
Abstract
Hypoxia (low oxygen) and Notch signaling are 2 important regulators of vascular development, but how they interact in controlling the choice between arterial and venous fates for endothelial cells during vasculogenesis is less well understood. In this report, we show that hypoxia and Notch signaling intersect in promotion of arterial differentiation. Hypoxia upregulated expression of the Notch ligand Dll4 and increased Notch signaling in a process requiring the vasoactive hormone adrenomedullin. Notch signaling also upregulated Dll4 expression, leading to a positive feedback loop sustaining Dll4 expression and Notch signaling. In addition, hypoxia-mediated upregulation of the arterial marker genes Depp, connexin40 (Gja5), Cxcr4, and Hey1 required Notch signaling. In conclusion, the data reveal an intricate interaction between hypoxia and Notch signaling in the control of endothelial cell differentiation, including a hypoxia/adrenomedullin/Dll4 axis that initiates Notch signaling and a requirement for Notch signaling to effectuate hypoxia-mediated induction of the arterial differentiation program.
Introduction
D
In addition to Notch signaling, the cellular response to hypoxia is an important regulator of vascular development. At the onset of vascularization, the embryo relies solely on diffusion of oxygen, which means that the early phases of vascularization take place in a hypoxic environment. Cells respond to hypoxia through a molecular machinery, in which the HIF transcription factors (HIF-1α or HIF-2α) become stabilized under low oxygen conditions. HIF, together with its partner HIF-1β (ARNT), activates downstream genes, including VEGF [10]. The important role for hypoxia in the vasculature is underscored by the effects of ablating key factors in the cellular hypoxic response. Arnt−/− mice die within 10.5 days of embryonic development (E10.5) and are characterized by defective vascular remodeling of the yolk sac, brachial arteries, and placenta [11]. A similar vascular phenotype is observed in Hif-1α −/− embryos that die at E11 with regressed vascular endothelium [12,13].
Notch signaling is initiated by activation of transmembrane Notch receptors by ligands on juxtaposed cells. Ligand activation leads to proteolytic cleavages of the Notch receptor, and the final cleavage, which liberates the Notch intracellular domain (Notch ICD), is executed by the γ-secretase complex, and can thus be blocked by γ-secretase inhibitors [14]. Notch ICD translocates to the nucleus, where it interacts with the DNA-binding protein CSL to control expression of Notch downstream genes, including the genes encoding the Hes and Hey transcription factors. Mouse embryos deficient for CSL or the Jagged1 ligand die around embryonic day 9 (E9) with vascular collapse, and removal of only one copy of the Dll4 ligand is sufficient to induce severe vascular deformations and death around the same time [15 –18]. Notch signaling and the cellular hypoxic response intersect at several levels in the signaling cascades. This includes the interaction between HIF and Notch ICD, and the enzymatic modification of both HIF and Notch ICD by FIH [19 –21]. There are also several indications that the 2 signaling pathways may interact during vascular development. A role for hypoxia in arterial differentiation has been proposed and linked to an intersection with the Notch signaling pathway; that is, a hypoxia-induced upregulation of the Notch downstream genes Hey1 and Hey2, coupled with a repression of COUP-TFII, was observed [22]. In another study, a link between HIF-1α and Notch via VEGF was established in choroidal neovascularization [23].
In light of the important roles of both Notch and the cellular hypoxic response during vascular differentiation and because the 2 signaling pathways interact, we have studied their roles and possible intersection during the choice between the arterial and venous fates in endothelial cells. In this report, we identify a hypoxia/adrenomedullin/Dll4 axis, in which hypoxia induces expression of Dll4 and elevates Notch signaling in an adrenomedullin-dependent manner. We also show that active Notch signaling is required for a significant portion of all hypoxia-induced genes to be upregulated by hypoxia. The set of hypoxia-induced genes that depend on Notch signaling included a set of arterial marker genes: Depp, Dll4, Connexin40 (Gja5), Cxcr4, and Hey1. We propose that the hypoxia/adrenomedullin/Dll4 axis initiates Notch signaling, which is then required for the hypoxia-induced activation of a larger arterial program. In conclusion, these data provide novel insights into the process of arterial differentiation and identifies an intricate interaction between Notch and the cellular hypoxic response in this process.
Materials and Methods
Embryonic stem cell culture and differentiation
Wild-type mouse CCE embryonic stem (ES) cells (a gift from Dr M. J. Evans, Welcome/CRC Institute, Cambridge, UK), Vegf−/−
[24], and Csl−/−
ES cells were a kind gift from Dr. Tasuku Honjo [18]. Notch1ΔETetOn ES cells have been previously described [25,26]. ES cells were cultured on gelatin-coated plates in the Glascow MEM (Sigma) supplemented with 15% fetal bovine serum (FBS) (Hyclone), 1,000 IU/mL leukemia inhibitory factor, 0.1 mM 2-mercaptoethanol, and 0.1 mM nonessential amino acids (Gibco). The medium was changed daily and cells passaged every second day. For differentiation, 104 ES cells/mL were aggregated as embryoid bodies (EBs) in 20 μL drops and differentiated 4 days in the IMDM, 15% FBS (Gibco), 0.15 mM monothioglycerol, and 50 μg/mL ascorbic acid (Sigma). On day 4, EBs were harvested and dissociated, stained for Flk1, and sorted to purity using flow cytometry. About 5×104 Flk1+ cells/mL were plated on collagen type IV (BD Bioscience)-coated plates and differentiated for 4 additional days with 50 ng/mL hrVEGF (R&D Systems). The endothelial CD31+ cells were isolated by flow cytometry for further analysis. Adrenomedullin was used at 1 μM (H-2932; BachemAG), and to block Notch signaling, the gamma-secretase inhibitor (GSI
Flow cytometry
Day 4 EBs were dissociated to single cells with collagenase (Sigma-Aldrich) treatment for 20 min, followed by 2-min trypsinization and passed 3 times through a 20G needle. Day 8 monolayer cultures were dissociated by brief trypsinization followed by trypsin inactivation with ice cold PBS with 4% fetal calf serum (FCS). Staining was performed as previously described [5] on ice in PBS with 4% FCS and analyzed using a FACSAria cell sorter (Becton Dickinson). Propidium iodide was used to exclude dead cells. Antibodies used include Flk1-PE (1:200) and CD31-APC (1:200), (PharMingen).
Real-time PCR
104 CD31+ cells from each biological sample were sorted by flow cytometry directly into a 350 μL RLT-lysis buffer and total RNA was isolated using the RNeasy Micro Kit (Qiagen). cDNA was synthesized using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer's instructions. The real-time PCR was performed using the SYBR-GREEN PCR Master Mix and analyzed using a 7300 Real-Time PCR System (Applied Biosystems). Values were normalized to Actb housekeeping transcript levels. Primer sequences are available upon request.
Transfections and luciferase reporter assay
Flk1+ cells were allowed to attach over 24 h before transfection in 6-well plates using the FuGENE HD Transfection Reagent (Roche). For the reporter assay, a beta-gal construct was cotransfected as an internal control of transfection efficiency. After an additional 3 days of differentiation, the CD31+ cells were sorted and lysed, and their luciferase and beta-gal activity was measured on a NOVOstar microplate reader (LabVision, Beckman Coulter). HIF-1α-PPA and HIF-2α-PPA were both stabilized against normoxic prolyl hydroxylation and subsequent ubiquitin-mediated proteolytic degradation by point mutations [27].
siRNA
Cultures were transfected with siRNA following Flk1 sorting using DharmaFECT1 (T-2001–02; Thermo Scientific) according to the manufacturer's specifications. Two μL DarmaFECT and 100 μL siRNA (2 μM) were added to each well of a 6-well plate in a total volume of 2 mL. To avoid cell toxicity, the transfection media was replaced after 24 h. We used ON-TARGETplus SMARTpool oligos against adrenomedullin (L-042880-01; Thermo Scientific) and scramble oligos as control.
Immunoblotting
EBs were plated in tissue culture dishes at day 4 in a differentiation medium with 50 ng/mL VEGF. At day 10, the cultures were stimulated for 60 min with either VEGF-A165 at 50 ng/mL or adrenomedullin at 1 μM (H-2932; BachemAG). The cells were lysed in 20 mM Tris HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 1% NP40, 2 mM EDTA, 500 μM Na3VO4, 1% aprotinin, 10 μg/mL leupeptin, and 1 mM phenylmethyl sulfonylfluoride. For immunoprecipitation, lysates were incubated with goat anti-mouse VEGFR-2 (R&D) at 4°C followed by incubation with fast-flow protein-G-Sepharose (Amersham Biosciences), end over end, at 4°C. Proteins were released by boiling in a sample buffer [59 mM Tris-HCl (pH 6.8), 1.5% sodium dodecyl sulfate (SDS), 4.35% glycerol, 4% β-mercaptoethanol, 0.0025% bromophenol blue], and separated with SDS-PAGE. Receptor activation was detected with mouse anti-phospho-tyrosine (4G10; Cell Signaling) [28]. Immunoreactivity was visualized by enhanced chemiluminescence. Quantification of immunoreactive bands was made in ImageJ.
Statistical analysis
Each experiment consists of a minimum of 3 biological replicates. The statistical significance was determined using the Student's t-test. Values are significant at ***P<0.001, **P<0.01, and *P<0.05, as indicated in the figures. Error bars represent SEM.
Transcriptome analysis
The microarrays were performed in 3 biological replicates for each treatment. >50,000 CD31+ cells were sorted from each sample and total RNA extracted using the RNeasy Micro kit (Qiagen). About 300 ng of total RNA from each sample was reverse transcribed into cDNA and in vitro transcribed into biotin-labeled cRNA using the Illumina TotalPrep RNA Amplification kit (Ambion). About 750 ng of cRNA from each sample was hybridized to MouseRef-8 v2.0 Expression BeadChip microarrays (Illumina) and scanned on the BeadArray Reader (Illumina) at scan factor 1. Raw intensity values were subjected to a background subtraction performed on the BeadStudio Data Analysis software (Illumina) and normalized using the cross-correlation method [29]. Differentially expressed transcripts were identified based on the average of Log2-fold change in expression values compared to the normoxia control for each biological replicate experiment, with a minimum cutoff at 1.5-fold change. The microarray data were deposited into NCBI GEO with the accession number GSE35894 (
Results
A significant portion of the hypoxia-induced transcriptome in CD31+ endothelial cells requires functional Notch signaling to be upregulated by hypoxia
To study how hypoxia and Notch signaling affect gene expression in endothelial cells, we took advantage of the fact that ES cells can be in vitro differentiated to vascular progenitors (angioblasts), which can be further matured toward arterial or venous fates [5,30]. Mouse ES cells were cultured as EBs for 4 days to generate Flk1+ vascular progenitors that were sorted to purity using flow cytometry. The Flk1+ cells were plated on collagen type IV-coated plates and differentiated with 50 ng/mL of human recombinant VEGF for 4 days to generate CD31+ endothelial cells that were sorted to purity and subjected to transcriptome analysis (Fig. 1A). To investigate the interaction between hypoxia and Notch signaling, the Flk1+ cells were cultured in normoxia or hypoxia (1.5%–2% O2) for 4 days before the CD31+ sorting, combined with normal or inhibited Notch signaling. Inhibition of Notch signaling was accomplished by treating the cells with the γ-secretase inhibitor (L-685.485), which inhibits the proteolytic processing of the Notch receptor and thus the release of the Notch ICD following ligand activation. The microarray data have been deposited in NCBI GEO with the accession number GSE35894 (
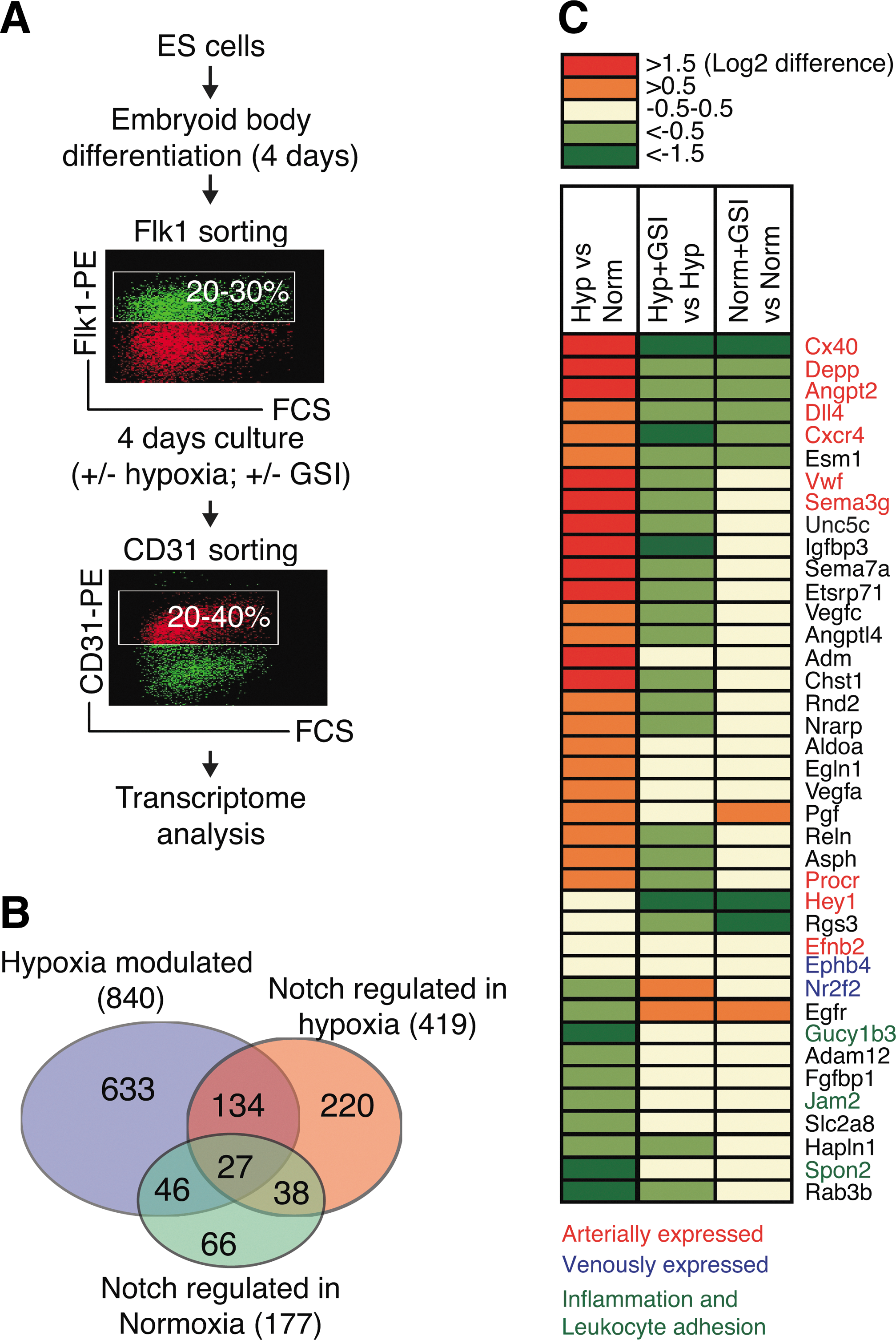
A significant portion of the hypoxia-induced transcriptome in CD31+ endothelial cells requires functional Notch signaling.
GO analysis identified an enriched number of genes associated with metabolic processes in the hypoxia-regulated only group, mitotic processes in the Notch regulated in normoxia only group, and vascular processes such as patterning of blood vessels in the dataset of 161 genes regulated by hypoxia in a Notch-dependent manner (Supplementary Table S1; Supplementary Data are available online at
Hypoxia-promoted arterial differentiation does not require endogenous VEGF
Hypoxia induces VEGF expression during angiogenesis, and VEGF acts upstream of Notch signaling during the early stages of vascular differentiation [9]. We have previously shown that not only is exogenous VEGF critical for endothelial differentiation, but also titrated VEGF dosage controls the arterial–venous differentiation in this system [5]. In these studies, we established that 2–10 ng/mL exogenous VEGF drives the venous endothelial differentiation, whereas 50 ng/mL promotes the arterial differentiation. Since the in vitro differentiation in the current study was already performed with arterial-promoting levels of VEGF (50 ng/mL), it was unlikely that hypoxia-induced VEGF could further drive the observed arterial response. To functionally eliminate effects of endogenous hypoxia-induced VEGF, we examined the endothelial differentiation of Vegf− /− ES cells [24]. These cells were differentiated with 50 ng/mL exogenous VEGF in normoxic or hypoxic conditions. Expression of the Dll4, Notch4, Connexin 40, and EphrinB2 genes was induced by hypoxia to a similar extent in Vegf− /− and wild-type ES cells, whereas Hey1 was activated to a more moderate extent in the Vegf− /− ES cells. Expression of EphB4 was not altered and COUP-TFII was downregulated in the CD31+ Vegf− /− ES cells (Fig. 2), similar to what was observed in the VEGF wild-type cells. This indicates that the hypoxia-driven arterial response is not mediated by hypoxic induction of endogenous VEGF, but through an alternative mechanism.

Hypoxia-induced arterial differentiation does not require endogenous VEGF. Transcriptional response to hypoxic culture during endothelial differentiation of wild-type (WT) and Vegf −/− ES cells. Graphs show real-time PCR quantification of mRNA expression. Values are expressed as transcript levels relative to that of the Actb housekeeping gene. Significant values from t-test: ***P<0.001, **P<0.01, and *P<0.05.
Hypoxia-induced expression of arterial genes, with the exception of Dll4, requires functional Notch signaling
We next addressed in more detail whether activation of the hypoxia-induced genes from the transcriptome analysis required functional Notch signaling and whether hypoxia activated the Notch signaling pathway. To first assess Notch activation, we transfected a Notch reporter construct, 12XCSL-luc, which contains multimerized binding sites for the DNA-binding protein CSL [41], into the Flk1+ cells 24 h after plating and analyzed the reporter activity 3 days later, in the CD31+ cells, after exposure to normoxic or hypoxic conditions. Hypoxic culture conditions robustly increased the reporter signal, as compared to normoxia (Fig. 3A).
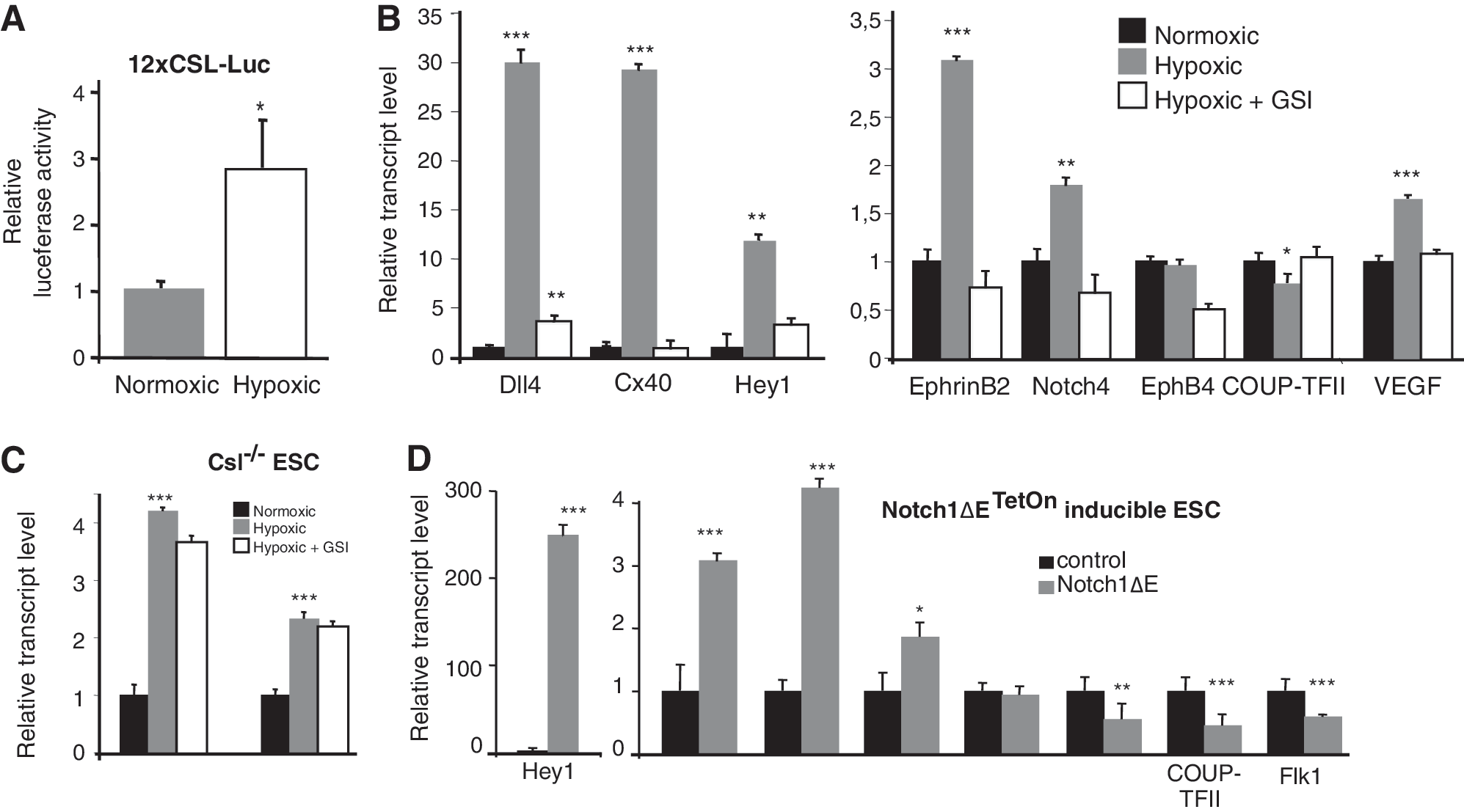
Hypoxia-induced expression of arterial genes, with the exception of Dll4, requires functional Notch signaling.
The hypoxia-induced activation of the arterial genes Notch4, Hey1, Connexin40, and EphrinB2 was blocked by GSI (Fig. 3B). However, Dll4, a critical factor for angiogenesis [15,16], posed an exception, as hypoxia-induced Dll4 expression was not completely blocked by GSI. To test in a more stringent manner whether hypoxia could indeed upregulate Dll4 expression in the absence of Notch signaling, we analyzed the effect of hypoxia in Csl−/− ES cells (Fig. 3C), which are unable to transduce canonical Notch signaling [18]. Both Flk1+ vascular progenitors and CD31+ endothelial cells were generated in similar numbers to wild-type cells (data not shown). Dll4 expression was upregulated by hypoxia in the Csl−/− ES cells, as expected (Fig. 3C). Expression of the venous marker COUP-TFII was increased in the Csl−/− ES cells, as compared to wild-type cells (Fig. 3C), which may suggest that CSL represses expression of COUP-TFII, in keeping with a previous report [42]. Together, these data indicate that hypoxia activates the immediate Notch signaling response and that hypoxia-induced expression of arterial genes, with the exception of Dll4, requires functional canonical Notch signaling.
Notch signaling can substitute for hypoxia in promotion of arterial gene expression
While Notch signaling was necessary for the hypoxia-induced upregulation of arterial markers with the exception of Dll4, we asked whether it was also sufficient to induce arterial gene expression under normoxic conditions. To activate Notch signaling in a temporally controlled manner, we used a recently developed ES cell line, Notch1ΔETetOn, in which a ligand-independent membrane-tethered constitutively active form of Notch, Notch1ΔE, is induced by doxycycline via the TetOn system [25,26]. Doxycycline stimulation of Flk1+ Notch1ΔETetOn cells, followed by CD31+ sorting, yielded a robust activation of Hey1 and Hey2 expression, as verification of Notch induction, and an increase in expression of the arterial markers Dll4 and Connexin40, whereas the venous markers EphB4 and COUP-TFII as well as Flk1 were downregulated (Fig. 3D). In conclusion, the data show that specific activation of Notch at the Flk1+ stage was sufficient to mimic the hypoxia-induced upregulation of several arterial genes.
Adrenomedullin is specifically upregulated by HIF and activates Dll4 expression in a Notch-independent manner
To gain further insights into how hypoxia regulates Dll4 expression, we first tested whether HIF transcription factors directly regulated Dll4 expression. To this end, we transfected normoxically stabilized versions of HIF-1α and 2α, HIF-1α-PPA and 2α-PPA, respectively [27], into the Flk1+ ES cells. In agreement with the published data, HIF activated expression not only of Flk1 and VEGF, but also of Hey1 and Hey2 [22]. Dll4, however, was not directly regulated by introduction of HIF-1α-PPA and 2α-PPA (Fig. 4A). This may indicate that some intermediate component is required for activation of Dll4, and as Notch signaling was dispensable for the hypoxia-induced Dll4 upregulation, we began the search for candidate genes involved in the Dll4 induction by examining the list of the 633 genes that were modulated by hypoxia in a Notch-independent manner (Fig. 1B, C). Within the top 20 most highly induced genes, we found Adrenomedullin, which encodes a vasoactive hormone that has recently been implicated in the arterial/venous specification [43]. Mice deficient for adrenomedullin or its putative receptors, the receptor activity-modifying protein 2 (RAMP2), and the calcitonin receptor-like receptor (Crlr), show blood and lymphatic vascular defects [43,44], and in zebrafish, loss of crlr leads to vascular abnormalities and a loss of arterial identity [45]. In the light of these reports, we decided to explore whether hypoxic adrenomedullin induction played a role in Dll4 activation. We found that HIF-1α-PPA, and more potently HIF-2α-PPA, upregulated transcription of Adrenomedullin in the CD31+ endothelial cells (Fig. 4A). Adrenomedullin was also induced by hypoxia and, importantly, this upregulation was not blocked by addition of GSI (Fig. 4B), indicating that the upregulation of adrenomedullin by hypoxia was Notch independent. Notch induction in Notch1ΔETetOn cells also did not alter Adrenomedullin expression (Supplementary Fig. S1).
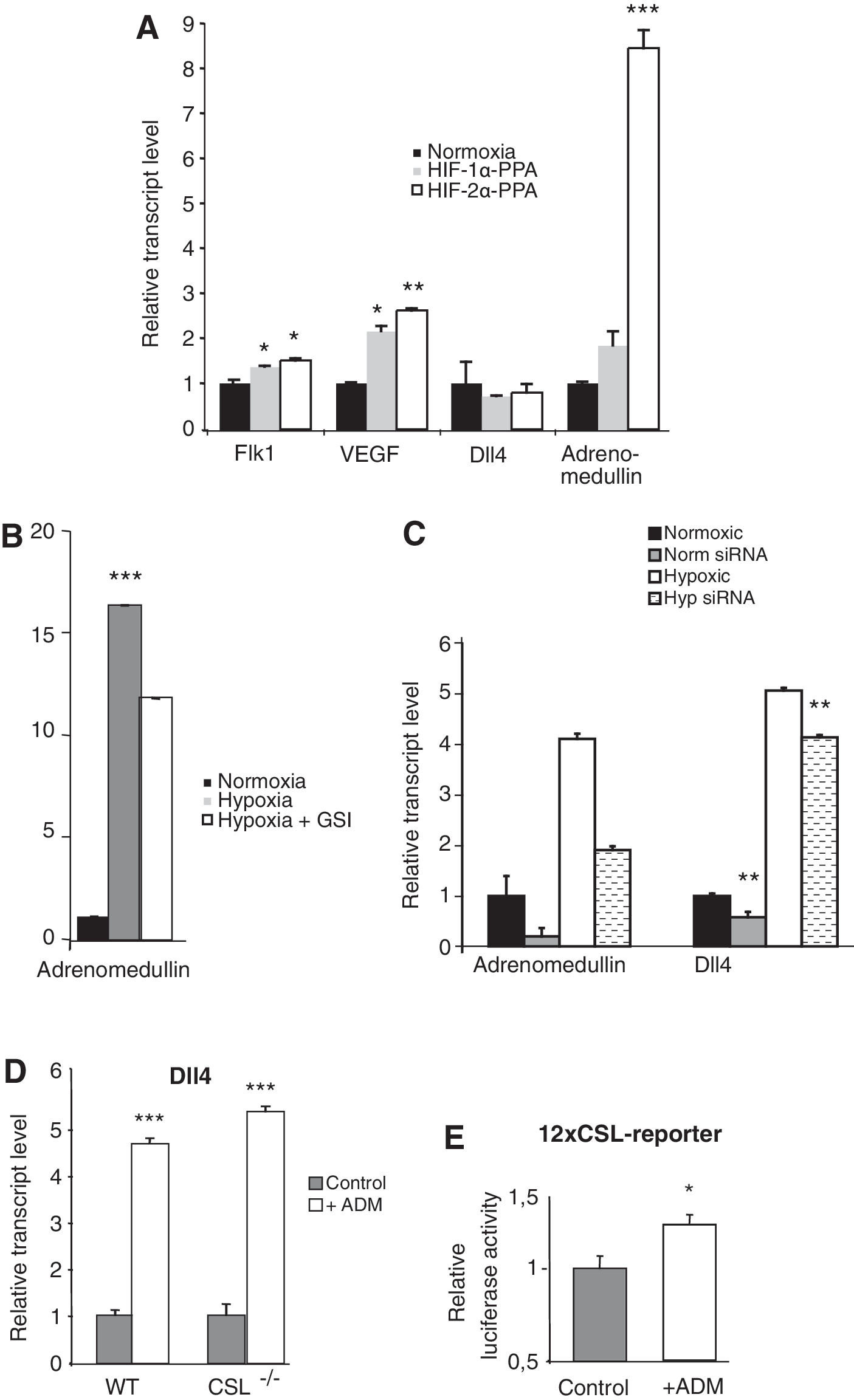
Adrenomedullin is specifically upregulated by HIFs and activates Dll4 expression in a Notch-independent manner.
To address whether adrenomedullin serves as a link between hypoxia and Dll4 regulation, we knocked down Adrenomedullin expression using siRNA (Fig. 4C). Downregulation of Adrenomedullin mRNA levels resulted in reduced Dll4 expression (Fig. 4C). Conversely, addition of recombinant adrenomedullin during vascular differentiation led to a more than 4-fold increase in Dll4 expression (Fig. 4D). Other Notch ligands were not affected by adrenomedullin treatment (Supplementary Fig. S2). A similar increase in Dll4 expression by adrenomedullin was observed in CD31+ cells derived from Csl−/− ES cells (Fig. 4D), providing further credence to the notion that Notch signaling is not required. Addition of adrenomedullin also increased the immediate Notch downstream signaling response (Fig. 4E), suggesting that the elevated Dll4 expression led to activated Notch signaling in the endothelial cells. Adrenomedullin has been suggested to act through transactivation of VEGFR2 in HUVECs [46], but we could not detect any activation of VEGFR2 following adrenomedullin stimulation in our system (Supplementary Fig. S3). In sum, these experiments show that the hypoxia target gene Adrenomedullin is necessary and sufficient to induce Dll4 expression as well as immediate downstream Notch signaling.
Discussion
The cellular hypoxic response and Notch signaling are 2 signaling cascades important for vascular development, and in this report we have addressed how they direct arterial versus venous differentiation in endothelial cells. We unravel a hypoxia/adrenomedullin/Dll4 axis, by which hypoxia leads to upregulation of Dll4 in an adrenomedullin-dependent manner, and show that hypoxia activates an arterial expression signature in a Notch-dependent manner. A model for how the hypoxia/adrenomedullin/Dll4 axis and the Notch-dependent hypoxic induction of arterial gene expression may be linked is presented in Fig. 5.

A model —for the interplay between hypoxia and Notch signaling in arterial differentiation. Hypoxia upregulates expression of adrenomedullin, which is necessary and sufficient for activation of Dll4 expression and Notch signaling. Notch signaling, in turn, positively feeds back by upregulating and sustaining Dll4 expression (gray arrows). Notch signaling is also required for hypoxia-mediated activation of arterial markers (black arrows).
That hypoxia can activate Dll4 expression and Notch signaling via adrenomedullin receives support from a number of observations. First, hypoxia was shown to upregulate adrenomedullin and as this also occurred after GSI treatment or in Csl− /− cells, we conclude that this upregulation did not require Notch signaling. Inhibition of adrenomedullin by siRNA abrogated the hypoxia-induced upregulation of Dll4 expression, and conversely, addition of adrenomedullin led to upregulation of Dll4 expression and the Notch immediate downstream response during normoxia. This demonstrates that adrenomedullin is both necessary and sufficient for induction of Dll4 expression. Dll4 was, however, upregulated not only by hypoxia and adrenomedullin, but also by active Notch signaling (Fig. 5). Thus, specific expression of Notch1ΔE in the Flk1+ vascular progenitors during normoxia was sufficient to upregulate Dll4 expression in the CD31+ endothelial cells. This dual regulation of Dll4 by hypoxia and Notch signaling could serve to (i) first induce Dll4 expression by hypoxia in a Notch-independent manner and (ii) sustain Dll4 expression by the activated Notch signaling, which in turn maintains Notch signaling in neighboring cells, resulting in a Dll4-Notch signaling positive feed forward loop (Fig. 5). Once Notch signaling is established and sustained, it can serve to mediate the hypoxia-induced upregulation of the other arterial markers (Fig. 5; black arrows). This notion is based on our finding that functional Notch signaling was required for hypoxia to induce expression of Depp, Connexin40, Cxcr4, Hey1, and EphrinB2, in contrast to Dll4, which as discussed above was upregulated independently of the Notch signaling status. Specific upregulation of Notch signaling was also sufficient to induce expression of the arterial markers, indicating that Notch signaling was sufficient and necessary for arterial marker expression.
The model in Fig. 5 is also corroborated by several earlier reports. A critical role for hypoxia, adrenomedullin, and Dll4 is in line with phenotypes resulting from targeting of the respective genes [12,15,16,43 –45]. In all 3 cases, gene knockouts lead to embryonic death with a severely compromised vasculature, and interestingly, both HIF-1α −/−, Adrenomedullin−/− and Dll4+/− embryos fail to remodel the vasculature from an initially, apparently, and normally formed vascular plexus. Failure to remodel the vasculature is in keeping with the roles in arterial versus venous differentiation demonstrated here, and the fact that the loss of only one Dll4 allele is sufficient to cause the phenotype suggests that regulation of Dll4 levels is critical. The hypoxia/adrenomedullin/Dll4 axis is corroborated by reports demonstrating that hypoxia regulates adrenomedullin expression [47,48]. Furthermore, adrenomedullin has been shown to control Notch signaling [36], and hypoxic induction of Dll4 has also been observed in tumor angiogenesis [49]. Moreover, hypoxia resulted in upregulation of arterial markers and downregulation of COUP-TFII [22], further supporting that the arterial fate was promoted at the expense of venous differentiation by hypoxia.
In conclusion, the data presented in this report unravel the hypoxia/adrenomedullin/Dll4 axis as an important initiator of Notch signaling during vasculogenesis, and show that Notch signaling is important for sustaining Dll4 expression and is also required for mediating the hypoxia-mediated activation of a larger set of arterial genes. This provides new insights into the complex interplay between hypoxia and Notch signaling and on the function of these 2 signaling mechanisms in the early steps of the formation of a functional vascular system.
Footnotes
Acknowledgments
We thank Dr. Henry Yang for the use of the cross-correlation method for microarray data normalization. This study was supported by grants from the Swedish Medical Research Council, the Swedish Cancer Society, Knut och Alice Wallenbergs Stiftelse, and the EC projects EuroSystem and NotchIT (UL).
Author Disclosure Statement
The authors declare no conflict of interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.