
Abstract
Human mid-trimester amniotic fluid stem cells (AFSC) have promising applications in regenerative medicine, being broadly multipotent with an intermediate phenotype between embryonic (ES) and mesenchymal stem cells (MSC). Despite this propluripotent phenotype, AFSC are usually cultured in adherence in a serum-based expansion medium, and how expansion in conditions sustaining pluripotency might affect their phenotype remains unknown. We recently showed that early AFSC from first trimester amniotic fluid, which endogenously express Sox2 and Klf4, can be reprogrammed to pluripotency without viral vectors using the histone deacetylase inhibitor valproic acid (VPA). Here, we show that mid-trimester AFSC cultured under MSC conditions contained a subset of cells endogenously expressing telomerase, CD24, OCT4, C-MYC, and SSEA4, but low/null levels of SOX2, NANOG, KLF4, SSEA3, TRA-1–60, and TRA-1–81, with cells unable to form embryoid bodies (EBs) or teratomas. In contrast, AFSC cultured under human ESC conditions were smaller in size, grew faster, formed colonies, upregulated OCT4 and C-MYC, and expressed KLF4 and SOX2, but not NANOG, SSEA3, TRA-1–60, and TRA-1–81. Supplementation with VPA for 5 days further upregulated OCT4, KLF4, and SOX2, and induced expression of NANOG, SSEA3, TRA-1–60, and TRA-1–81, with cells now able to form EBs and teratomas. We conclude that human mid-trimester AFSC, which may be isolated autologously during pregnancy without ethics restriction, can acquire pluripotent characteristics without the use of ectopic factors. Our data suggest that this medium-dependant approach to pluripotent mid-trimester AFSC reflects true reprogramming and not the selection of prepluripotent cells.
Introduction
A
When cultured under mesenchymal stem cell (MSC) conditions in fetal bovine serum (FBS), amniotic fluid stem cells (AFSC) are broadly multipotent, being able to differentiate into mesoderm-derived lineages such as bone, fat, and cartilage, but also into nonmesodermal lineages such as hepatic [9], endothelial [10], and neuronal [7]. The latter findings however relied on expression of genes upregulated during early differentiation, and functional in vivo lineage differentiation toward all 3 germ layers has not yet been fully demonstrated. In addition, AFSC are nontumorigenic [7], and are unable to form teratomas in vivo when injected into immunodeficient mice, thereby failing to fulfil one of the stringent characteristics of pluripotent stem cells. Together, these data highlight the plasticity of AFSC, but fail to substantiate earlier claims of pluripotency. This lack of tumorogenicity in a primitive stem cell type and the presence of progenitors of a wide range of lineages render AFSC attractive candidates for regenerative medicine. Mid-trimester AFSC have been shown to be amenable to reprogramming using the Yamanaka factors, upon which they show overlapping, but distinct, gene expression patterns compared to human embryonic stem cells (hESC) [11]. We recently showed that c-KIT+ stem cells isolated from first-trimester amniotic fluid can be reprogrammed to functional pluripotency in a transgene-free approach using valproic acid (VPA) [12,13]. However, these early amniotic cells are more likely to be of germ origin, appear phenotypically more primitive, and are not as clinically accessible as mid-trimester AFSC [13]. In addition, chemical epigenetic reprogramming is only translatable if applied to mid-trimester AFSC, which can be isolated during amniocentesis. It is still not clear whether first- and second-trimester c-KIT+ cells share common potential, and if this this chemical approach would be sufficient to revert a more mature cell type to an earlier state of pluripotency.
An increasing number of publications report advantageous characteristics of mid-trimester AFSC over other neonatal and postnatal stem cell sources. Mid-trimester AFSC are typically expanded in serum-based culture media, as standard for MSC expansion. We investigated whether culture under conditions that maintain pluripotency in first-trimester AFSC would favor the induction of pluripotency in mid-gestation amniotic cells. We show that human mid-trimester AFSC cells can be reprogrammed using a chemical approach, that VPA action relies on ES rather than MSC culture conditions, and that this is not the result of selecting propluripotent cells, but true reprogramming.
Materials and Methods
Ethics
Amniotic fluid collection for research purposes was approved by the Research Ethics Committee of Hammersmith and Queen Charlotte's Hospitals. National guidelines (Polkinghorne) were complied with in relation to the use of fetal tissues for research. All animals were handled in strict accordance with good animal practice as defined by the UK Home Office Animal Welfare Legislation, and all animal work was approved by the Institutional Research Ethics Committee (Institute of Child Health, University College London, UK) and performed under the project license number 70/7024.
Cell isolation and culture
Amniotic fluid was collected during 15–18 weeks of gestation (mid-trimester) (n=10) under ultrasonography guidance in women with otherwise uncomplicated pregnancies undergoing clinically indicated prenatal diagnosis. Cytospun cells were first plated in Dulbecco's modified Eagle's medium (DMEM) high-glucose (Sigma-Aldrich,
Human adult MSC were obtained from the Tulane Center for Gene Therapy (Tulane University, New Orleans, LA). The immortalized human embryonic kidney cell line 293T was purchased from ATCC (CRL-11268; Teddington,
The hESC lines H1 and H9 (WiCell Research Institute,
Real-time quantification
For real-time protocols, we used SYBR Green dye fluorescence (Applied Biosystems) and the ABI Prism 7700 Sequence Detection system (Applied Biosystems). As the cycle threshold value (Ct) to reach fluorescence is directly proportional to the log of the initial amount of input cDNA, we estimated the amount of target sequence in the experimental sample (ABI Prism 7700 Sequence detection System User Bulletin #2, 2001, Relative quantification of gene expression) by referencing the Ct of each experimental sample to a standard curve created with a serial dilution of a reference template. All samples were run at least in duplicate. Standard curves were generated with ABI Prism 7700 SDS 1.7 software, and R 2 values were always ≥0.998. Primers used are listed in Table 1.
Relative telomere length and telomerase activity
Relative telomere length and telomerase activity were assessed by quantitative real-time reverse transcription
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (PFA) in 125 mM HEPES (pH 7.6; 10 min, 4°C), or 8% PFA in the same buffer (50 min, 4°C) and permeabilized in 0.5% Triton X-100 in PBS (30 min, gentle rocking). After fixation, cells were rinsed in PBS, blocked (1 h) with PBS+ [PBS supplemented with 1% bovine serum albumin (BSA), 0.2% fish skin gelatin, 0.1% casein; pH 7.6], incubated (2 h) with primary antibodies in PBS+, washed (5× over 1.5 h) in PBS+, incubated (1 h) with secondary antibodies in PBS+, washed (overnight, 4°C) in PBS+, rinsed in PBS, and stained with TOTO-3 before being mounted in VectaShield (Vector Labs) and visualized immediately. For fluorescence confocal laser-scanning microscopy, images were collected sequentially on Leica TCS SP5 (X1000 PL APO oil objective) and transferred to Adobe Photoshop (Adobe Systems). All primary antibodies (listed in Table 1) were used at 1:10–1:100 dilution. Secondary antibodies for immunofluorescence were donkey anti-mouse and anti-rabbit IgG conjugated with FITC (1:100 dilution; multiple-labeling grade; Jackson ImmunoResearch Laboratories,
IF, immunofluorescence; FC, flow cytometry.
Flow cytometry
Monolayers of adherent cells were detached and resuspended in an FACS buffer (PBS, 1% BSA; 5×105 cells/100 μL FACS-buffer). For cell surface FACS staining, cells were stained with specific antibodies (PE-conjugated or unconjugated) for 1 h at 4°C. When using unconjugated primary antibodies (mouse anti-human SSEA3, TRA-1–60, and TRA-1–81, and rabbit anti-human NANOG), cells were subsequently washed 3 times in the FACS buffer, and a secondary goat anti-mouse IgM PE-conjugated or goat anti-rabbit IgG FITC-conjugated antibody was added and incubated for 30 min. Afterward, cells were washed and resuspended in the FACS buffer. For intracellular FACS staining, cells were washed once in the FACS buffer, fixed for 10 min in 0.01% PFA, washed twice with PBS, resuspended in a permeabilization buffer (PBS, 1% Triton), and stained as above. All samples were analyzed by an FACScalibur flow cytometer (Becton Dickinson) and analyzed using CellQuest software. Positive controls were hESC, and negative controls were IgG or IgM primary antibody-specific isotype controls. Antibodies used are listed in Table 2.
RT and RT-PCR
Total RNA was extracted from cells using the RNeasy Mini RNA isolation kit (Qiagen) and eluted from minicolumns with 50 mL RNase-free water. cDNA was subsequently synthesized using Pd(N)6 random hexamers (Amersham) and 1 mL of 200U M-MLV Reverse Transcriptase in the presence of dNTPs (Promega Corp.). The reaction was performed for 10 min at 25°C, for 60 min at 42°C, and for 10 min at 75°C, and stored at −20°C until use. The cDNA was amplified by 30 cycles of denaturation (60 s at 94°C), annealing (30 s, 60°C), and elongation (30 s at 72°C), followed by a final step at 72°C for 5 min. Positive controls were ES cells and negative controls adult MSC. Primer sequences are listed in Table 2.
Cell size
The size of passage-5 cells in a suspension stained with Trypan blue was determined using ImageJ software and calibrated with a hemocytometer. The relative size of live cells was assessed under a microscope magnification ×20. A total of 250 cells per sample were counted and averaged.
Growth kinetics
Cells were plated in triplicate at 1×104/cm2 in 10-cm plates in a growth medium. Cells were trypsinized when subconfluent and counted in Trypan blue in a hemocytometer to exclude dead cells before replating at similar density. The doubling times were calculated over 50 days via the formula, DT=t/(Log2[y/m]), with DT=doubling time, t=time in culture, y=number of cells at end of culture, and m=number of cells at beginning of culture.
To compare the growth rate of cells expanded on Laminin or Matrigel, we seeded 35,000 cells in triplicates in wells of a 6-well plates. The number of cells was counted every 2 days over a 2-week period, and the growth rates were determined by plotting the number of cells against time in culture.
Embryoid body formation and analysis
Undifferentiated cells were induced to differentiate in vitro into embryonic bodies (EBs) by incubating cells from five to six confluent wells of a 6-well plate with a differentiation medium containing 80% knockout DMEM (KO-DMEM; Gibco BRL Life Technologies) supplemented with 1 mM
Teratoma formation assay
Immunodeficient mice were housed in microisolator laminar flow-caging systems and were supplied with sterile water, food, and bedding. About 2×106 cells resuspended in Matrigel were injected subcutaneously into the dorsal flank of 8–12-week-old common γ chain −/−, RAG2−/−, C5−/− immunodeficient mice without preconditioning. Mice were observed for the growth of solid tumors up to 8 weeks or up to 10-mm3 tumour volume. All experiments were performed according to the Home Office animal welfare legislation. Positive controls were hESC.
Viability and apoptosis detection by flow cytometry
To detect cell death and apoptosis of cells grown in hESC conditions, cells were plated on Matrigel in an MSC medium, allowed to attach, and the medium was changed to Nutristem. Cells were trypsinized at different time intervals after medium change and collected into centrifuge tubes. The supernatant medium and PBS used to wash cells, containing any dead cells, were also collected in the same tube. Cells were pelleted and stained with 10 μM DioC6 (Sigma, detected on the FL-1 channel) and 10 μg/mL propidium iodide (PI; Sigma, detected on the FL-3 channel) for 15 min at 37°C, before flow cytometric analysis for the detection of cell death/apoptosis. For the analysis, the cells were plotted on an FL-1/FL-3 dot-plot graph and separated in quadrants depending on detection. The upper left quadrant represented alive and healthy cells. The upper right quadrant represented cells that are in the process of dying. The lower left quadrant represented cells that are apoptotic, and the lower right quadrant represented cells that are dead. Three samples were assayed for each condition, and cells treated with 1-μm staurosporine (Sigma) for 24 h were used as a positive control of apoptotic cells.
Single-cell cloning
Single-cell cloning was carried out by serial dilution of cells, and 1 cell was plated in each well of a 24-well plate. Each well was visually inspected to ensure that a single cell was present per well, and wells containing more than 1 cell were excluded from analysis. Clones were expanded under MSC culture conditions, and the medium was changed twice per week. Ten clones were analyzed in total.
ALP staining
Alkaline phosphatase (ALP) staining was performed using the ALP kit from Sigma, according the manufacturer's instructions. MEF cells were used as a negative control and hES cells as positive control.
Statistical analysis
Statistical analysis was performed using InStat Version 3.0a statistical software (GraphPad software). Descriptive statistics for parametric data included standard deviation (SD) or standard error of the mean (SEM), with 2-tailed t-tests, were used to compare means in independent samples. P<0.05 was considered significant.
Results
Human mid-trimester AFSC cultured under MSC conditions express telomerase, CD24, OCT4, C-MYC, KLF4, and SSEA4
We isolated mid-trimester AFSC (15–18-week gestation, n=10) under ultrasonography guidance and confirmed their fetal, as opposed to maternal, origin using RT-PCR for the SRY gene and human chromosomes X and Y probes for fluorescence in situ hybridization (FISH, data not shown). Their nonhematopoietic phenotype was evidenced by the absence of CD14, CD34, and CD45 expression, and low/null expression of HLAI and HLAII (data not shown). AFSC were subsequently cultured under MSC conditions, that is, the DMEM supplemented with 10% FBS on noncoated plastic dishes. First, we show that 85% of the AFSC population contains a subset of cells positive for CD24, which is a cluster-of-differentiation marker associated with pluripotent hESC, not expressed in MSC [16 –18] (Fig. 1A). Using quantitative real-time-based assays, as previously described [15], we showed that AFSC had active telomerase (Fig. 1B) and longer telomeres than adult bone marrow MSC (Fig. 1C).

Expression of pluripotency markers by amniotic fluid stem cells (AFSC) cultured under mesenchymal stem cells (MSC) conditions.
We used real-time RT-PCR to quantify basal expression levels of pluripotency-associated markers under MSC culture conditions. The relative expression of each gene was normalized by its expression in hESC (H9 line) after normalization to GAPDH (primers listed in Table 1). Results showed that AFSC expressed C-MYC at significantly higher levels than hESC (2863%±0165%), but lower levels of OCT4A (0.51%±0.12%), and null/low (<0.1) levels of NANOG (0.02%±0.002%), KLF4 (0.04%±0.02%), or SOX2 (0.017%±0.01%) (Fig. 1D). At the protein level (antibodies listed in Table 2), flow cytometry (Fig. 1E, F) and confocal immunofluorescence (Fig. 1G) showed that the AFSC population contained cells positive for the MSC markers CD105 (71.8%±0.8%), CD90 (82.4%±0.1%), CD44 (78.8%±1.1%), CD73 (76.7%±0.6%), and CD29 (77.2%±0.7%) (Fig. 1E). The AFSC population was negative for the pluripotency markers NANOG, SOX2, KLF4, SSEA3, TRA-1–60, and TRA-1–81, but contained a distinct subset of cells positive for OCT4A (70.2%±4.3%), SSEA4 (65.2%±7.3%), and C-MYC (83.2%±7.3%). To further investigate the heterogeneity of the population, we analyzed 10 clones (Fig. 1H, I). We found 7 clones that contained a distinct subset of cells expressing high levels of SSEA4 and 3 clones where this subset was absent (referred subsequently as SSEA4+ and SSEA4− clones) (Fig. 1H). Results showed that SSEA4+ clones had a similar profile compared to SSEA4- clones for expression of CD105, CD90, CD44, CD73, CD29, OCT4, and C-MYC (Fig. 1I).
Culture of human mid-trimester AFSC under hESC conditions affects their morphology and growth
When cultured under MSC conditions, AFSC grew as a monolayer of cells with a mesenchymal morphology, that is, a large spindle-shape cytoplasm, with daughter cells moving away from each other upon cell division (Fig. 2A). In contrast, when the cells from the same samples and passage were cultured under hESC conditions, they lost their fibroblastic appearance and formed colonies of packed cells upon cell division (Fig. 2B). Nuclear area was similar in AFSC cultured under MSC and in hESC conditions (20.3±0.8 N=43 vs. 21.4±0.7 N=44, respectively, P=0.30) (Fig. 2C). To further investigate changes in overall cell size, we detached the cells and resuspended in the medium before visualizing them under a microscope in a hematocytometer. Cell diameter measure showed that AFSC expanded in Nutristem were significantly smaller than those cultured under MSC conditions (17.45±0.3053 N=42 vs. 15.42±0.4817 N=43, P<0.001) (Fig. 2D).
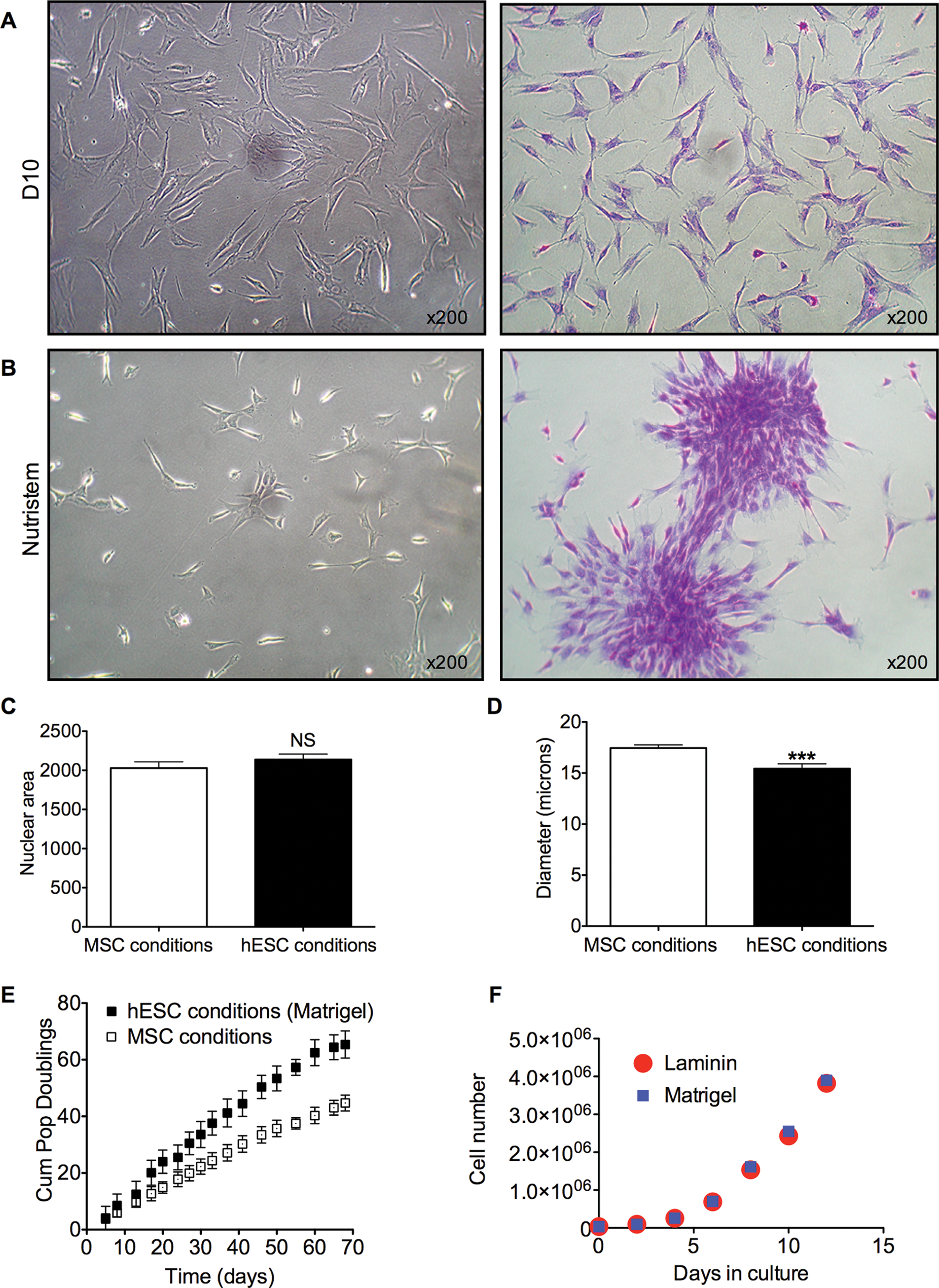
Growth and kinetics of AFSC cultured under MSC and human embryonic stem cells (hESC) conditions.
We also found that AFSC cultured under hESC conditions showed higher cell-division kinetics (Fig. 2E), with lower average doubling time (22.8±1.9 h N=6 vs. 33.2±2.5 h N=6, P<0.001). Cells expanded in an hESC condition on Matrigel or Laminin over 12 days showed a similar growth rate (Fig. 2F).
AFSC cultured under hESC conditions show upregulation of OCT4, C-MYC, KLF4, and SOX2
AFSC expanded in hESC conditions on Matrigel showed a 24-fold upregulation of OCT4 (to 12.2%±0.5%), 11-fold upregulation of C-MYC (to 32120%±210%), now also expressing KLF4 (to 0.7%±0.2%), and SOX2 (to 0.4%±0.05%) (Fig. 3A). In comparison, cells expanded on Laminin did not upregulate OCT4, SOX2, C-MYC, and KLF4 (Fig. 3A). In addition, AFSC still did not express the hESC marker ALP (Fig. 3B). At the protein level, AFSC expanded under MSC conditions showed a bimodal distribution of expression for OCT4, SOX2, C-MYC, and SSEA4 on flow cytometry, but were negative for NANOG, SSEA3, TRA-1–60, and TRA-1–81 (Fig. 3C). Because the whole population of AFSC is heterogeneous, we next investigated whether hESC conditions were detrimental for a subset of fibroblastic cells and favoring the survival of more propluripotent cells. To assess the viability and apoptosis of the cells in an MSC medium and upon transfer of the cells to the hESC medium, we used DiOC6 staining, a dye selective for the mitochondria of live cells, and propidium iodide (or PI) staining, a dye that binds tightly to the nucleic acids of necrotic cells, but does not penetrate the plasma membranes of live cells or early apoptotic cells. The cells were cultured in the MSC medium for 1 week before the medium was replaced by an hESC medium, and cell death was analyzed 3, 6, 12, 24, and 72 h later. Results showed that hESC conditions did not trigger cell death, with the percentage of live cells remaining stable 3 to 72 h after the change of culture conditions and the percentage of dead cells not increasing significantly (Fig. 3D, E). Together, these results indicate that it is unlikely that the different phenotypes of AFSC obtained under MSC or hESC conditions are based on selection of a subset of cells.
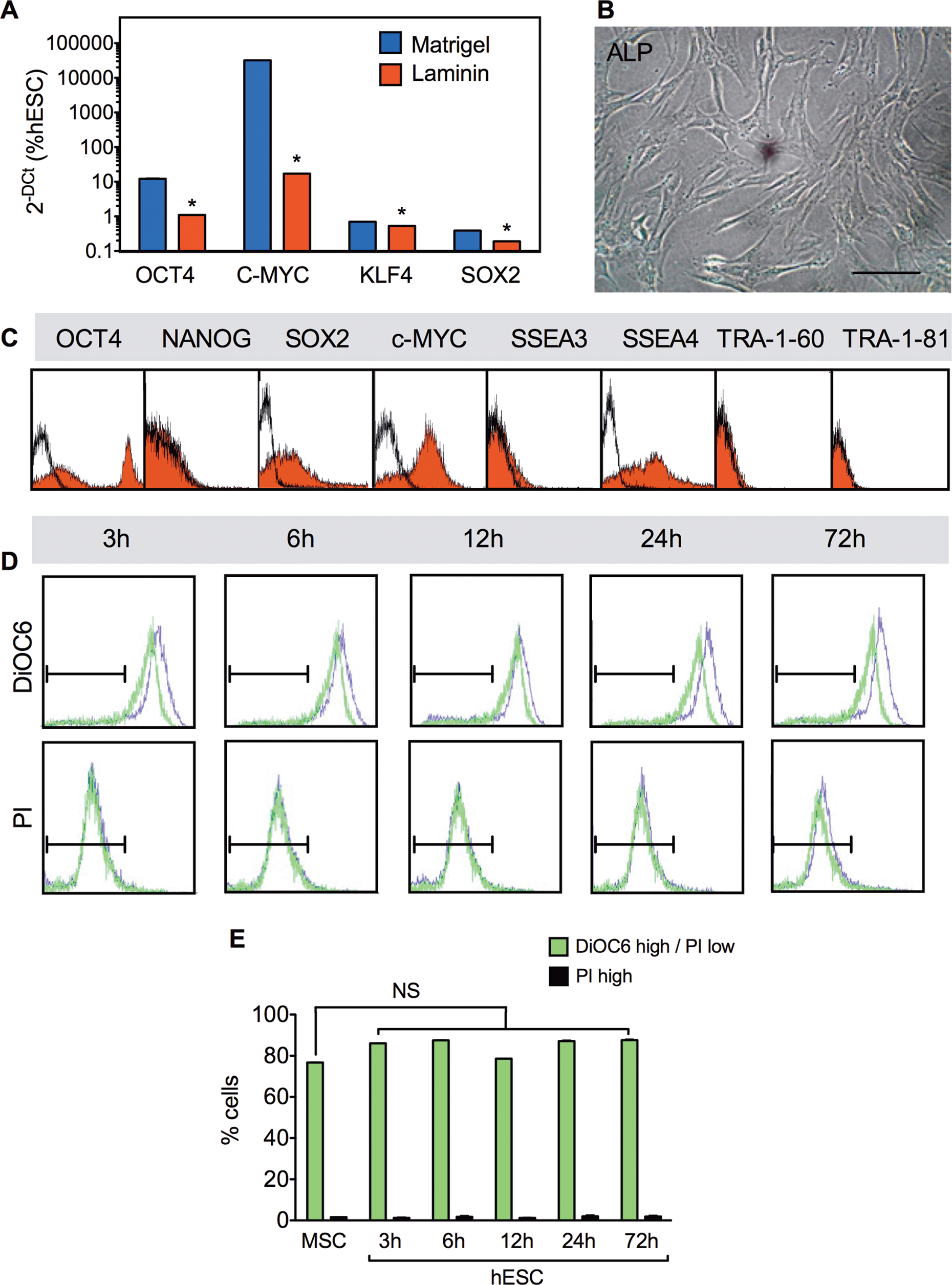
Expression of pluripotency markers in AFSC cultured under hESC conditions.
When grown in a suspension under low-attachment conditions in a medium containing 80% KO-DMEM supplemented with 1 mM
Adding the histone deacetylase inhibitor VPA for 5 days to AFSC under hESC conditions upregulates pluripotency markers further
In addition to its widespread use as an antiepileptic drug for bipolar disorders, VPA has been successfully employed to increase reprogramming efficiency of primary human fibroblasts for the generation of induced pluripotent stem cells (iPSC). VPA acts as an epigenetic modifier by remodeling chromatin through direct inhibition of histone deacetylase (HDAC) [19,20]. Interestingly, it was recently shown that increased pluripotency of hESC by HDAC inhibition acts to upregulate OCT4A expression through factors targeting a proximal hormone-response element and to enhance the activity of target genes involved in ECM production to support pluripotency [19 –21]. Optimal VPA supplementation (concentration, i.e., 0.5–10 mM, and length of supplementation, that is, 1 to 10 days, data not shown) was determined to 1 mM VPA for 5 days. This resulted in significant upregulation of OCT4 (75%±12.5%), SOX2 (20.8%±4.4%), KLF4 (21%±3.2%), and C-MYC (32100%±320%), but not in changes in gene expression for the housekeeping genes ACTIN and HPRT (1.36-fold and 1.64-fold upregulation, respectively), indicating that the effect of VPA was affecting primarily genes determining stemness (Fig. 4A). In comparison, cells expanded on Laminin showed a significantly lower upregulation of OCT4, SOX2, C-MYC, and KLF4 (Fig. 4A). Culture of the cells in hESC conditions without VPA did not significantly upregulate hTERT expression compared to levels found in cells expanded in MSC conditions, but VPA supplementation led to a 4-fold upregulation of hTERT (Fig. 4B). This was concomitant with AFSC now expressing SOX2 (53.5%±1.3%), SSEA3, TRA-1–60, and TRA-1–81 (Fig. 4C).
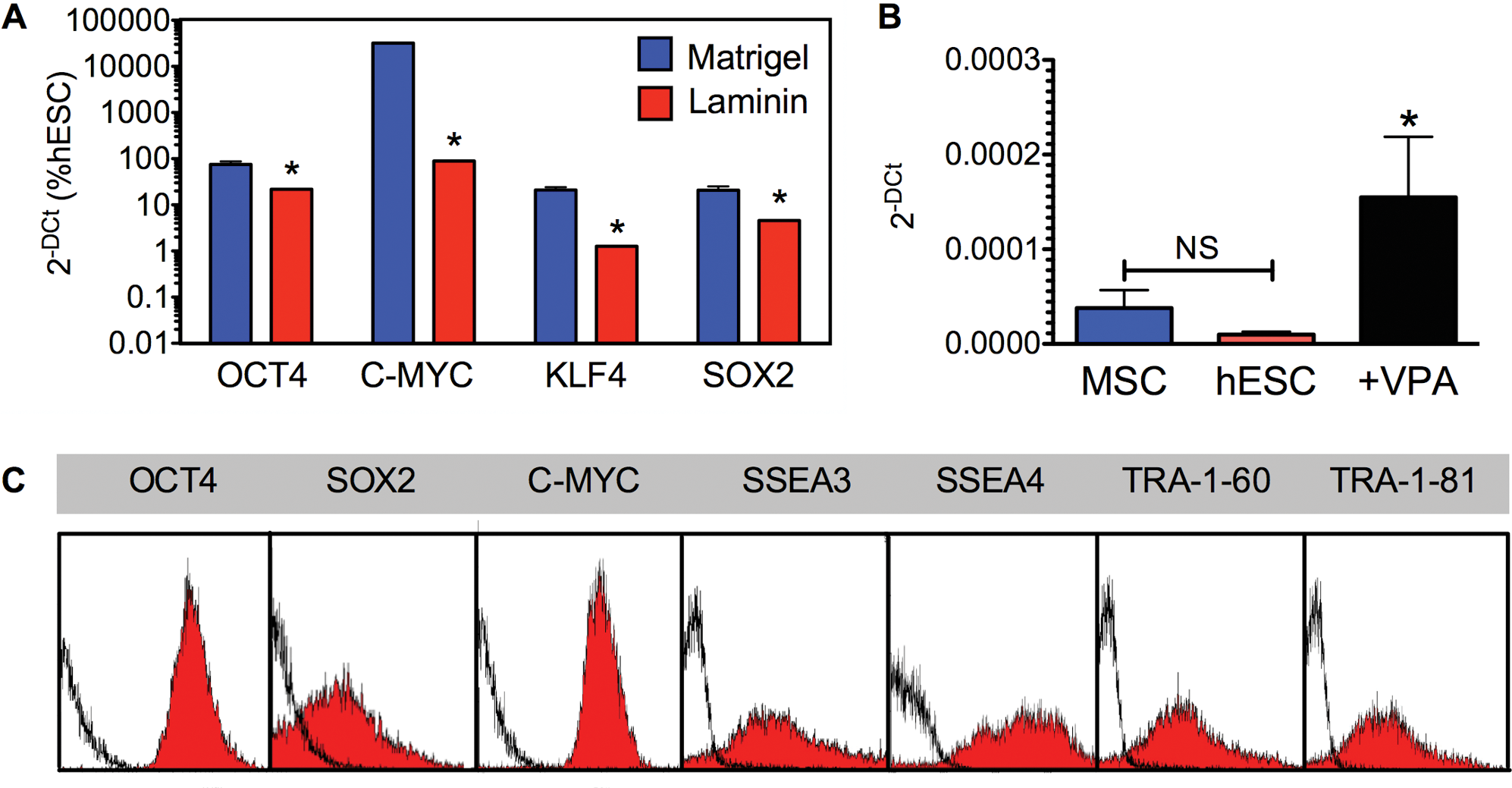
AFSC cultured in hESC conditions supplemented with Valproic acid (VPA) express pluripotency markers and form embryoid bodies.
To investigate whether it is essential to prime AFSC with the hESC medium condition before the induction of pluripotency through application of VPA, we directly added VPA to D10 for 5 days. Quantification of pluripotency gene expression showed that supplementation of D10 with VPA did not significantly induce upregulation of OCT4 (1.34-fold upregulation compared to levels obtained in D10), SOX2 (1.65-fold upregulation), C-MYC (0.87-fold downregulation), and KLF4 (1.4-fold upregulation), indicating that priming with the hESC medium is required to induce pluripotency via VPA supplementation.
Analysis of clonal populations reveals interchangeable features when the medium conditions are changed from one to the other type
To investigate whether the changes in pluripotency gene expression were interchangeable when the medium conditions were changed from one to the other type, we analyzed 4 independent clonal populations. Cells were expanded in D10 in MSC conditions before the medium conditions were switched to hESC and subsequently either supplemented with VPA or replaced by D10 (Fig. 5A). Analysis of pluripotency gene expression using quantitative real-time RT-PCR showed in 4 independent clones (data presented for 2 representative clones) that levels of expression for OCT4A, SOX2, and C-MYC were upregulated upon switch of the culture medium from D10 to Nutristem, and reverted to original D10 levels when the medium was further reverted to D10 one week later, or were further upregulated upon VPA supplementation (Fig. 5B), as observed in the whole population. Interestingly, although the levels of KLF4 lowered when the Nutristem medium was replaced by D10, the levels remained higher than in the first phase of D10 culture, possibly because it takes longer for the cells to fully revert to the original phenotype. Together, these data show that pluripotency gene expression is reversible upon switch of culture conditions and also suggest a phenotypical switch of the same cell type rather than a selection of different cell types.
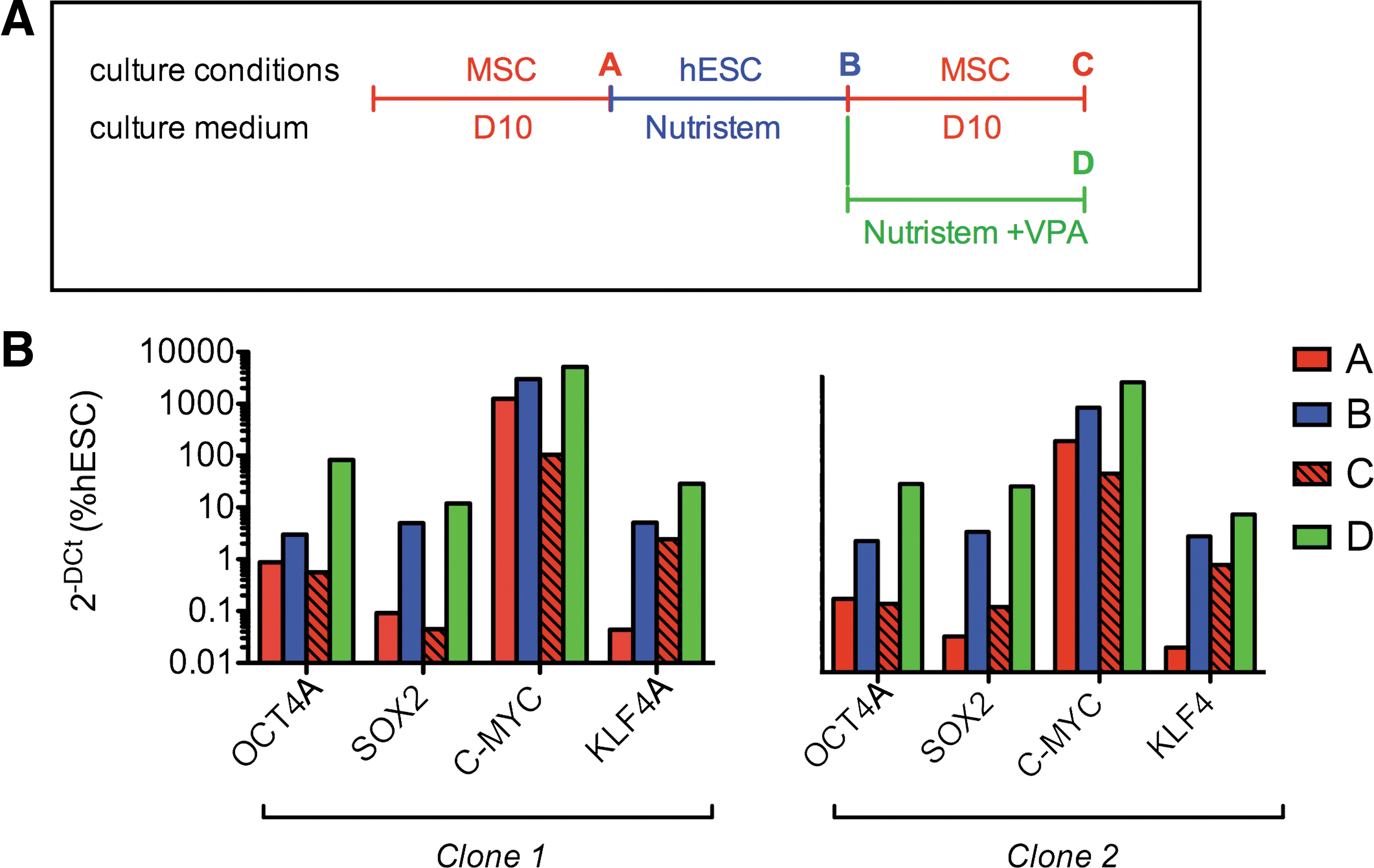
Interchangeablility of gene expression at clonal level following switch of culture conditions.
VPA supplementation bestows EB formation competence in vitro and teratoma formation in vivo
When cultured in low-attachment plates in a medium containing 80% KO-DMEM supplemented with 1 mM
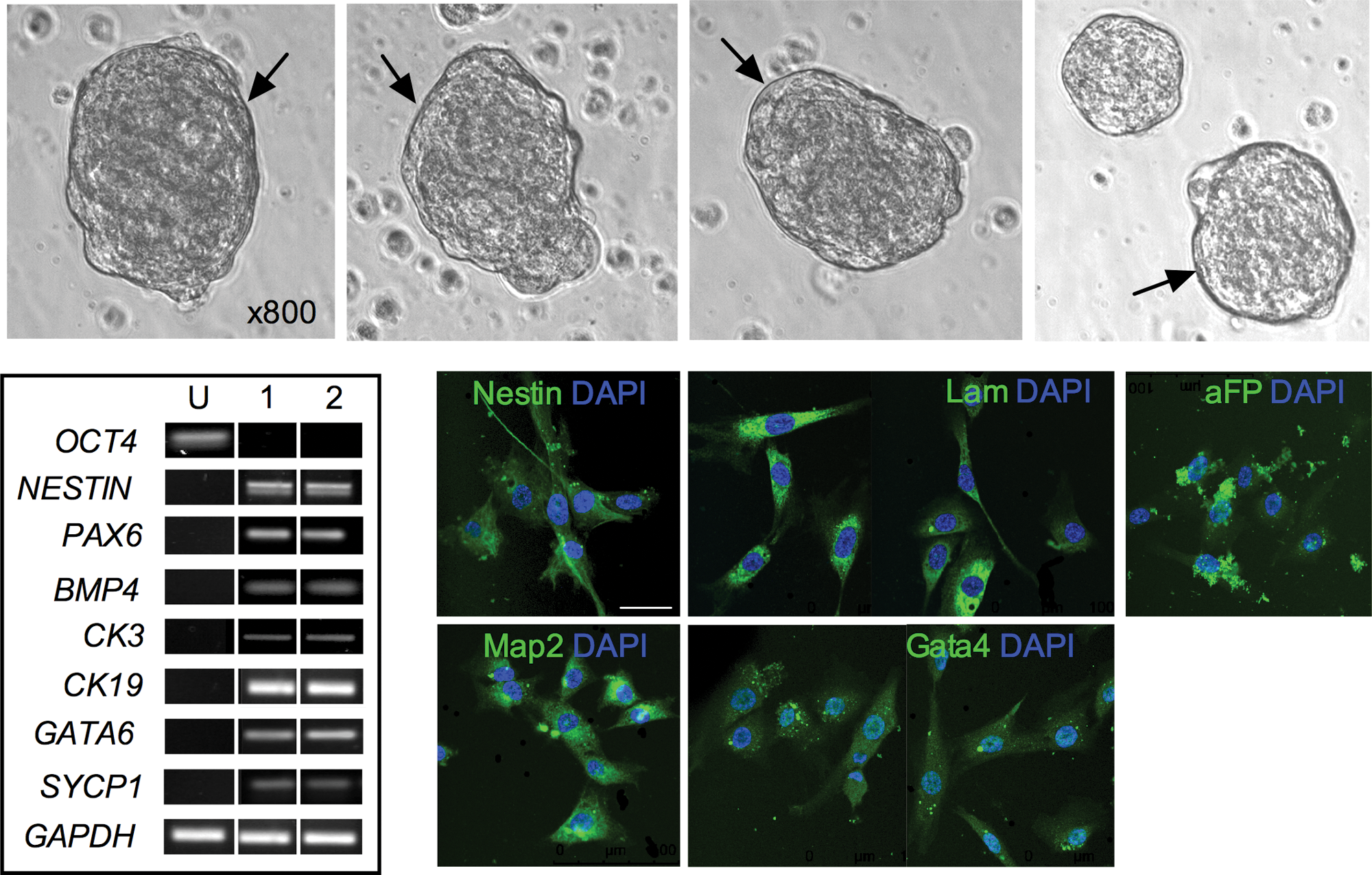
Embryoid body (EB) formation. Phase contrast image of EBs from AFSC showing peripheral lining, indicated by arrow. RT-PCR for OCT4A, NESTIN, PAX6, BMP4, CK3, CK19, GATA4, SYCP1 and GAPDH in AFSC cultures (undifferentiated, U) and in 2 embryoid bodies (EB1 and EB2). Confocal immunofluorescence staining of EBs embedded in paraffin for GATA4, aFP, NESTIN, LAMININ (LAM), and MAP2. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. Color images available online at
VPA-treated AFSC suspended in Matrigel were injected subcutaneously into the dorsal flank of immunodeficient mice. Solid tumors were visible 8 weeks later and collected for immunohistochemical analysis (Fig. 7A). Teratomas contained solid tissues of human origin, and histological analysis revealed the presence of derivates from 3 germ layers, including neural tube and squamous epithelium (ectoderm), collagenous tissue and blood vessels of human origin (mesoderm), and alveolar and gut epithelium (endoderm) (Fig. 7B). Immunohistochemical staining for differentiation markers confirmed the presence of germ-layer-specific antigens (Fig. 7C).

Teratoma formation of AFSC cultured under hESC conditions supplemented with valproic acid for 5 days.
Human mid-trimester AFSC express some primordial germ cell markers
To investigate the cellular origin of AFSC, we examined the expression of primordial germ cell (PGC) markers. Flow cytometry revealed that the AFSC population contained a small subset of cells positive for all markers, that is, FRAGILIS, SSEA1, TNAP, NANOS3, BLIMP1, PUM2, STELLA, DAZL, and VASA (Fig. 8A). Confocal microscopy localization showed that SSEA1 was expressed on the cell surface of AFSC, and that VASA, BLIMP1, TNAP (tissue-nonspecific ALP), PUM2, NANOS3, and DAZL were localized in the cytoplasm [22], with FRAGILIS and STELLA having both nuclear and cytoplasmic expression [23,24] (Fig. 8B).
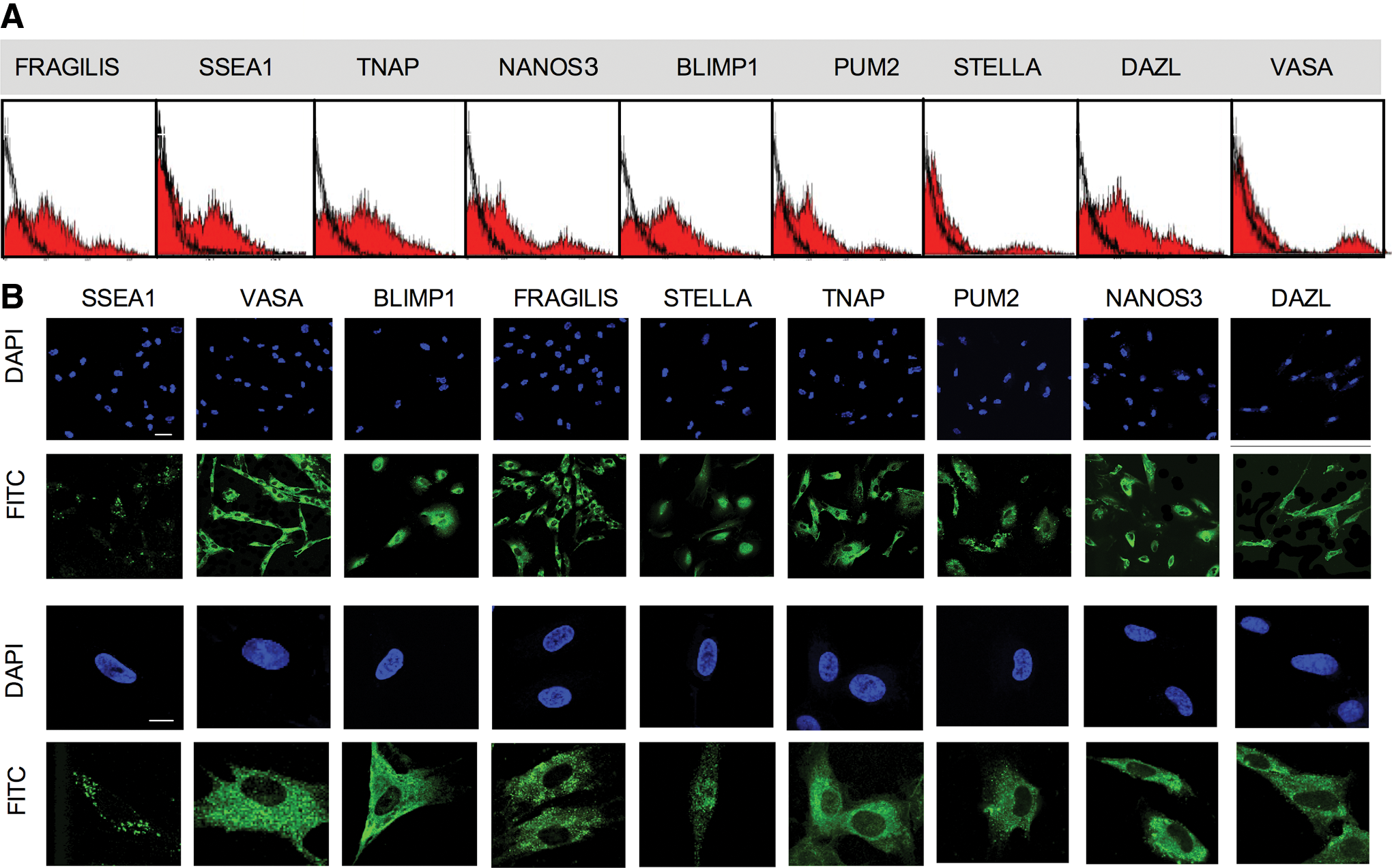
AFSC express markers of primordial germ cells.
Subcellular protein localization of PGC markers is indicative of their individual stage of migration from the allantois to the genital ridge. For example, BLIMP1, a transcriptional repressor expressed in the nucleus of PGCs, progressively relocates to the cytoplasm during migration of PGC to the genital ridge and is absent in EG cells [25]. Similarly, DAZL (Deleted in Azoospermia-Like) relocates from the nucleus to the cytoplasm, paralleling downregulation of OCT4 and the onset of expression of VASA [26], while PUM2 (Pumilio-2) colocalizes with DAZL [24]. We found that DAZL, PUM2, BLIMP1, and VASA were expressed in the cytoplasm, consistent with a subset of mid-trimester AFSC having originated from migratory PGCs deposited throughout the embryo on their way to the genital ridges, as previously hypothesized [27].
Discussion
Human mid-trimester AFSC hold promise in regenerative medicine, because they can be isolated during ongoing pregnancy without ethics restriction, although with a small risk of miscarriage, and because they demonstrate an intermediate phenotype between ES and MSC, expressing markers of both and capable of differentiating into lineages of all 3 germ layers. However, AFSC are typically expanded in serum-based culture conditions. We recently showed that stem cells of possible germ origin present in early amniotic fluid obtained at first-trimester termination could be reverted to functional pluripotency using VPA. However, in contrast to mid-trimester AFSC [7], first-trimester AFSC were expanded under ES conditions, where they grow as compact colonies and contain a subset of ALP+ cells, which also express NANOG, SSEA3, TRA-1–60, and TRA-1–81 [13]. Here, we investigated whether the same methodology could be applied successfully to clinically relevant cells with a more mature phenotype.
We showed that mid-trimester AFSC, like other fetal stem cells when expanded under MSC conditions, expressed the MSC markers CD105, CD90, CD73, CD44, and CD29 along with a subset of cells expressing OCT4A, C-MYC, and SSEA4. However, unlike first-trimester AFSC, mid-trimester AFSC in MSC media showed low/null levels of NANOG, SOX2, KLF4, SSEA3, TRA-1–60, or TRA-1–81. Culture in ES conditions combined with VPA supplementation for 5 days induced major upregulation of OCT4, SOX2, C-MYC, and KLF4, with cells expressing NANOG, SSEA3, SSEA4, TRA-1–60, and TRA-1–81, gaining EB and teratoma formation competency, showing that a chemical approach can also be used on this cell type. In the present article, we also show that there is no passage number dependence for the development of pluripotency. Moreover, analysis of the heterogeneity of the population revealed that OCT4+/SSEA4+/C-MYC+ cells present in the population also express the MSC markers present on the surface of the OCT4− cells, indicating that it is unlikely that pluripotent cells were present in the parental population.
In line with this, we showed here that pluripotency gene expression is reversible upon switch of culture conditions, and the different phenotype of AFSC obtained under MSC or ES conditions reflects a phenotypical switch of the same cell type rather than selection of different cell types. While it was not clear whether the VPA effect was dependent on culture conditions in our study on first-trimester AFSC, we show here that it is essential to prime mid-trimester AFSC with the ES medium condition before the induction of pluripotency through application of VPA, and that this cannot be achieved with cells held under the MSC medium condition.
Although VPA induced C-MYC expression to high levels, we previously showed that C-MYC levels dropped to undetectable levels upon differentiation [13], and therefore the derivates of AFSC reprogrammed to pluripotency using VPA potentially used in cell-based therapies may not be oncogenic. Interestingly, we found that reprogramming efficiency was stable within the exponential phase of the growth rate of the cells (until passage 20) (data not shown). Nutristem has been developed to sustain pluripotency and viability of pluripotent cells without the need for feeder cells in combination with Matrigel, a commonly used purified gel matrix that consists of a mixture of extracellular matrices, proteoglycans, and growth factors. A possible mechanism in which Matrigel predisposes the cells to pluripotency is unknown, but its porosity might trap growth factors and release them slowly in the culture, as it has been shown with Noggin [14].
VPA is an HDAC inhibitor, which can induce pluripotency genes in hESC [19,21] and enhances the efficiency of iPSC production [20]. Interestingly, it was recently shown that increased pluripotency of hESC after HDAC inhibition acts to upregulate OCT4A expression through factors targeting a proximal hormone-response element and to enhance the activity of target genes involved in ECM production to support pluripotency [21]. Our study showed similar results in human cells, indicating that prepluripotent cells that endogenously express some, but not all, pluripotency markers can generate pluripotent cells without viral vectors, but by manipulation of culture conditions. Interestingly, while human AFSC endogenously expressed OCT4, C-MYC, and SSEA4, mouse myoblats expressed another set of pluripotency markers, that is, Sox2, Klf4, and c-Myc, with c-Myc being the common marker expressed by both cell types when cultured under an FBS-based medium. This indicate that endogenous expression of Sox2 might not be the condition sine que non for the capacity of the cells to revert to pluripotency using small-molecule compounds.
Compared to our previous data on first-trimester AFSC showing that majority of the cells expressing all the PGC markers investigated, that is, FRAGILIS, SSEA1, TNAP, NANOS3, BLIMP1, PUM2, STELLA, DAZL, and VASA, mid-trimester AFSC contain a very small fraction of cells expressing STELLA and VASA. It is possible that a small subset of PGC-derived cells present in the first trimester remain in the parental populations at mid-trimester.
We conclude that surplus of amniotic fluid specimens from prenatal diagnostics could be used to isolate and bank stem cells for autologous patient-specific use that can be reprogrammed to pluripotency using a chemical method.
Footnotes
Acknowledgments
This research was funded by the Genesis Research Trust, Newlife Foundation, Action Medical Research, Henry Smith Charity, and Kidney Research United Kingdom. We wish to thank A. Ditadi for helpful comments on the manuscript. DM was supported by Kidney Research United Kingdom. GJ was supported by the Medical Research Council. PDC was supported by Great Ormond Street Hospital Children's Charity.
Author Disclosure Statement
The authors declare that they have no competing or financial interests.