
Abstract
The presence of mesenchymal stem cells (MSCs) has been described in various organs. Pericytes possess a multilineage differentiation potential and have been suggested to be one of the developmental sources for MSCs. In human liver, pericytes have not been defined. Here, we describe the identification, purification, and characterization of pericytes in human adult and fetal liver. Flow cytometry sorting revealed that human adult and fetal liver contains 0.56%±0.81% and 0.45%±0.39% of CD146+CD45−CD56−CD34− pericytes, respectively. Of these, 41% (adult) and 30% (fetal) were alkaline phosphatase-positive (ALP+). In situ, pericytes were localized around periportal blood vessels and were positive for NG2 and vimentin. Purified pericytes could be cultured extensively and had low population doubling times. Immunofluorescence of cultures demonstrated that cells were positive for pericyte and mesenchymal cell markers CD146, NG2, CD90, CD140b, and vimentin, and negative for endothelial, hematopoietic, stellate, muscle, or liver epithelial cell markers von Willebrand factor, CD31, CD34, CD45, CD144, CD326, CK19, albumin, α-fetoprotein, CYP3A7, glial fibrillary acid protein, MYF5, and Pax7 by gene expression; myogenin and alpha-smooth muscle actin expression were variable. Fluorescence-activated cell sorting analysis of cultures confirmed surface expression of CD146, CD73, CD90, CD10, CD13, CD44, CD105, and ALP and absence of human leukocyte antigen-DR. In vitro differentiation assays demonstrated that cells possessed robust osteogenic and myogenic, but low adipogenic and low chondrogenic differentiation potentials. In functional in vitro assays, cells had typical mesenchymal strong migratory and invasive activity. In conclusion, human adult and fetal livers harbor pericytes that are similar to those found in other organs and are distinct from hepatic stellate cells.
Introduction
P
Pericytes were isolated by fluorescence-activated cell sorting (FACS) as CD146high (melanoma cell adhesion molecule), CD45− [leukocyte common antigen, CD56− (neural cell adhesion molecule)], and CD34− (hematopoietic progenitor cell antigen) cells, and have been described as negative for typical endothelial, hematopoietic, or myogenic cell markers, such as CD144 (vascular endothelial-cadherin), vWF (von Willebrand factor), CD34, CD31 (platelet endothelial cell adhesion molecule), CD56, CD45, or Pax7 (paired box protein 7) [1,5]. CD146 is a cell surface glycoprotein and is used in pathology as a classical marker associated with melanoma [16]. Although it has been identified on some mesenchymal progenitor populations [3], it also has been detected on the endothelium and subsets of T and B lymphocytes [17,18]. The function of CD146 is not well understood, but was suggested to be associated with multipotency, because mesenchymal progenitors with a higher differentiation potential demonstrated a stronger CD146 expression [19]. Also, interaction of CD146 of endothelium with CD146 on activated T cells was proposed as a mechanism for the recruitment of the T cells to inflammation [20].
Crisan et al. described the characteristics of pericytes in detail [1,5]. Depending on the tissue of origin, frequencies of pericytes were between 0.29% and 1.79%, with the highest percentage in the stromal vascular fraction of adipose tissue (14.6%). The majority of pericytes were positive for alkaline phosphatase (ALP), a marker associated with highly proliferative cells. Cells could be cultured over the long term. Cultures demonstrated low population doubling times (PDT) of about 60 h (between 5–13 passages), with higher PDT at initial and late cultures times (162 and 106 h, respectively). Cells exhibited migratory activity, multilineage differentiation potential in vitro and osteogenesis and myogenicity in vivo.
In this work, we asked if perivascular progenitors also exist in human liver. We identified hepatic pericytes in human adult and fetal liver that are similar to those found in other organs and are distinct from hepatic stellate cells. Hepatic pericytes exhibit multilineage differentiation potential and exponential growth rates in culture.
Materials and Methods
Immunohistochemistry
To localize and analyze marker expression of pericytes in vivo, we investigated fetal and adult human liver sections by immunohistochemistry. Sections incubated with isotype-matched immunoglobulins served as negative controls. Human fetal muscle tissue was used as a positive control (BioChain, Hayward, CA). Unfixed liver tissue was embedded in OCT compound (Sakura Finetek, Torrance, CA) and flash frozen in liquid nitrogen-cooled isopentane followed by freezing in liquid nitrogen. Five- to 10-μm sections were cut, air-dried, fixed in ice-cold acetone/methanol [1:1 (v/v)] at −20°C for 10 min, and air-dried. Washing steps between stainings always included 3 washes with phosphate-buffered saline (PBS) 0.1% Triton X-100. Sections were incubated with 1% hydrogen peroxide for 1 h at room temperature (RT) for quenching endogenous peroxidases. Sections were blocked with 10% goat serum, 10% FcR block in PBS 0.1% Triton X-100. Primary antibodies to CD146, CD34, vWF (Santa Cruz Biotechnolgy, Santa Cruz, CA), glial fibrillary acid protein (GFAP), vimentin, desmin (Fisher Scientific, Pittsburgh, PA), NG2 (Becton Dickinson, Bedford, MA), and α-SMA (Dako, Glostrup, Denmark) were applied in a blocking buffer. CD146 immunoreactivity was visualized with goat TSA-AF488 or—AF555, secondary antibodies, included goat anti-rabbit or mouse IgG AF555 and AF488, and were applied in a blocking buffer, including DAPI (Invitrogen, Carlsbad, CA). Images were acquired by confocal microscopy using an Olympus Fluoview 1000 system (Center Valley, PA).
Liver cell isolation
Human fetal livers of 10–20 weeks of gestational age were obtained as anatomical gifts provided by the Allegheny Reproductive Health Center (Pittsburgh, PA). Organs were retrieved from abortions after informed consent of the donor and approval by the local Institutional Review Board. Liver cells were isolated as described previously [21], with slight modifications. Briefly, complete liver tissue (including smaller intrahepatic vessels) was minced with scalpels and digested with collagenase type IV (Sigma-Aldrich, St. Louis, MO) at 37°C. Cell suspensions were washed 2 times by centrifugation at 300 g and filtered through 40-μm pore size cell strainers (Becton Dickinson). Total cell suspensions were depleted of red blood cells by adding the red blood cell lysis buffer for 10 min at RT (Roche, Mannheim, Germany). Cells were resuspended in a high-glucose Dulbecco's modified Eagle's medium (DMEM) free of phenol red and supplemented with 20% fetal bovine serum (FBS), a mix of penicillin, streptomycin, amphotericin, and 2 mM Glutamax (Invitrogen).
Human adult liver cells of different ages and sex were purchased from Becton Dickinson. Freshly isolated cells in suspension were shipped overnight on ice. Cells were centrifuged at 300g and resuspended in a supplemented DMEM.
Cells were counted in a Neubauer chamber. Cell viability was determined by trypan blue (Invitrogen) exclusion.
Sorting by flow cytometry
Freshly isolated liver cell suspensions were used to sort out pericytes by FACS. We used a similar sorting strategy as published by Crisan et al. [1,5] to be able to compare our results to those obtained on other tissues. Cells were incubated in the blocking buffer (20% human FcR block; Miltenyi Biotec, Auburn, CA), 0.5% bovine serum albumin (BSA) (Sigma-Aldrich), and 2 mM EDTA (Sigma-Aldrich) in DPBS without calcium and magnesium (Invitrogen; pH 7.2) and incubated with the following antibodies: CD56-PE-Cy7 mouse IgG1 (No. 557747), CD45-APCCy7 mouse IgG1 (No. 557833), CD34-AF700 mouse IgG1 (No. 561440), ALP-AF647 mouse IgG1 (No. 561500) (all Becton Dickinson), and CD146-PE mouse IgG1 (No. 130-092-853) (Miltenyi Biotec). Negative controls included nonstained cells and isotype-control stained cells. Isotype controls were mouse IgG1-PE-Cy7, -APC-Cy7, -AF700, -AF647, and -PE (all Becton Dickinson). Compensation beads (Becton Dickinson) were used to compensate fluorochrome spectral overlap. Cells were sorted using a FACS Aria II (Becton Dickinson). After exclusion of cell debris and cell doublets by applying a forward versus side-scatter gate, a second gate was applied to sort the CD45− and CD56− populations. A third gate was applied to select the CD146+ and CD34− population, such that the finally obtained population was CD45−CD56−CD146+CD34−. This population was analyzed in parallel for its expression of ALP and α-SMA. For analysis of intracellular α-SMA, expression cells were stained for CD surface markers with antibodies or isotype controls as described, and subsequently fixed and permeabilized for 2 h at −20°C with 75% ethanol, including 1% FBS. Cells were washed twice, and stained with α-SMA-FITC rabbit IgG (No. 29533-FITC) (AnaSpec, Fremont, CA) or isotype control rabbit IgG-FITC (No. ab37406) (Abcam, Cambridge, MA), repectively.
Cell culture and PDT
FACS-sorted fetal pericytes were cultured as described [1]. In detail, cells were plated on gelatin-coated dishes (Becton Dickinson) at about 10,000 cells/cm2 in the supplemented EGM-2 medium (Lonza, Walkersville, MD) and cultured until they reached about 70% confluence (8–13 days). Cells were subcultured long term on plastic ware when they reached about 70% confluence in the supplemented DMEM [high-glucose DMEM with phenol red, supplemented with 20% FBS, mix of penicillin, streptomycin, amphotericin, and 2 mM Glutamax (Invitrogen)] and replated at about 10,000 cells/cm2, corresponding to a ratio of about 1:4–1:6. Cell numbers were counted in a Neubauer cell counting chamber, and PDT were calculated as described [22]. Cultures were observed and photographed using a phase-contrast light microscope (InvertoskopC), equipped with a digital camera (AxioCam MRc) and software (Axiovision Vs40, V4.2.0.0) (Zeiss, Thornwood, NY).
Cell migration and invasion in vitro
Pericytes were analyzed for their capability to migrate and invade in vitro. Assays were performed with the xCelligence Real-Time Cell Analysis (RTCA) DP instrument, RTCA software version 1.2.0.0909 (Roche Applied Science, Indianapolis, IN), and CIM-Plates 16, representing a modified Boyden chamber that allows monitoring of cell adherence through resistance measurement in real time. Cells in culture were starved by reducing the serum concentration from 20% to 0.1% for 20 h. 20,000 cells were plated per well in the upper chambers with a serum-free medium; chambers were either uncoated for cell migration or coated with collagen 1 for cell invasion analysis (Becton Dickinson). Lower chambers were filled with a medium containing 20% or 0% FBS. Background controls did not include any cells. Cell migration into the bottom chamber was monitored in real time over a 24-h period via embedded microelectrodes, measuring every minute for the first 10 h and every 5 min for the following 14 h.
Flow cytometric analyses of cultures
Cells propagated in culture for 2 or 3 passages were analyzed for their expression of various lineage specific surface markers. Single-cell suspensions were incubated with a blocking buffer containing 20% FcR block (Miltenyi Biotec), 0.5% BSA (Sigma-Aldrich), and 2 mM EDTA (Sigma-Aldrich) in DPBS without calcium and magnesium (Invitrogen), pH 7.2. Controls included nonstained cells, and cells incubated with corresponding isotype controls (Becton Dickinson). Labeled antibodies were ALP-AF647 (No. 561500), CD10-PECy7 (No. 341092), CD13-PerCPCy5.5 (No. 561361), CD44-V450 (No. 561292), CD73-PE (No. 550257), CD90-PerCPCy5.5 (No. 561557), CD105-FITC (No. 561443), CD34-AF700 (No. 561440), CD45-APCCy7 (No. 557833), CD56-PECy7 (No. 557747), and human leukocyte antigen (HLA)-DR-FITC (No. 556643), all from Becton Dickinson, and CD146-PE (No. 130-092-853), CD326-PE (No. 130-091-253), CD133/1-APC (No. 130-090-826), from Miltenyi Biotec. For analyses of intracellular α-SMA expression of cells at different passages in culture, cells were fixed, permeabilized, and stained for α-SMA as described above for the sorting of pericytes by flow cytometry. Cells were analyzed with a FACS Aria II (Becton Dickinson). Compensation beads (Becton Dickinson) were used to compensate potential spectral fluorochrome overlap. Cell debris and cell doublets were excluded by applying a forward versus side-scatter gate.
Differentiation in vitro
Cells were induced in culture to myogenic, adipogenic, chondrogenic, and osteogenic differentiation, as well as epithelial transition.
For myogenic differentiation, cells were cultured essentially as described previously [1]. Cultures were fixed with 4% paraformaldehyde (PFA) and washed; washing steps between stainings always included 3 washes with PBS 0.1% Triton X-100 (Alfa Aesar, Ward Hill, MA). Cells were incubated with a blocking buffer containing 10% goat serum (Invitrogen), 10% FcR block (Miltenyi Biotec), 0.1% Triton X-100 in PBS for 1 h. Cells incubated with an isotype-matched immunoglobulin served as negative controls. Cells were stained with a primary antibody to desmin (Dako) and secondarily with AF488 goat anti-mouse IgG, including DAPI for nuclei stain (Invitrogen). Images were acquired by confocal microscopy using an Olympus Fluoview 1000 system (Center Valley, PA).
For adipogenic differentiation, cells were cultured for about 2 weeks in the StemXVivo Adipogenic Medium (R&D Systems, Minneapolis, MD) and fixed with 4% PFA. Lipids were detected with an oil red O Stain Kit (ScyTek Laboratories, Logan, UT). Images were acquired with a phase-contrast light microscope (InvertoskopC), equipped with a camera (AxioCam MRc) and software (Axiovision Vs40, V4.2.0.0) (Zeiss).
For chondrogenic differentiation, 5×105 cells were pelleted at 300 g for 2 min and cultured in the StemXVivo Chondrogenic Medium (R&D Systems) for 2–3 weeks. Pellets were fixed with 4% PFA, embedded in paraffin, and sections were stained with alcian blue and nuclear fast red.
For osteogenesis, cells were cultured in the StemXVivo Osteogenic Medium (R&D Systems) for 2–3 weeks, and calcium was detected using a NovaUltra Alizarin Red Stain Kit (IHC World, Woodstock, MD).
For epithelial induction, 20,000 cells/cm2 were plated onto 3D, 1-mm-thick collagen I gels in 35-mm-diameter dishes (Becton Dickinson). After 24 h of attachment, the medium was switched to the William's Medium E supplemented with insulin, transferrin, selenium, hydrocortisone, glutamax, mix of penicillin, streptomycin, amphotericin, and 10% FBS (Invitrogen). The culture medium was changed after 1, 4, and 7 days. Supernatants were collected for analyses of secreted α-fetoprotein (AFP) and albumin. Cultures were observed and photographed using a phase-contrast light microscope. At day 7, cultures were either lysed with the RLT-buffer and subjected to gene expression analyses, or fixed with 4% PFA to perform immunocytochemistry for protein expression analyses.
AFP and albumin enzyme-linked immunosorbent assays
Cell culture supernatants of epithelial-induced cells were analyzed for secreted albumin and AFP by sandwich enzyme-linked immunosorbent assays (ELISA). Briefly, MaxiSorp Immunoplates (Nunc, Rochester, NY) were coated with the anti-human albumin antibody (Bethyl Laboratories, Montgomery, TX) or the anti-human AFP antibody (Abcam), respectively, incubated with samples or standards (albumin (Bethyl Laboratories), AFP (MP Biomedicals, Solon, OH), and conjugated with a goat anti-human albumin horseradish-peroxidase-conjugated antibody (Bethyl Laboratories) or a mouse anti-human AFP horseradish-peroxidase-conjugated antibody (Abcam), respectively. The Tetramethylbenzidine substrate solution was incubated for up to 10 min, the enzymatic reaction was stopped with 2M sulfuric acid (Fisher Scientific), and absorbance was read at 450 nm with a Power Wave 340 microplate reader equipped with KCjunior software version 1.6 (Bio-Tek, Winooski, VT).
Gene expression analyses
Gene expression was studied from short-term expanded, nondifferentiated perivascular cell cultures of passages 2 or 3, as well as from cultures that were subjected to epithelial induction. Cells in culture were lysed directly with the RLT-buffer, and RNA was isolated using shredder and isolation columns of RNeasy-mini kits (Qiagen, Valencia, CA). RNA was reverse transcribed to cDNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). Gene expression was analyzed by real-time polymerase chain reaction (PCR) using the StepOnePlus system and software, predesigned TaqMan probe assay mixes, and gene expression master mixes (Applied Biosystems). Beta-actin served as a housekeeping gene for internal normalization. Total human fetal liver and muscle RNA (Biochain) served as a relative quantitative normalizer. Expression was quantified using the ddCt method.
Immunocytochemistry
Cells in regular culture without induced differentiation were investigated for their protein expression of CD146 and α-SMA. Cell clusters in culture that underwent epithelial inductive conditions were analyzed for their expression of cytokeratin 8/18, cytokeratin 19, and E-cadherin. After fixation with 4% PFA, endogenous peroxidases were quenched for 1 h at RT with 1% hydrogen peroxide (Fisher Scientific). Washing steps between stainings always included 3 washes with PBS 0.1% Triton X-100. Cells were incubated with a blocking buffer containing 10% goat serum (Invitrogen), 10% FcR block (Miltenyi Biotec), and 0.1% Triton X-100 in PBS for 1 h. Cells incubated with isotype-matched immunoglobulins served as negative controls. Total human fetal liver cell cultures served as positive controls for cytokeratin 8/18, cytokeratin 19, and E-cadherin stainings. Cells were stained with primary antibodies to α-SMA (AnaSpec) and CD146 (Abcam), or cytokeratin 8/18, cytokeratin 19, and E-cadherin (all from Fisher Scientific). Secondary antibodies were AF488 goat anti-rabbit or -mouse, AF568 goat anti-rabbit or -mouse, and HRP goat anti-mouse or -rabbit in combination with TSA-AF555 or AF488; DAPI was included for nuclei stain (Invitrogen). Images were acquired by fluorescence microscopy using a Nikon Eclipse TE300 microscope (Tokyo, Japan) equipped with a ProgRes MF camera and software (Jenoptik, Jena, Germany).
Statistics
Data are given as means from biological repeats±standard deviation. The Student's t-test was used to analyze statistically significant differences in gene expression.
Results
Sorting of pericytes by flow cytometry
To investigate the frequency of pericytes in fetal and adult liver, we sorted out pericytes by FACS. We used a similar sorting strategy as described for other organs to ascertain comparability of results [1]. After exclusion of debris and cell doublets by applying a forward and side-scatter gate, we gated successively to obtain a CD146+CD34− fraction from a CD45−CD56− population. In parallel, we also analyzed the percentages of cells positive for ALP or α-SMA in this CD146+CD34−CD45−CD56− cell subset (Fig. 1). In fetal livers of gestational age between 10 and 20 weeks, we observed an average of 0.45%±0.39% (n=9) CD146+CD34−CD45−CD56− cells (fetal liver cell suspensions were depleted beforehand of red blood cells by lysis). In adult liver (donor ages 18, 56, and 75 years), we determined an average of 0.56%±0.81% (n=3) CD146+CD34−CD45−CD56− cells. By average, 30% (fetal) and 41% (adult) of liver CD146+CD34−CD45−CD56− cells were ALP+. 76.2%±22.8% (n=9) of fetal liver CD146+CD34−CD45−CD56− cells were also positive for α-SMA.

Sorting of pericytes from fetal
Morphology and PDT of pericytes in culture
At initial plating on gelatin, FACS-sorted pericytes exhibited a rather flat morphology, and cells grew to near confluence within 10 days (Fig. 2). Subsequently, passaged cells were cultured directly on plastic, and cells uniformly exhibited an elongated, spindle-shaped morphology, typical of purified pericytes. After initial passage, cells demonstrated a low average PDT of 63±16 h (n=7), which increased for the next passage to 849±162 h; an example of growth curves from 3 populations is given in Fig. 3. After this lag phase, however, subsequent passages demonstrated exponential cell growth with low average PDT of 51±27 h.
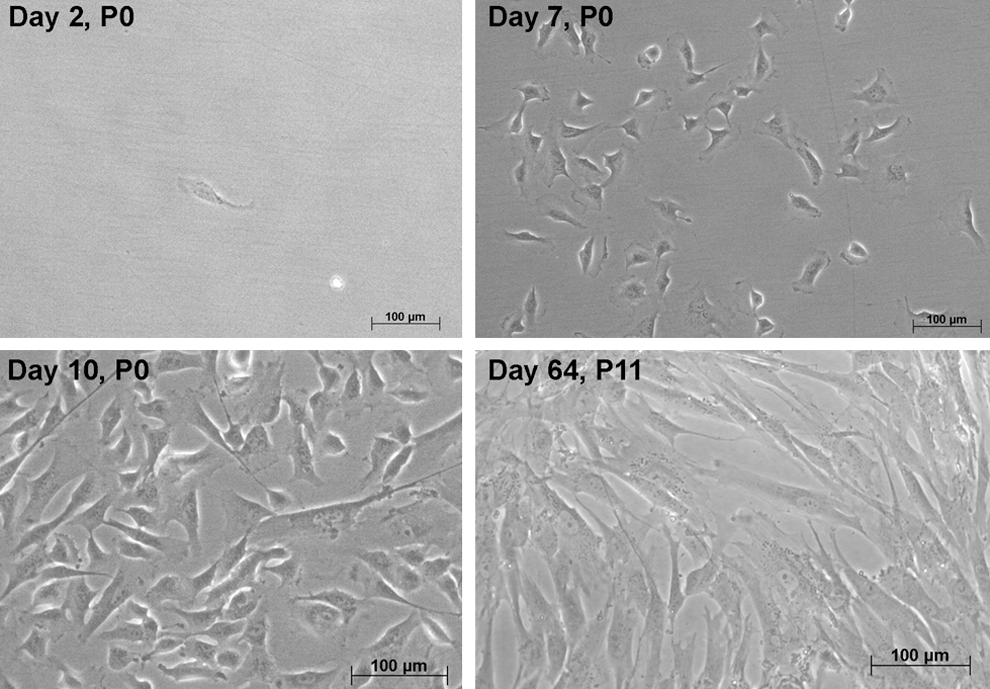
Cell morphology of cultured pericytes isolated by FACS. After initial attachment on gelatin-coated dishes, cells grew to near confluence within 10 days. In subsequent culture on plastic ware, cells exhibited more of a typical elongated, spindle-shaped morphology. Cells could be grown long term. P, passage.

Population doubling of pericytes in culture. Pericytes from human fetal livers were sorted out by FACS and cultured for 16 passages by average. Cell numbers were recorded after each passage; cell population doubling times were calculated from these data as described in the Materials and Methods section, and averaged 51 h. Example cultures derived from 3 different fetal livers are shown, each line representing cell numbers from one culture.
Immunohistochemical detection of hepatic pericytes in liver tissue
We next analyzed the in vivo localization and marker expression of pericytes on human fetal and adult liver tissue sections (Fig. 4). We found pericytes in fetal and adult liver sections. In both, pericytes were localized around blood vessels. Pericytes were detected in periportal vascular structures, but not pericentral ones. In adult and fetal, CD146-positive pericytes coexpressed NG2 (Fig. 4E, L), vimentin (Fig. 4A, H), and desmin (Fig. 4B, I), but did not coexpress the stellate cell protein GFAP (Fig.4C, J), the endothelial marker vWF (Fig. 4F, M), or hematopoietic CD34 (Fig. 4G, N). In immunohistochemistry analyses, CD146-positive pericytes were almost exclusively α-SMA-negative; α-SMA-positive cells did not express CD146 in fetal and adult tissue (Fig. 4D, K); we could detect, however, α-SMA coexpression in FACS analyses and variable α-SMA gene expression by PCR. Only in one tissue section from a 70-year-old donor, we found coexpression of α-SMA and CD146 (not shown).

Immunohistochemistry for pericytes in vivo. Sections of human adult (45–65 years donors) and fetal (16–20 weeks of gestation) livers were stained for coexpression of CD146 (green) with vimentin
Gene expression of pericytes
To analyze the expression of various mesenchymal, hematopoietic, endothelial, hepatic progenitor, and liver-specific genes, we used short-term cultured (passages 2 or 3), nondifferentiated pericytes isolated from fetal liver (Table 1). We also investigated gene expression in 3D epithelial-induced spheroids that formed in culture on collagen I gel. Expression is given relative to that detected in total fetal liver cell suspension for all genes. In case of MYF5 and PAX7, we could not detect any gene expression in fetal liver; therefore, these are given relative to total fetal muscle gene expression. We included total human fetal muscle tissue to enable comparison to published data on fetal muscle-derived human pericytes [1]. In undifferentiated cultures, we detected no or extremely low expression of hematopoietic, endothelial, myogenic, or epithelial/hepatic genes, for example, CD34, CD31, CD144, vWF, MYF5, PAX7, albumin, AFP, C3A7, CK19, CD326, and CD45. We detected strong expression of genes expressed in pericytes in other organs, that is, CD146, NG2, CD90, and CD140b, as well as the typical mesenchymal protein vimentin. α-SMA and myogenin gene expressions were quite variable between cultures; differences were not statistically significant, but on average were higher in pericyte cultures than in total fetal liver cell suspensions. GFAP, typically expressed by human liver stellate cells, could not be detected at all. Other neuronal or myogenic cell associated markers, such as nestin and TUBB3, could be only detected at a low level that was not statistically significantly different from that detected in total liver. We also investigated expression of CD44h, the hyaluronic acid-binding receptor, and CD44v, a variant form with abrogated hyaluronic acid binding. CD44h is expressed on human hepatic progenitors and liver mes-endodermal progenitors [23,24]. We could detect CD44h and CD44v expressions that were higher and lower, respectively, compared to total fetal liver, but these differences were not statistically significant.
Human fetal liver cell suspensions were sorted by FACS for CD45−CD56−CD146+CD34− pericytes. Cells were cultured short term in nondifferentiation conditions (HFLP undifferentiated) or in epithelial induction conditions (HFLP spheroids), and compared to human fetal muscle and liver tissue. Gene expression was analyzed by quantitative real-time PCR. Beta-actin gene expression served as endogenous normalizer, and data are given relative to human fetal liver (or muscle, for MYF5 and PAX7 as gene expression could not be detected in fetal liver). Gene expression of undifferentiated pericyte cultures is significantly higher (bold) or lower (italic) compared with total human fetal liver cells. Significant differences of gene expressions of undifferentiated pericyte cultures versus total human fetal liver cells, and of spheroid cultures versus undifferentiated pericyte cultures were determined by Student's t-test with * P<0.05 and ** P<0.01. Data are given as means from n=6 biological repeats (each measured in triplicate)±standard deviation.
This is not a complete listing of expressions, but it rather intends to point out typical positive cell/tissue types or lineages.
TaqMan assay used binds to exons 1 and 2, which are constant exons expressed by all isoforms, including CD44h and CD44v, that is, NM_001001392.1, NM_001001391.1, NM_000610.3, NM_001001390.1, NM_001001389.1, NM_001202557.1, NM_001202555.1, and NM_001202556.1.
TaqMan assay used binds to exons 10 and 11, which are variant exons that abrogate hyaluronic acid binding and are expressed only by variant (CD44v) isoforms NM_000610.3 and NM_001001389.1, but not CD44h [37].
vWF, von Willebrand factor; ASMA, alpha-smooth muscle actin; MELCAM, melanoma cell adhesion molecule; AFP, α-fetoprotein; GFAP, glial fibrillary acid protein; LHX, Lim-homeobox gene; HFM, human fetal muscle; HFL, human fetal liver; HFLP, human fetal liver pericytes; PCR, polymerase chain reaction; CNS, central nervous system.
Furthermore, we asked whether the mesenchymal pericytes could be induced to epithelial transformation, which is supported by their spontaneous and immediate formation of spheroidal structures strikingly resembling clusters of hepatic stem cell colonies (formed after epithelial induction of cultures of pericytes on 3D collagen 1 gel with the hepatic medium). We investigated changes in gene expression in these structures, compared to undifferentiated cultures of pericytes. Induced cultures demonstrated increased expression of cytokeratin 19, an epithelial-specific cytokeratin expressed by the liver biliary epithelium and hepatic stem cells. In addition, we observed an increase of genes expressed by undifferentiated pericytes and described to be expressed also by mes-endodermal progenitors, such as vimentin, CD90, and CD140b.
Immunocytochemistry of pericytes in culture
In culture of undifferentiated pericytes, we found variable α-SMA protein expression. Most cells expressed α-SMA and CD146 (Fig. 5).
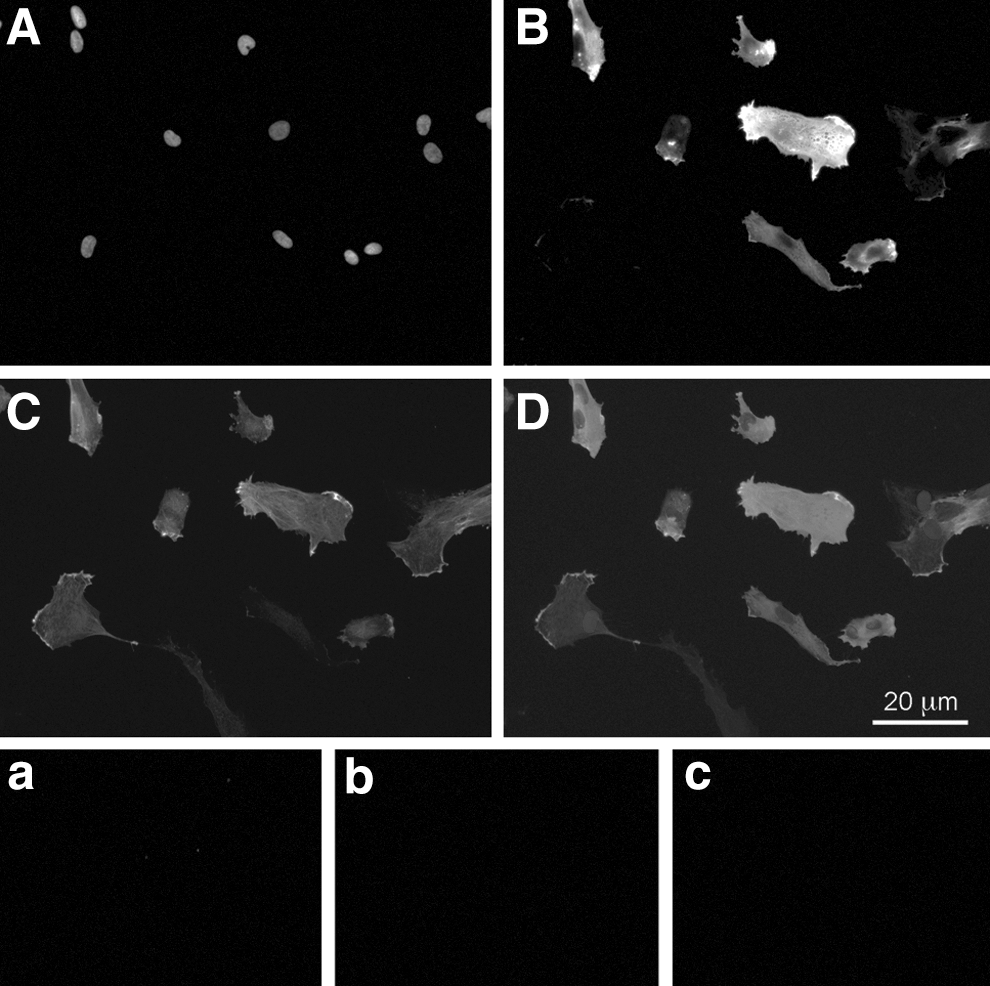
Immunocytochemistry of undifferentiated cultures of pericytes. Pericytes isolated from human fetal liver in undifferentiated culture were analyzed for their coexpression of CD146 and α-SMA. Single fluorescence stains
Further, we investigated if pericytes could be induced in culture to epithelial transition and myogenic, adipogenic, chondrogenic, and osteogenic differentiation (Fig. 6). Pericytes demonstrated considerable osteogenic (Fig. 6A) and myogenic (Fig. 6B), but low adipogenic (Fig. 6C) and low chondrogenic (Fig. 6D) differentiation potential. Cells subject to epithelial induction (Fig. 6E) formed instantly 3D spheroidal structures within one day of culture that remarkably resembled clusters of hepatic stem cell colonies, and demonstrated induced gene expression of epithelial-specific cytokeratin 19 (Table 1). Pericytes that were cultured under epithelial induction conditions on a collagen I gel with the hepatic medium were also investigated for secretion of hepatic serum proteins, albumin, and AFP, and were analyzed for expression of epithelial-specific proteins cytokeratin 19, cytokeratin 8/18, and E-cadherin by immunocytochemistry. We could not detect any secretion of albumin or AFP, or expression of epithelial-specific proteins in induced cultures.
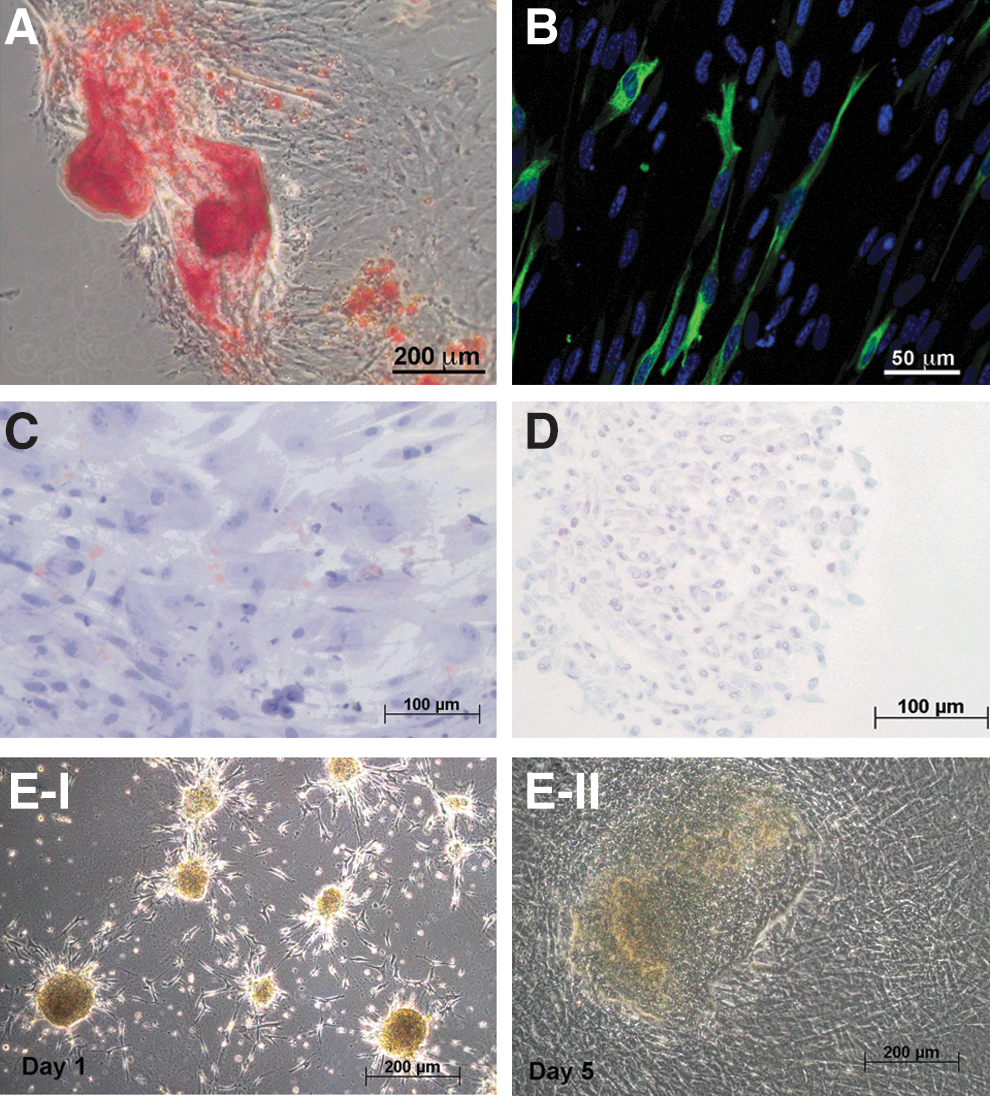
Differentiation of pericytes in culture. Cells in culture were subject to differentiation protocols.
FACS analysis of pericyte cultures
We analyzed by FACS a broad panel of surface marker expressions of cultured pericytes (Fig. 7). All cells expressed CD146, CD73, CD90, CD10, CD13, CD44, CD105, and were mostly ALP-positive. Cells lacked expression of HLA-DR. The majority of cells were negative for CD45 and CD34. Unexpectedly, some cells acquired CD56 expression. Very few cells were weakly positive for CD133 and CD326.

FACS of cultured pericytes. Cultures of pericytes of passage 4 were analyzed for surface expression of CD146, CD73, CD90, CD10, CD13, CD44, CD105, ALP, CD45, human leukocyte antigen (HLA)-DR, CD56, CD326, CD34, and CD133. Isotype controls are displayed as open black histogram, positive-stained cells as light gray filled histogram. Graphs are representative for 3 biological repeats.
We also analyzed the coexpression of CD146 and α-SMA (Fig. 8) of pericytes at different passages in culture to monitor potential changes over culture time. In the original, the sorted perivascular cell fraction (P0) 76.2%±22.8% (n=9) of fetal liver CD146+CD34−CD45−CD56− cells were also positive for α-SMA. After 2 passages, 82.1% of CD146-positive cells coexpressed α-SMA; after 4 passages, 87.0% of CD146-positive cells coexpressed α-SMA; at passage 13, 87.3% of CD146-positive cells coexpressed α-SMA. Although a slight increase in the percentage of α-SMA coexpressing cells could be observed from original sorted P0 cells (76.2%) to subsequent passages, this increase was not statistically significant (P=0.68, Student's t-test).
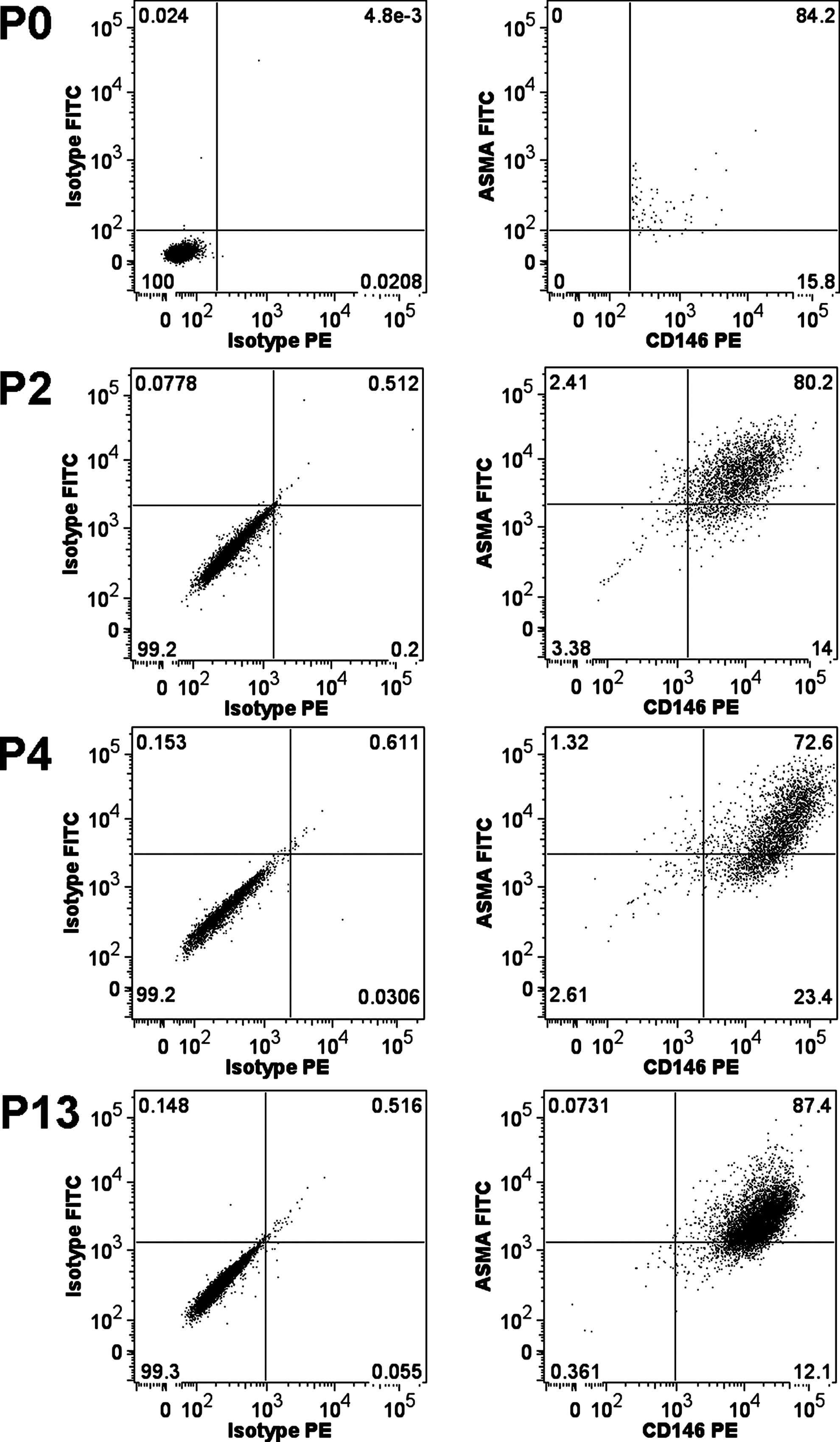
FACS for CD146 and α-SMA coexpression. Cell suspensions of freshly isolated, sorted pericytes (P0), as wells as cultures of pericytes from passages 2 (P2), 4 (P4), and 13 (P13), were analyzed for coexpression of CD146 and α-SMA. Isotype controls are displayed left, positives right.
Cell migration and invasion
The capability to migrate and to invade extracellular matrices in vitro and in vivo has been associated typical to MSCs. Therefore, we tested if pericytes are able to migrate and to invade collagen. We used the RTCA instrument, which represents a modified Boyden chamber that allows monitoring of cell adherence through resistance measurement in real time. Serum-starved progenitors in culture demonstrated a strong migratory activity (Fig. 9A) and invasion capability (Fig. 9B) toward 20% FBS containing a medium as attractant. Even without added FBS to the bottom chamber, cells demonstrated considerable migration and invasion toward the bottom chamber.

Cell migration and invasion. Sorted pericytes in culture were analyzed for their capability for migration
Discussion
Pericytes with multilineage differentiation potential have been isolated from several human tissues, including fetal muscle, pancreas, heart, skin, bone marrow, gut, lung, eyes, and adult pancreas, as well as umbilical cord and placenta [1,5], but not liver. These pericytes were defined by distinct surface molecule and intracellular marker expressions; sorting strategies included selection of a CD45− CD56− CD146+and CD34− population, which has been described as negative for typical endothelial, hematopoietic, or myogenic markers, such as CD144, vWF, CD34, or CD31, CD56, CD45, or Pax7. Depending on the tissue type, frequencies were between 0.29% and 1.79%, with a higher percentage in adipose tissue (14.6%).
We were able to identify, purify, and characterize pericytes from human fetal and adult livers using the same sorting strategies, resulting in recovery of pericytes at a similar percentage as previously described for other organs. Our data suggest that pericytes are distinct from hepatic stellate cells. In situ, CD146-positive cells were localized in the periportal region, but not pericentral or within the space of Disse, where hepatic stellate cells are located [25]. In addition, stellate cells in vivo are variably α-SMA-positive, and become strongly α-SMA-positive in fibrosis or in cell culture [8,10], acquiring a myofibroblast phenotype. Our immunohistochemical analyses demonstrated that CD146-positive cells in situ were α-SMA-negative. We could detect, however, by FACS, that 76.2% of freshly isolated CD146+CD34−CD45−CD56− cells coexpressed α-SMA, which is probably due to the more sensitive detection method of flow cytometry used to detect the intracellular expression of α-SMA. Cultures of FACS sorted pericytes revealed that α-SMA expression is maintained, although not homogeneously, as detected by immunocytochemical stains and gene expression analyses. Gene expression analysis and immunohistochemistry for the typical marker for mature stellate cells in liver, GFAP demonstrated no expression in pericytes.
A common mesodermal origin of stellate cells and perivascular mesenchymal cells was indicated in mouse liver [13,14]. During early liver development, stellate cells are assumed to originate in the septum transversum mesenchyme [26]; cells positive for the Lim-homeobox gene (Lhx2) migrate into the forming liver bud and become desmin- and Lhx2-positive stellate cells. We could detect Lhx2 expression in pericytes. Overall, pericytes have been suggested [1] to be one of the sources of MSCs [27 –29] and multipotent adult progenitor cells [30,31] found in the bone marrow and other organs. In the present work, we confirmed that liver pericytes shared phenotypical characteristics typically associated with MSC. Liver pericytes exhibited exponential growth rates, typical MSC surface marker and gene expression profiles and multilineage differentiation potential.
Additionally, we investigated if liver pericytes showed plasticity toward epithelial transition. Cells in culture on collagen-I gels formed 3D aggregates. These resembled human hepatic stem cell colonies by a phenotype [24] and human fetal liver multipotent mes-endodermal progenitors that were capable of differentiating into hepatocytes and bile duct cells as well as mesenchymal lineages [23]. Pericyte that were cultured under epithelial inductive conditions demonstrated induced gene expression of cytokeratin 19, CD44h, and CD90, but did not show albumin or AFP gene expression or protein secretion, nor did they express epithelial-specific proteins, such as cytokeratin 19, cytokeratin 8/18, or E-cadherin. Both, epithelial-to-mesenchymal transition (EMT) (for review, see Ref. [32]) as well as mesenchymal-to-epithelial transition (MET) [33] have been described in liver studies. EMT of hepatocytes and cholangiocytes has been suggested to play a role in the development of liver fibrosis [34,35]; recent studies using lineage tracing in a mouse, however, demonstrated that hepatocytes and cholangiocytes do not undergo EMT in various models of induced hepatic fibrosis [36]. The occurrence of MET in liver is under debate; only indirect evidence of MET has been demonstrated, verified by an acquired epithelial phenotype of fibroblasts after ectopic expression of HNFalpha [33]. While our results indicate a potential capacity of pericytes to undergo MET in vitro based on induced epithelial gene expression and morphology, we could not confirm this hypothesis based on more detailed analyses of epithelial and hepatic functionality (i.e., cytokeratin 19, cytokeratin 8/18, and E-cadherin protein expression by immunocytochemistry as well as albumin and AFP secretion). Further in vivo studies will be necessary to corroborate these findings.
In conclusion, we found that human adult and fetal livers harbor pericytes, which are similar to pericytes found in other organs and are distinct from hepatic stellate cells.
Footnotes
Acknowledgments
Financial support for this study was granted by the University of Pittsburgh Medical Center (UPMC). We thank Lynda Guzik of the Flow Cytometry Facility at the McGowan Institute for her excellent technical support. We thank Emma Rehm, Claire Keyes, Rebecca McDermott, Sarah Dittoe, and the rest of the staff of the Allegheny Reproductive Health Center for their enthusiasm and diligence in supporting the project.
Author Disclosure Statement
No competing financial interests exist.