tek) on excitation wavelength at 485 nm and emission wavelength at 528 nm. The data are expressed in percent of the unexposed control.
Estimation of intracellular glutathione levels
Glutathione (GSH) levels were assessed in the cells exposed to MCP (10−5 M), camptothecin (3 μg/mL), NAC (10 μM), and NAC+MCP (10 μM) for 6 h using a commercially available kit (Glutathione Detection Kit, Catalog No. APT250; Chemicon). In brief, after respective exposures, cells were collected through centrifugation at 700 g for 2 min at 4°C and lyzed in a lysis buffer. The samples were recentrifuged at 12,000 g for 10 min at 4°C, and supernatants were collected. To estimate the GSH levels, the lyzed samples (90 μL/well) were transferred to 96-well black-bottom plates, mixed with a freshly prepared assay cocktail (10 μL), and incubated for 2 h. Thereafter, plates were read on an excitation wavelength at 380 nm and an emission wavelength at 460 nm using a Multiwell Microplate Reader (Synergy HT; Bio-Tek). The standard curve was plotted using the GSH standard supplied in the kit and used to calculate the experimental values.
Terminal deoxynucleotide transferase dUTP nick-end labeling assay
Apoptosis was detected by terminal deoxynucleotide transferase dUTP nick-end labeling (TUNEL) assays using the APO-BrdU TUNEL Assay Kit with Alexa-Fluor-488 anti-BrdU (Molecular Probes, Invitrogen Detection Technologies; Catalog No. A23210) by a flow cytometer (BD-LSRII) and analyzed by Cell Quest 3.3 software. Debris was excluded by forward and sideway light scattering. Positive and negative controls were provided along with the kit. Cells exposed to camptothecin (3 μg/mL) for 6 h were also used as positive controls.
Detection of mitochondrial membrane potential
Mitochondrial membrane potential, an early marker of apoptosis induction, was assessed using a Mitolight™ Apoptosis Detection Kit (APT142; Chemicon). Cells in culture (4×104 cells/well in a PLL-coated black-bottom 96-well plate) were exposed to MCP (10−5 M for 6 h). The medium was then aspirated, and cells were washed with DPB. Cells were then reincubated with 100 μL of prediluted Mitolight™ dye solution for 15 min (37°C, 5% CO2–95% air) and allowed to equilibrate at room temperature for 10 min at dark. The fluorescence intensities were measured at an excitation wavelength 485 nm and an emission wavelength 530 nm to monitor monomer and wavelength 580 nm for aggregated molecules, respectively.
Transcriptional changes
Cells were exposed to MCP (10−5 M) for 3 h. Cells were then harvested and processed for real-time PCR. TaqMan Low Density Array of 48 genes was designed in 384-well plate formats and procured from Applied Biosystems (Table 1). MCP-induced alterations in the expression of mRNA of markers genes of stemness, proliferation, apoptosis, oxidative stress, and metabolism were analyzed using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). The whole procedure was carried out by following the protocol as described by us earlier [22,23]. In brief, RNA was isolated from both MCP-exposed and unexposed cells using a GenElute mammalian total RNA Miniprep Kit (Catalog No. RTN-70; Sigma). The purity and yield of RNA were assessed by a Nanodrop ND-1000 Spectrophotometer V3.3 (Nanodrop Technologies, Inc.), and the quality was assessed by running RNA onto a 2% denaturing agarose gel. Total RNA (2 μg) was reverse-transcribed into cDNA using a SuperScript III first-strand cDNA synthesis Kit (Catalog No.18080-051; Invitrogen Life Science) and treated with RNase-free DNase I to remove any potential DNA contamination. cDNA was first preamplified using the primer pool provided by Applied Biosystems with a customized TaqMan Low Density Array as per the manufacturer's protocol. Quantitative real-time PCR was performed by using the ABI PRISM®7900HT Sequence Detection System (Applied Biosystems). Real-time reactions were carried out in triplicate. GAPDH was used as internal control to normalize the data. MCP-induced alterations in mRNA expressions of different genes are expressed in relative quantity compared with unexposed control groups.
Translational studies
Cells were exposed to MCP (10−5 M) for 6 h. The altered expression of marker proteins of apoptosis and cell death pathways was studied in both experimental and control groups. Proteins harvested from experimental and control groups were processed for western blot analysis following the protocol described earlier by us [22,23]. In brief, MCP-exposed and unexposed cells were pelleted and lyzed using the CelLytic™ Cell Lysis Reagent (Catalog No. C2978; Sigma) in the presence of 1×protein inhibitor cocktail (Catalog No. P8340; Sigma) and 1 mM sodium orthovanadate. After protein estimation by BCA Protein Assay (Catalog No. G1002; Lamda Biotech, Inc.), equal amounts (50 μg/well) of proteins were loaded in a 10% Tricine–SDS gel and blotted on a polyvinylidene fluoride membrane using a wet transfer system. After blocking for 2 h at 37°C, the membranes were incubated overnight at 4°C with specific antiprotein primary antibodies of PI3K (1:1,000), pGSK3β (Ser9), GSK3α/β, β-catenin, pAkt (Thr308) and pAkt (Ser473) (1:500), p-JNK (1:1,000); p53 and P21 (1:500), Bax (1:500), Bcl2 (1:1,000), and activated caspase-9 and 3 (1:500 and 1:200; Chemicon, Inc.; BD Biosciences; Santa Cruz) in a blocking buffer (pH 7.5). The membranes were then incubated for 2h at room temperature with a secondary antiprimary immunoglobulin-G conjugated with horseradish peroxidase (Calbiochem). The blots were developed using luminol (Cat. No. 34080; Thermo Scientific), and densitometry for protein-specific bands was done in a Gel Documentation System (Alpha Innotech) with the help of AlphaEaseTM FC StandAlone V. 4.0.0 software. Actin-β was used as the internal control to normalize the data. MCP-induced alterations in the expression of marker proteins are expressed in relative quantity compared with the unexposed control groups.
Activity of caspase-9 and 3
MCP-induced alterations in the activity of caspase-9 and caspase-3 were estimated using kits (Calbiochem; Catalog No. QIA72 and Biovision; Catalog No. K196, respectively). The exposure groups and conditions were identical to that of ROS and GSH. After respective exposures, cells were pelleted, resuspended in a prechilled extraction buffer (50 μL), and incubated for 10 min on ice. Then, the samples were centrifuged for 5 min at 500 g, and the clear supernatant (50 μL per well) was transferred to black-bottom 96-well culture plates for caspase-9, and transparent-bottom plates for caspase-3 activity. Assay buffer (50 μL) and substrate conjugate (10 μL) for caspase-9 and (5 μL) for caspase-3 were added and mixed well. Upon the completion incubation of 2 h at 37°C in dark, contents were mixed thoroughly and read for fluorescence quantification on excitation at 400 nm and emission at 505 nm for caspase-9, whereas caspase-3 activity was measured by taking absorbance at 400 nm. The values of experimental groups were compared with the unexposed control group, and data expressed in fold change in activity.
Statistical analysis
Results were expressed as mean±standard error of mean (SEM) for the values obtained from at least 3 independent experiments. The level of significance for comparison between samples was ascertained using the Student's t-test distribution. For recovery experiments, 1-way ANOVA, followed by post hoc Tukey's b test, was used. The data in the graphs are expressed as the mean±SEM. Graphpad Prism 3.0 software (Graphpad Software) was used for all the statistical analyses. The values P<0.05 were considered to be significant.
Results
In silico molecular docking analysis of MCP
The aim of the molecular docking study was to elucidate whether the test compound MCP modulates the cell survival protein kinase B (Pkb/Akt), and to study their possible mechanisms of action. The results of the molecular docking suggest that the studied compound binds strongly with the kinase and C-terminal regulatory domains of Akt at a region that also serves as an ATP-binding pocket. In the work presented here, we explored the orientation and binding affinity [in terms of the total score in −log10 (Kd) units to represent binding affinities] of camptothecin and MCP toward the anticancer target Akt kinase (PDB code: 3CQU). The docking reliability was validated using the known crystalline X-ray structure of Akt kinase complexed with the pyrrolopyrimidine inhibitor. The co-crystallized pyrrolopyrimidine inhibitor was redocked into the binding site, and the docked conformation with the highest total score of 7.7894 was considered as the most probable binding conformation. The low root mean-square deviation of 0.3769 Å between the docked and the crystal conformation indicates the high reliability of Surflex-dock software in reproducing the experimentally observed binding mode for this inhibitor. As shown in Fig. 1, redocked molecules were almost in the same position with co-crystallized at the active site of the pyrrolopyrimidine inhibitor (Fig. 1A).

In silico molecular docking studies elucidating the possible mechanisms of monocrotophos (MCP)-induced modulation of cell survival protein kinase B (Pkb/Akt). The docking studies were carried out using SYBYL-X 1.3, Tripos International.
On the other hand, docking results for camptothecin against the anticancer/survival target protein Akt kinase showed a high binding affinity docking score indicated by a total score of 5.7543 and forms a H-bond of length 2.1 Å to the hydrophobic aliphatic residue that is, Alanine 230. In the docking pose of the camptothecin–Akt complex, the chemical nature of binding site residues within a radius of 4 Å was basic (polar, hydrophobic, positive charged), for example, Arg-4 (Arginine) and Lys-276 (Lysine); aromatic (hydrophobic), for example, Phe-161, Phe-438 (Phenylalanine), and Tyr-229 (Tyrosine); acidic (polar, negative charged), for example, Glu-228 (Glutamic acid) and Asp-292 (Aspartic acid); polar amide type, for example, Asn-279 (Asparagine); hydrophobic, for example, Ala-177, Ala-230 (Alanine), Gly-157 (Glycine), Leu-156 (Leucine), Met-227, and Met-281 (Methionine); and nucleophilic (polar, hydrophobic), for example, Thr-211 and Thr-291 (Threonine); as a result, the bound compound showed a strong hydrophobic interaction with Akt kinase, thus leading to more stability and activity in this compound (Fig. 1B).
The binding affinity obtained in the docking study allowed a comparison between the activities of the MCP to be compared to that of the standard anticancer drug camptothecin. MCP showed a high binding affinity against the Akt kinase target protein. During the comparison of the nature of interaction between the binding pocket amino acid residues of target protein and the compound, it was found that the compound MCP showed molecular interaction with conserved hydrophobic amino acid residues, thus leading to more stability and potency (Table 2). The docking results for MCP showed that the compound docked on the anticancer target Akt kinase with a high binding affinity docking score indicated by its total score of 5.2748 and also showed the formation of a H-bond of length 2.3 Å to the hydrophobic nucleophilic (small, polar) residue Threonine, that is, Thr-291. The MCP-Akt-docked complex also showed a similar type of binding site residues within a radius of 4 Å of bound ligand such as basic (polar, hydrophobic, positive charged) residues, for example, Arg-4 (Arginine), Lys-158, and Lys-179 (Lysine); aromatic (hydrophobic) residues, for example, Phe-161, Phe-438, and Phe-442 (Phenylalanine); acidic (polar, negative charged) residues, for example, Glu-234, Glu-278 (Glutamic acid), Asp-292, and Asp-439 (Aspartic acid); polar amide type residue, for example, Asn-279 (Asparagine); hydrophobic residue, for example, Met-281 (Methionine); and nucleophilic (polar, hydrophobic) residue, for example, Thr-291 (Threonine), compared to camptothecin; therefore, the docked molecule also showed a strong hydrophobic interaction with Akt kinase, thus leading to more stability (Fig. 1C).
Surflex-Dock scores (total scores) were expressed in −log10 (Kd) units to represent binding affinities.
The docking results for the negative control molecule SP600125 (JNK inhibitor) with the anticancer/survival target protein Akt kinase showed a low binding affinity docking score, indicated by a low total score of 4.6662 without any H-bond (hydrogen bond) formation (Fig. 1D), in comparison to the docking score of anticancer known inhibitors such as pyrrolopyrimidine and camptothecin, which showed a total score of 7.7894 and 5.7543, respectively (Table 2). Thus, the docking procedure of Surflex-dock software (Sybyl-X 1.3) in reproducing the experimental binding affinity seems reliable, and therefore predicted as true positive.
In vitro validation studies
Isolation, purification, and characterization of umbilical cord blood stem cells
In the first instance, isolated stem cells were 60%–85% purified and having more than 85%–90% viability. Cells were showing the expression of surface markers CD34 (77.35%±13.04% cells) and Thy1 (69.77%±12.32% cells) (Fig. 2A, B). In a myelocult culture medium supplemented with the cocktail of specific growth factors and cytokines, the purity of stem cells showing these markers was increased with further passages. The purified population of stem cells could be maintained for more than 3 months up to 10–12 passages. During cultivation, a progressive cell proliferation with cluster formation was observed (Fig. 2C). Upon breaking, individual small rounded cells increased in size and formed large colonies within the time of a week. Some single cells and colonies were found to be adhered on to the substratum of the culture flask. Numerous cells within adhered clusters started to send out tiny processes, and later on, clearly distinct cell types appeared. They tightly adhered to the surface of the culture flask and frequently had numerous small cells on their surface or in close vicinity to the cell (Fig. 2D). The cells grown under deprivation of growth factors and cytokines were decreased in number and could survive.
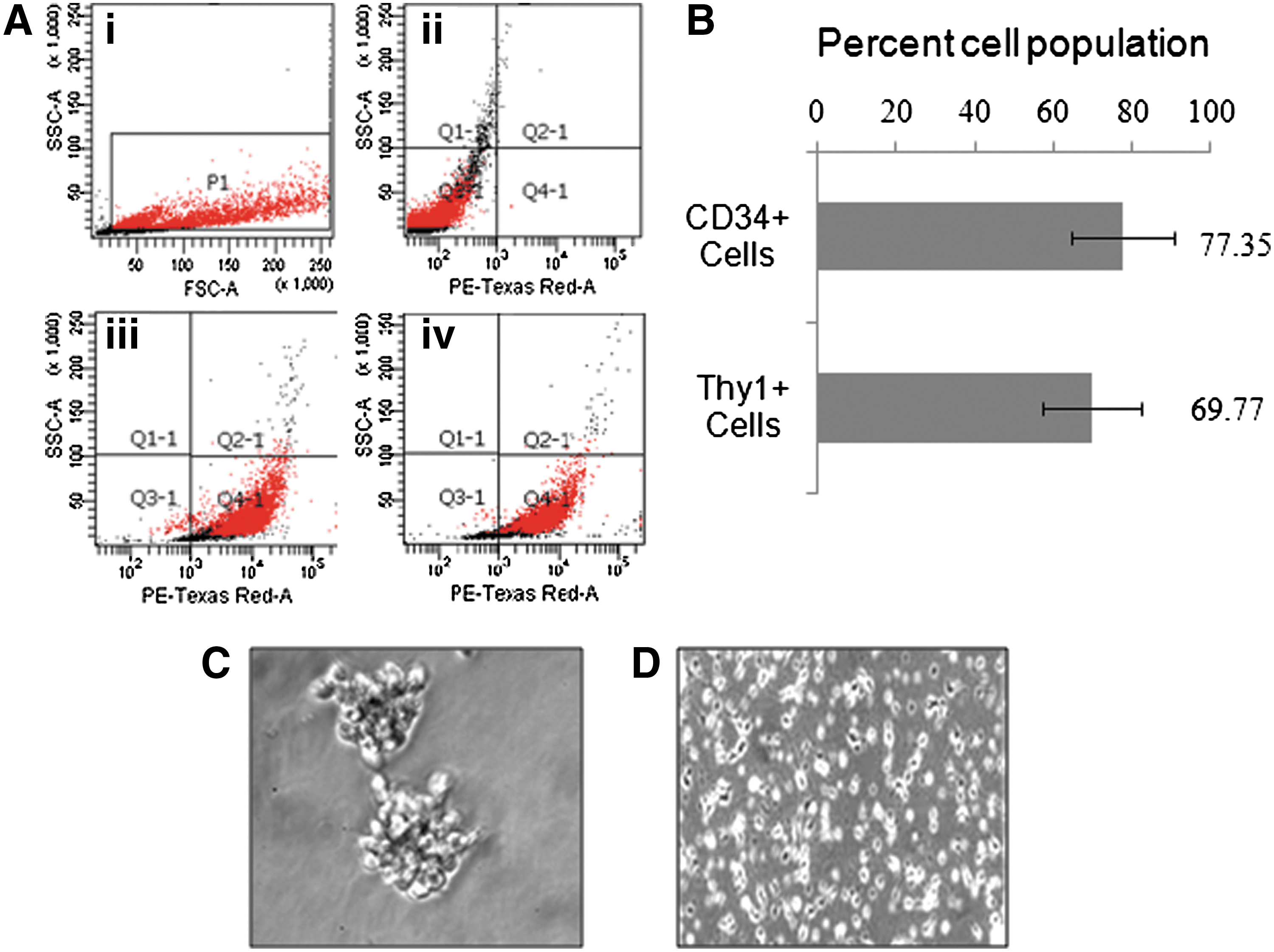
Purification and characterization of human umbilical cord blood stem cells.
Cytotoxicity studies
Prior using in the experiments, biologically safe doses of MCP were identified using tetrazolium bromide salt (MTT) and LDH-release assays. In MTT assay, no significant cytotoxic response was recorded until 24-h exposure, except for MCP 10−3 M concentration. Thereafter, a dose-dependent decrease in the percent cell viability was observed till the end of exposure, that is, 96 h. In general, MCP (10−3 and 10−4 M) exposure for 48 h and onward were found cytotoxic, whereas, rest of the concentrations of MCP used in the study were safe until 48 h (Fig. 3A). Data of LDH-release assay were also showing a similar trend (Fig. 3A, B). Based on these results, further experimentations were carried out using a safe dose of MCP (10−5 M) for 3 h (RNA expression studies) and 6 h (protein expression and apoptosis studies).

Identification of noncytotoxic doses of MCP, an organophosphate pesticide. Cells were exposed to MCP (10−3 to 10−8 M) for a period of 12–96 h. *P<0.05; **P<0.001.
Oxidative stress
ROS generation
MCP exposure for 6 h shows statistically significant (P<0.001) generation of ROS in cells, that is, 227.6%±6.3% of control. Similarly, the positive control camptothecin has also induced significant ROS generation 209.3%±4.6% of control. Cells receiving the co-exposure of MCP and NAC (known antioxidant) show the significant (P<0.001) restoration (126.3±5.21 of control) in ROS levels, when compared with MCP-alone exposure (Fig. 4A).

MCP-induced oxidative stress analysis and ameliorative responses of N-acetlylcysteine (NAC) in cultured human umbilical cord blood stem cells.
Intracellular GSH levels
A significant (P<0.05) reduction (23.2±0.6 mM) in the levels of intracellular GSH was observed in MCP- (10−5 M for 6 h) exposed cells when compared with the unexposed control cells (35.5±1.2 mM). NAC was found to counteract the GSH-diminishing activity of MCP, as cells receiving the coexposure of MCP and NAC show the significant (P<0.05) restoration (33.1±0.8) in GSH levels. As anticipated, a highly significant (P<0.001) reduction (18.5±0.8 mM) in the levels of GSH was recorded in cells exposed to the positive control camptothecin (Fig. 4B).
Induction of apoptosis
Our results from TUNEL assays indicate that MCP (10−5 M) exposure for 6 h induced significant apoptosis (19.14%±2.35%) in an exposed population of cells compared to 1.78%±0.93% control. Cells exposed to camptothecin (3 μg/mL) for 6 h have also shown the significant apoptosis (20.15%±2.34%) (Fig. 5A). A significant decrease (0.4-fold±0.12-fold of control) in the mitochondrial membrane potential was also observed using Mitolight™ dye in the cells exposed to identical conditions as exposed for the TUNEL assay (Fig. 5B).

Detection of apoptosis in cultured human umbilical cord blood stem cells exposed to MCP (10−5 M) for 6 h.
Transcriptional studies
The results of MCP- (10−5 M for 3 h) induced alterations in the expression of markers of stemness, cell signaling, MAP kinases, apoptosis, oxidative stress, and metabolism are summarized in Fig. 6A–E. MCP exposure induces significant downregulation [CD34 (0.068E-06-fold of control) and SHH (8.44E-04-fold of control)] and upregulation [KLF4 (3.75±0.43-fold of control) and POU5F1 (5.93±1.3-fold of control)] of stemness genes studied (Fig. 6A). All the examined signaling genes studied were also found to be upregulated [mTOR (2.46±0.21-fold), NFκB (1.81±0.17-fold), NOS (6.05±0.53-fold), STAT3 (1.87±0.13-fold), and STAT4 (3.32±0.14) of unexposed control], except in case of WNT1, where the expression was downregulated significantly (8.17±04-fold of control) (Fig. 6B). MCP exposure also induced upregulation of MAP kinases significantly, that is, MAPK10 (27.07±2.1-fold), c-JUN (4.95±0.32-fold), MAPK1 (4.14±0.27-fold), MAK3K5 (3.27±0.22-fold), MAPK9 (2.34±0.16-fold), and CREB1 (2.294.95±0.18-fold) of control (Fig. 6C). MCP-induced changes were also found to be associated with xenobiotic metabolizing cytochrome P450s and genes involved in oxidative stress as evidenced by significant (P<0.05) alterations in the expression of these genes, that is, upregulation of CYP2B1 (5.46±0.67-fold), CYP2B6 (4.33±0.58-fold), CYP1A1 (2.05±0.37-fold), GPX3 (6.40±0.72-fold), GSR (2.16±0.17), SOD1 (2.62±0.34) and downregulation of CAT (0.84±0.07-fold) (Fig. 6D). Similar to other genes, all the apoptosis marker genes were significantly upregulated (P<0.05), that is, Bad (10.45±1.10-fold), caspase-9 (4.99±0.62-fold), CAPN1/2 (2.10±0.16/4.03±0.32), Bcl2 (2.10±0.17-fold), Bax (1.93±0.11-fold), and CDKN1A (3.27±0.27-fold) of control, except caspase-3 (Fig. 6E).

Assessment of transcriptional changes in the selected marker genes of stemness, proliferation, signaling, apoptosis, oxidative stress, and metabolism in cultured human umbilical cord blood stem cells exposed to MCP (10−5 M) for 3 h. Real-time PCR was done using TaqMan Low Density Array designed in a 384-well plate format (Applied Biosystems). Analysis was done in ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). *P<0.05; **P<0.001.
Translational changes in marker proteins in differentiating cells exposed to MCP
The western blot analysis results of MCP- (10−5 M for 6 h) induced changes in the expression of markers involved in PI3K/Akt/GSK3β/β-catenin signaling and apoptosis in hUCBSCs are summarized in Fig. 7A–E. In general, MCP significantly downregulates the expression of pAkt (Thr308/Ser473)/pGSK3β (ser9)/β-catenin in hUCBSCs. The magnitude of alterations was also similar for camptothecin, a positive control used in the study. Although both MCP and camptothecin induce alterations in the expression of PI3k and nonphosphorylated GSK3α/β, the changes were statistically nonsignificant. However, a significant downregulation in the expression of survival protein β-catenin was observed in cells exposed to MCP/camptothecin. Stem cells responded significantly to MCP exposure in terms of apoptosis markers viz., pJNK1/2, c-JUN, P53, Bax, and caspase 9/3. Pretreatment of SP600125, an inhibitor of JNK1/2, was found effective to control the altered expression of proapoptotic marker proteins viz., JNK1/2, c-JUN, p53, p21, BAX, and CASP 9/3, in hUCBSCs exposed to MCP.
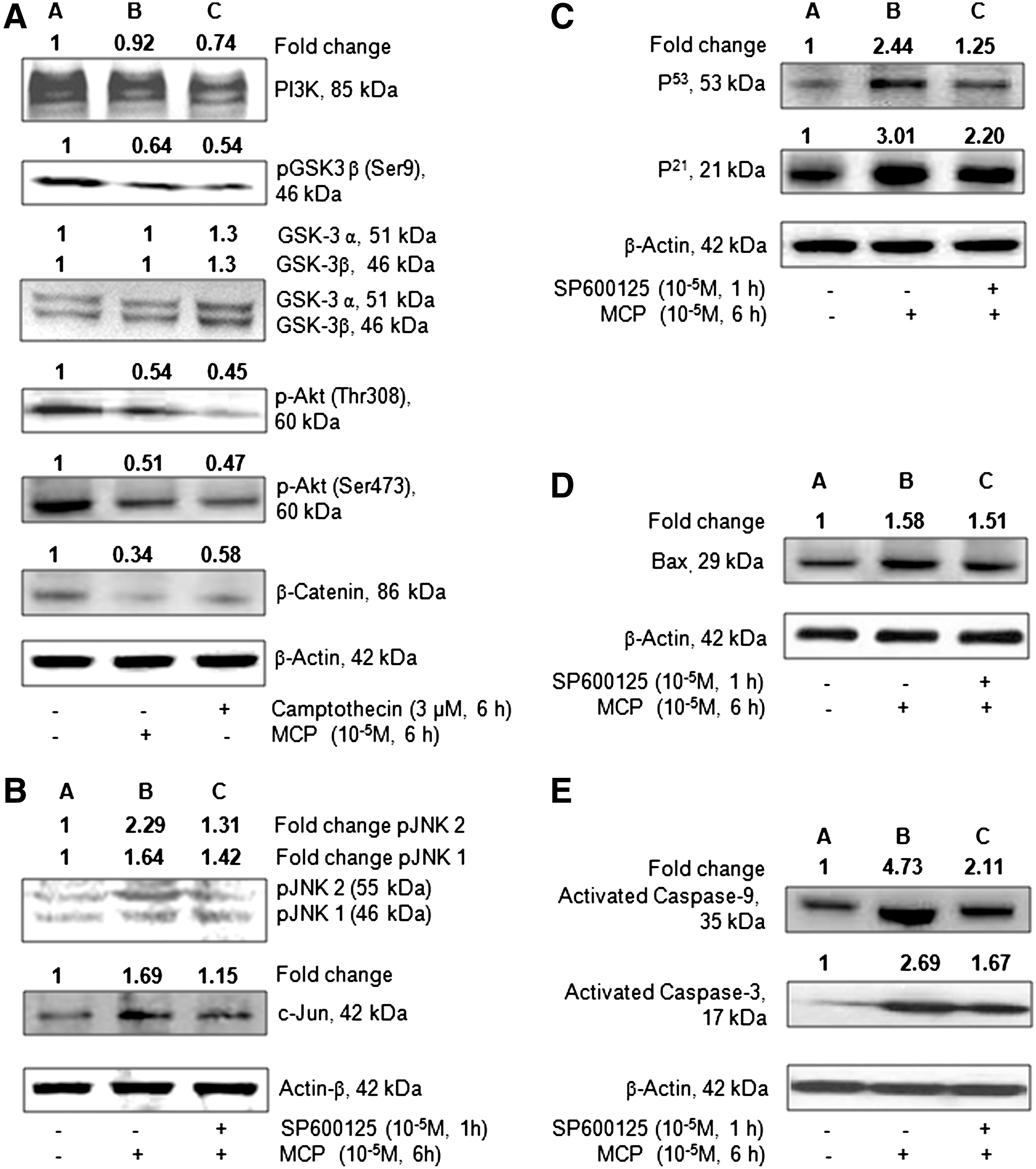
Western blot analysis to study the translational changes in the selected marker markers involved in phosphoinositide 3-kinase (PI3K)/Akt/GSK3β/β-catenin signaling and apoptosis in cultured human umbilical cord blood stem cells exposed to MCP (10−5 M) for 6 h.
Activity of caspase-9 and 3
The results of MCP-induced alterations in the activity of caspase-9 and 3 and ameliorative responses of NAC are summarized in Fig. 8A and B, respectively. MCP exposure (10−5 M for 6 h) significantly (P<0.001) induces the activity of caspase-9 (3.43±0.4-fold of control). NAC was found effective to ameliorate the activity responses significantly (P<0.05), as co-exposure of MCP+NAC brought down the caspase-9 activity near to the unexposed control, that is, 1.80±0.05-fold of control (Fig. 8A). The trend for the activity of caspase-3 was similar to that of caspase-9; however, the magnitude of induction was comparatively lower, that is, 2.63±0.11-fold of control. The ameliorative response of NAC to the activity of caspase-3 was also more or less similar (1.53±0.08-fold of control) to that of caspase-9 (Fig. 8B).

MCP-induced alterations and ameliorative responses of NAC on the activity of caspase-9
Discussion
In our earlier studies, we have shown that MCP induces mitochondrial-mediated apoptosis and association of xenobiotic-metabolizing P450s in rat pheochromocytoma (PC12) cells. We have also demonstrated the altered expression and possible association of selected xenobiotic-metabolizing cytochrome P450s with MCP-induced damages in the same cells [22,23]. In the present investigations, we used the primary cultures of human hematopoietic stem cells derived from cord blood to investigate the responsiveness of these cells against MCP exposure. Thus, with the background knowledge generated on apoptotic responses of MCP in PC12 cells, the present study is focused to explore the role of protein kinase-B in the regulation of apoptosis in human hematopoietic stem cells. Hematopoietic stem cells used in the study were isolated from human umbilical cord blood, as it is a noninvasive, rich source of CD34+CD38− stem cells [28 –30]. In general, a range from 0.6% to 1.35% (N=52) of CD34+ stem cells could be isolated with the viability of 90%–95%. A number of reasons might be responsible for the loss of 5%–10% viability during sample processing such as blood storage time, magnetic effects during processing, addition of an antiblood coagulant, etc. Also, the other factors like gestational age, mode of delivery, and positioning of neonate after delivery have also found to be associated with variation in the number of hematopoietic stem cells [31,32]. Using permutation of combination of various cytokines/growth factors, viz., TPO, FLT-3 ligand, SCF, and FGF-basic, cells could be cultured in a proliferative stage up to 4-month time. This suggests the extensive self-renewal and undifferentiated proliferation capacity of cultured hematopoietic stem cells for such a prolonged period of time.
In the present investigations, human hematopoietic stem cells responded against MCP exposure similar to that of PC12 cells [22,23], Caco-2 cells [33], and peripheral blood lymphocytes [34,35]. Since, the MCP-induced alterations in the expression of markers of oxidative stress, apoptosis, and xenobiotic-metabolizing cytochrome P450s in hUCBSCs were comparable to our earlier findings with PC12 cells [22,23]. Based on the findings, we hypothesized the involvement of Akt, also known as Pkb, in the regulation of signaling of the whole cascade of MCP-induced apoptosis and cell injury in hUCBSCs. The hypothesis was based on the established role of Akt in the suppression of apoptosis by inhibiting the proteins involved in cell death, including ASK1/caspase-9/Bad/P53 and P21 and promoting the cell survival pathway, including IGF-1R/PI3K/PTEN/mTOR [4]. However, such findings related to Akt are largely considered as its anticancerous phenomenon property and looked into cancerous-related studies only [5,12,14 –16]. Perhaps, the role of Akt in the regulation of xenobiotics-induced apoptosis and injury in hUCBSCs is being reported for the first time. To prove the hypothesis, molecular docking studies were carried out to elucidate whether the test compound MCP modulates the anticancer target Pkb/Akt, and to study the possible mechanisms of action. The docking results were further validated using the expression (mRNA and protein) studies for markers of apoptosis, cell injury, selected CYPs, and Akt signaling pathways.
The binding affinity obtained in the docking study allowed the activity of the MCP compared to that of the standard anticancer drug camptothecin. MCP showed high binding affinity against the Akt kinase target protein. When we compared how the binding pocket amino acid residues of target protein interacted with the compound, we found that the compound MCP has molecular interaction with conserved hydrophobic amino acid residues, thus leading to more stability and potency. The docking results for MCP showed that the compound docked onto the anticancer target Akt kinase with a high binding affinity docking score and also formed H-bond of length 2.3 Å to the hydrophobic nucleophilic (small, polar) residue Threonine, that is, Thr-291, similar to that of camptothecin. To better understand the binding nature of MCP to Akt, the MCP-Akt-docked structure was compared with the presolved X-ray co-crystal structure of the Akt–imidazopiperidine analog (8b) and anilinotriazole analog 5d [5]. Most notably, MCP interacts with Glu-234 and Asp-292 and forms a hydrogen bond between N-H and Thr-291 of Akt in the vicinity of where the ribose of ATP would normally occupy. Similar types of findings have been observed in the imidazopiperidine analog 8b, in which the protonated imidazole nitrogens involve in 2 hydrogen bonds between Glu-234 and Asp-292 [5]. In addition, Phe-161 was also found to interact with MCP as in the case of the imidazopiperidine analog (8b), which pi-stacked with the anilinotriazole analog 5d, resulting in a significant conformational change in this portion of the protein [5]. Gurbani et al. [36] has also reported a similar type of mechanism of inhibition of the ATPase domain of Human Topoisomerase IIα (TopoIIα) by different quinone analogs. They showed the interaction with Ser-148, Ser-149, Asn-150, and Asn-91 residues of the ATPase domain of Hu-TopoIIα with quinone analogs viz., 1, 4-benzoquinone, 1, 2-naphthoquinone, 1, 4-naphthoquinone, and 9, 10-phenanthroquinone [36]. The combined results of molecular docking and western blot analysis suggest that MCP significantly inhibits the activity of Akt kinase by hampering the phosphorylation at Thr308/Ser 473, which may be due to interactions at the ATP vicinity and works as a selective, ATP-competitive inhibitor of Akt [5,37].
Our wet laboratory data confirm the in silico finding that MCP significantly reduces the phosphorylation of Akt at both the domains at position Thr308 and Ser473 position. Phosphorylated/activated Akt inactivates the GSK3β by phosphorylating it at Ser9 [4]. In our studies, the reduced levels of phosphorylated GSK3-β (Ser9) and unaffected levels of nonphosphorylated GSK3β suggest the degradation of beta-catenin through an ubiquitin-dependent proteasome pathway, which leads to apoptotic cell death [38]. The activation of GSK3β is also known to inhibit the Wnt/frizzled/disheveled (DSH) pathway to promote cell survival and growth [38]. Thus, the reduced levels of pAkt, Wnt, and β-catenin observed in present study may also indicate to cause the impaired Wnt/Akt/β catenin signaling and subsequent induction of apoptosis in hUCBSCs exposed to MCP. Unaltered levels of PTEN and PI3K confirm the involvement of other routes of pAkt inhibition in MCP-induced cell death signals in hUCBSCs. Phosphorylation of Akt (Ser473) at the AGC-kinase C-terminal regulatory domain via mTOR plays an important role in triggering the signals of cell survival/growth [4]. Our in silico data show that MCP binds with the Akt AGC-kinase C-terminal regulatory domain, so causes the hindrance in phosphorylation of Akt (Ser473). This might be the reason that even after significant upregulation in the levels of mTOR/FRAP1, there was no sufficient phosphorylation in Akt (Ser473) in MCP-exposed hUCBSCs.
The protective role of pAkt is reported primarily by NFκB survival signaling, thereby inhibiting ASK1, which eventually blocks the phosphorylation of downstream JNK1/2 [4]. Such elevated levels of pJNK1/2 have been reported to induce apoptosis via the activation of downstream molecules viz., P53, P21, Bax, and caspases, in a variety of cells [22,23,39]. In agreement to the earlier findings, present investigations also confirm the upregulation of NFκB, ASK1, and p-JNK1/2 expression in MCP-exposed hUCBSCs. Our study related to a JNK inhibitor (SP600125) also indicates the involvement of ASK1 and p-JNK1/2 pathways in causation of apoptosis mediated through P53, P21, Bax, and caspases 9/3. Our findings are indicating that MCP-induced apoptosis also involve the pathways of oxidative stress and xenobiotic metabolism. Earlier, we have reported the association of oxidative stress and xenobiotic-metabolizing cytochrome P450s in the mitochondrial-mediated apoptosis induced by MCP in PC12 cells [22,23].
Bad, a soluble proapoptotic protein, interacts with the antiapoptotic proteins Bcl-2/Bcl-xL into the mitochondrial membrane, and prevents the antiapoptotic activity of these proteins via preventing the interaction of proapoptotic Bax with these proteins [4,39]. As a result, Bax creates homo-oligomeric channels in the mitochondrial membrane and leads to the release of cytochrome-c from the mitochondria to the cytoplasm. Cytochrome-c interacts with the adapter protein Apaf-1, which leads to caspase cascade-regulated and mitochondrial-mediated cell death [22,23]. Thus, the significant increased expression level of the Bad gene indicates the involvement of mitochondrial proteins in MCP-induced apoptosis and cell injury in hUCBSCs. There are indirect, but important, lines of evidence showing the effect of Akt signaling on cell survival in the maintenance of mitochondrial membrane potential [40,41].
In summary, Akt plays a significant role in the regulation of MCP-induced apoptosis in hUCBSCs. MCP shows strong binding with kinase and the AGC-kinase C-terminal regulatory domain of Pkb/Akt. This interaction impairs the phosphorylation of Pkb/Akt on the Thr309 and Ser473 positions. MCP-induced downregulation of pAkt in hUCBSCs was found to be associated with the impaired expression of the Wnt/GSKβ/β-catenin pathways. At the same time, upregulated expression of ASK1/pJNK1/2 and Bad could be associated with upregulated expression of P53, P21, Bax, and activated caspase-9 and 3 (Fig. 9). Our data suggest that the Akt protein plays an important role in MCP-induced apoptosis in hUCBSCs. We also identified that MCP induces the cellular responses through strong binding and inhibition of kinase and the AGC-kinase C-terminal regulatory domain of Akt protein.
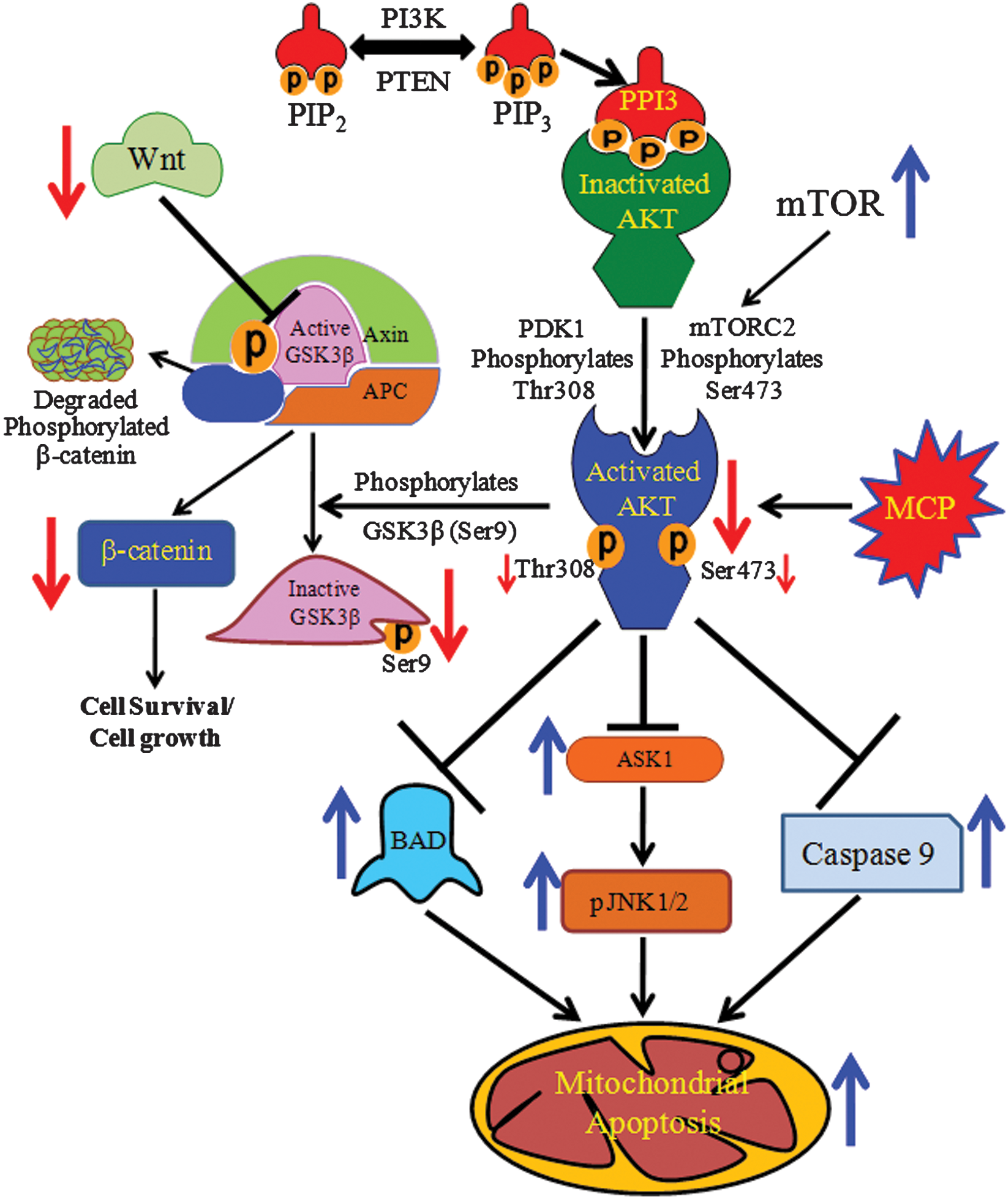
Schematic flow chart depicting the mechanisms involved in Akt-mediated regulation in MCP-induced apoptosis in cultured human umbilical cord blood stem cells. Color images available online at
Footnotes
Acknowledgments
Authors are grateful to the director, IITR, Lucknow, India, for his keen interest in the study. The work was supported by the Council of Scientific & Industrial Research (CSIR), New Delhi, India, (Grant Supra Institutional Project SIP-08) and the Department of Biotechnology (DBT), New Delhi, India (grant no. 102/IFD/SAN/PR-1524/2010-201). The University Grant Commission (UGC), New Delhi, India, is acknowledged for providing the fellowship to Dr. M.P. Kashyap. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
Authors of this article have no conflicts of interest among them or anybody else regarding the scientific contents, financial matters, and otherwise.