
Abstract
In mammalian ovaries, many immature follicles remain after the dominant follicles undergo ovulation. Here we report the successful production of rabbit embryonic stem cells (ESCs) from oocytes produced by in vitro culture of immature follicles and subsequent in vitro maturation treatment. In total, we obtained 53 blastocysts from oocytes that received intracytoplasmic sperm injection followed by in vitro culture. Although only weak expression of POU5f1 was observed in the inner cell masses of in-vitro-cultured follicle-derived embryos, repeated careful cloning enabled establishment of 3 stable ESC lines. These ESC lines displayed the morphological characteristics of primed pluripotent stem cells. The ESC lines also expressed the pluripotent markers Nanog, POU5f1, and Sox2. Further, these ESCs could be differentiated into each of the 3 different germ layers both in vitro and in vivo. These results demonstrate that immature follicles from rabbits can be used to generate ESCs. Moreover, the use of rabbit oocytes as a cell source provides an experimental system that closely matches human reproductive and stem cell physiology.
Introduction
O
In vitro ovarian follicle culture has been used to produce developmentally competent oocytes from immature follicles isolated from mouse [4] and bovine ovaries [5,6]. In other species it is possible for embryos to develop to morula or blastocyst stages following in vitro culture of immature follicles [7 –12]; however, these embryos do not progress to later developmental stages.
Recently, it was reported that mouse ESC lines could be established from oocytes obtained from immature follicles. In these studies, the oocytes were cultured in vitro, subsequently maturated [in vitro maturation (IVM)], and finally either fertilized in vitro or parthenogenetically activated [13 –15]. If these technologies can be translated to other mammalian species, ESC use may become more widespread and progressive in medical/clinical research and in the livestock industry. However, there are currently no reports of successful ESC production from immature follicle cultures, except in mice.
A number of recent studies have provided evidence that there are dramatic differences between mouse ESCs and human ESCs [16 –18]. For example, mouse ESCs form dome-shaped colonies and require leukemia inhibitory factor (LIF) to maintain their pluripotency. In contrast, human ESCs form flat colonies and undergo basic fibroblast growth factor (bFGF)-MEK/Erk- and Activin-Smad-dependent self-renewal. At present, 2 stem cell types have been defined: one being the naive type (eg, mouse ESCs) and the other being the primed type (eg, human ESCs) [17]. Therefore, it is usually assumed that the results obtained from mouse studies cannot be extrapolated to humans. Thus, it is essential to develop technologies to generate, characterize, and utilize ESCs from species that show human-ESC-type characteristics.
Some recent studies have established that rabbit ESCs express human-ESC-like phenotypes in terms of colony morphology, expression of specific markers, and dependence on bFGF-MEK/Erk and Activin-Smad signaling for maintenance of pluripotency [19,20]. Moreover, rabbits have been used frequently as models for human osteoarticular [21,22], circulatory [23,24], and metabolic disorders [25,26]. Further, there are some practical advantages associated with rabbit models. Their larger body size enables certain surgical operations to be performed that are not possible with smaller rodents, such as mice and rats. Additionally, rabbits can be housed without the need for large expensive facilities that are required for other animal species that possess pluripotent stem cells with human-like characteristics (eg, pigs and monkeys). Therefore, the rabbit model is currently considered one of the most useful models for stem cell research [27]. In this report, we demonstrate that rabbit ESC lines can be generated from embryos produced by intracytoplasmic sperm injection (ICSI) and IVM of oocytes obtained from in vitro culture of immature rabbit ovarian follicles.
Materials and Methods
Animal use and care
All animal use complied with the regulations of the Institutional Animal Use and Care Committee of the Kinki University (IACUC, No. KABT-20-003). Each rabbit was housed in an individual pen with food and water ad libitum. Animals were exposed to an artificially controlled light–dark regime with 14 h of lighting and 10 h of darkness. Temperature was maintained between 20°C and 25°C in a ventilated room.
Collection of ovarian follicles, in vitro culture, and IVM
Female New Zealand White rabbits (Kitayama Labes Co. Ltd., Nagano, Japan) were superovulated by a subcutaneous injection of 80 IU of pregnant mare serum gonadotropin (PMSG; Sankyo Life Tech, Kanagawa, Japan) and then intravenously administered 60 IU of human chorionic gonadotropin (hCG; Teikoku Zoki, Tokyo, Japan) 72 h later. Rabbit ovaries were recovered 14 h after the hCG injection. Follicles of 200–399 μm in diameter at pre-antral to early antral stages were removed from the ovarian surface using a scalpel. To perform in vitro culture, oocyte-cumulous/granulosa cell complexes (OCGCs; Fig. 1A) were recovered from the follicles using 26-gauge needles (Fig. 1B). In our previous study, we confirmed that follicles <399 μm in diameter collected from rabbit follicles did not possess competence to resume meiosis after IVM without in vitro follicle culture [28]. Follicles <200 μm in diameter were too difficult to isolate without causing damage and could not be dissected for in vitro OCGC culture.
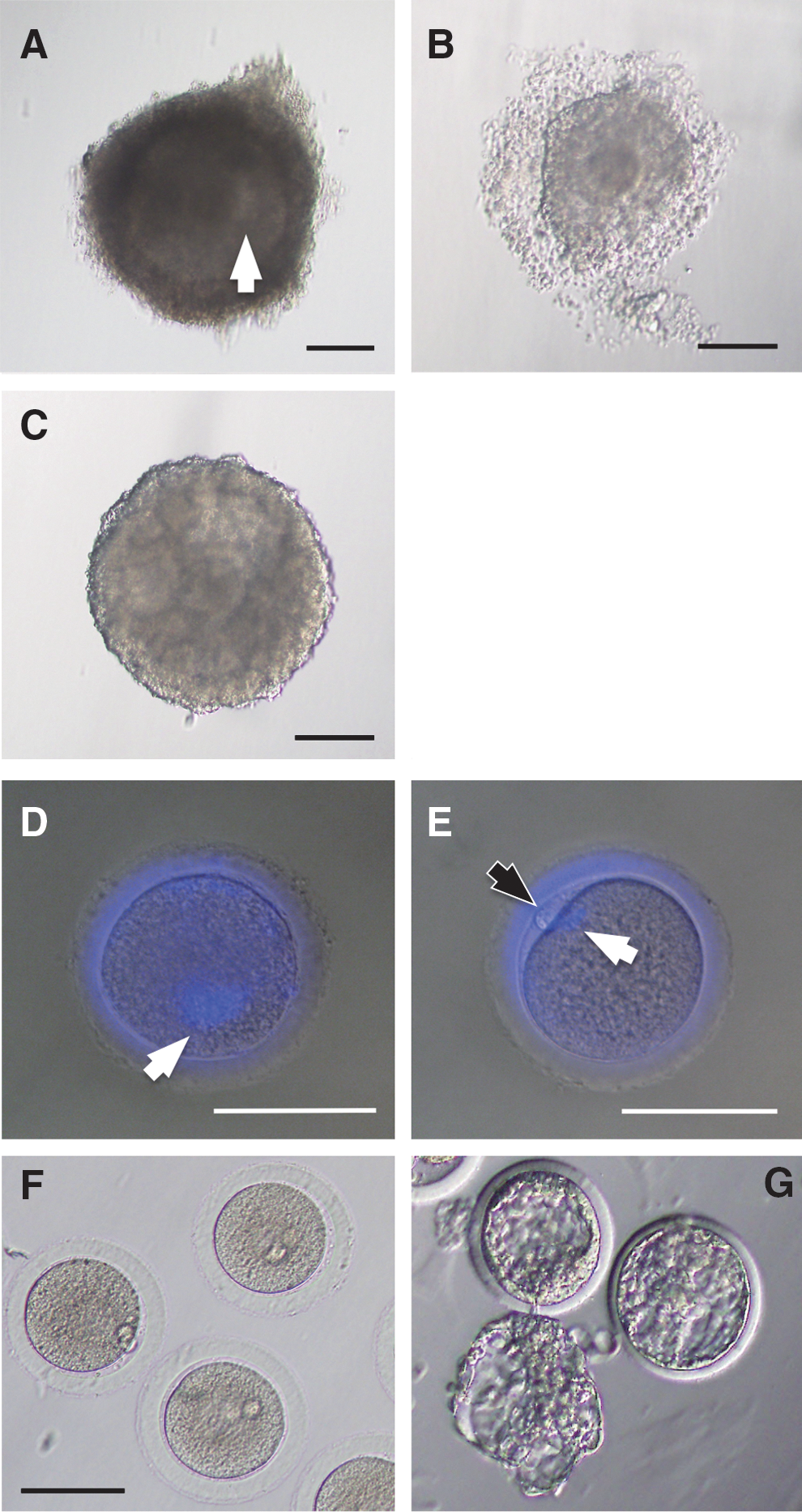
Morphology of follicles and cultured oocytes.
OCGCs were cultured individually in 50 μL droplets of follicle culture medium consisting of 0.05% fetal calf serum (FCS; Hyclone; Thermo Fisher Scientific, Waltham, MA), α-minimum essential medium (Invitrogen, Carlsbad, CA), 3 mg/mL bovine serum albumin (Sigma-Aldrich, St. Louis, MO), 50 μg/mL ascorbic acid (Sigma-Aldrich), 1% antibiotic/antimycotic solution (Sigma-Aldrich), and 1% insulin-transferrin-selenium-A solution (Invitrogen) under mineral oil at 37°C in a humidified atmosphere of 5% CO2 and 95% air for 8 days. Medium refreshment was performed every day. For inducing resumption of meiosis, denuded oocytes were transferred into 50 μL drops of maturation medium consisting of TCM 199 (Nissui Pharmaceutical, Tokyo, Japan), 0.2724 mg/mL water-soluble β-estradiol (Sigma-Aldrich), 0.1 mg/mL polyvinyl alcohol (average molecular weight 30,000–70,000; Sigma-Aldrich), and 10 ng/mL epidermal growth factor (Sigma-Aldrich) and cultured for 16 h at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
ICSI and embryo culture
ICSI has been well established in rabbits [29]. For oocytes that develop in vitro, it is necessary to assess their stage, that is, germinal vesicle (GV), metaphase I (MI), or metaphase II (MII) stage. To make such an assessment, granulosa/cumulus cells must be denuded. Therefore, conventional in vitro fertilization (IVF) was not appropriate for our study.
Seminal fluid was obtained from fertile male Dutch Belted rabbits (Kitayama Labes Co. Ltd.) using an artificial vagina. The seminal fluid was washed once in 5 mL of M2 medium followed by centrifugation at 750 rpm for 1 min. Medium was aspirated from the resulting sperm pellet that sank to the bottom in 2 mL of fresh M2 medium. Sperm that were capable of swimming upward from the pellet were selected and transferred to fresh M2 medium. Microinjection was conducted using a piezo-driven micromanipulation system (PRIME Tech Ltd., Ibaraki, Japan). The sperm that had translatory movement were transferred to 10% polyvinylpyrrolidone (SIGMA) and the sperm heads were isolated. The oocytes were positioned on the microinjection system with the first polar body at either 6 or 12 o'clock. An injection needle was then inserted at 3 o'clock and pushed across 3-quarters of the oocyte diameter to puncture the oocyte membrane by faint piezo-pulse. An isolated sperm head was then gently injected into the ooplasm.
Injected oocytes were washed twice in 50 μL drops of CMRL1066 medium (Invitrogen) supplemented with 30% FCS (Thermo Fisher Scientific K.K.), 0.55 mg/mL sodium pyruvate (SIGMA), 0.146 mg/mL
Establishment of the ESCs
Inner cell masses (ICMs) were obtained from blastocysts using immunosurgery. After removal of the zonapellucidae by treatment with 0.05% Pronase (Roche Diagnostics, Basel, Switzerland), embryos were cultured in 50 μL of 10% FBS-DMEM containing 5 μL of anti-rabbit guinea pig anti-serum for 20 min. The embryos were then transferred to 50 μL of guinea pig serum for 20 min. The isolated ICMs were individually seeded onto mitomycin-C-treated (10 μg/mL in medium for 90 min; Invitrogen) mouse embryonic fibroblast (MEF) feeder cells and cultured in rabbit ESC medium (ESM) consisting of 20% Knockout serum replacement (Invitrogen), DMEM/F12 (Invitrogen), 2 mM
Immunofluorescence evaluation
The cell cultures were fixed with Mildform 10N (Wako Pure Chemical Industries) at room temperature for 1 h. Fixed cells were washed with PBS, blocked with Block Ace (Dainippon Sumitomo Pharma, Osaka, Japan) for 1 h, washed twice more, and incubated with each primary antibody (Table 1) overnight at 4°C. The samples were then washed twice and incubated with secondary antibodies (FITC or Texas red-conjugated polyclonal antibodies; all were purchased from Santa Cruz Biotechnology, Santa Cruz, CA). Samples were counterstained with DAPI (1 μg/mL in PBS) prior to direct observation. Negative controls were prepared omitting the primary antibodies (data not shown).
IF, immunofluorescence; WB, western blot analysis.
Western blot analysis
Cells were collected by scraping, homogenized in SDS buffer (4% SDS, 125 mM tris-glycine, 10% 2-mercaptoethanol, and 2% bromophenol blue in 30% glycerol), and then subjected to polyacrylamide gel electrophoresis (PAGE) in the presence of SDS (SDS/PAGE), followed by electrotransfer onto PVDF membranes (Hybond-P; Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom). Feeder layers (MEF) were used as a negative control. The blotted membranes were blocked overnight with Block Ace (Dainippon Pharmaceutical, Osaka, Japan) and treated with each primary antibody (Table 1) overnight at 4°C. Detection was realized by enhanced chemiluminescence with an ECL plus western blotting detection system (Amersham Pharmacia Biotech) and horseradish peroxidase (HRP)–conjugated secondary antibodies (all were purchased from Santa Cruz Biotechnology) corresponding to each primary antibody. The lumino-labeled membranes were analyzed using an LAS 4000 CCD-based chemiluminescent analyzer (Fujifilm, Tokyo, Japan).
RNA extraction, RT-PCR, and quantitative real-time polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. One microgram of total RNA was used for cDNA synthesis with random hexamers. Products were filled up to 100 μL with Tris-EDTA (TE), and used at 1 μL per reaction for RT-PCR or quantitative real-time PCR (qRT-PCR). RT-PCR was carried out using a Thermal Cycler Dice® (Takara Bio, Inc., Shiga, Japan). Nanog, POU5f1, Sox2, and GAPDH amplifications were performed with a denaturation step at 95°C for 10 min, followed by 35 cycles of denaturation at 95°C for 20 s, primer annealing at 58°C for 20 s, and extension at 72°C for 20 s. qRT-PCR was carried out using Perfect real-time SYBR green II (Takara Bio, Inc.) with rabbit-specific primers (Table 2) in a Thermal Cycler Dice Real Time System (Takara Bio, Inc.). Reactions were performed with a denaturation step at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s and primer annealing and extension at 60°C for 30 s. Reactions were run in duplicate in 3 independent experiments. To quantify the relative expression of each gene, Ct (threshold cycle) values were normalized to an endogenous reference (ΔCt=Ct target – Ct internal control) and compared with the mean score from the calibrator (control sample) using the ΔΔCt method (ΔΔCt=ΔCt sample – ΔCt mean of calibrator). The mean score of the 28sRNA housekeeping gene was used as an internal control to normalize the variability in expression levels because of its sensitivity. Stability of gene expression across cell lines and validation of the internal control are provided in Supplementary Fig. S1 (Supplementary Data are available online at
For comparison of pluripotency-related gene expression levels between the ICMs from in vitro follicular-culture-derived embryos and the ICMs from control embryos, ICMs were paired to increase the total amount of RNA available for analysis. ICMs were collected by immunosurgery as described previously, and 3 pairs (in total 6 blastocysts) were analyzed for each group. For qRT-PCR of ICMs, quantification of total RNA was not performed and all extracted RNA was used for reverse transcription. In this experiment, cDNA products were filled up to 20 μL in TE and used at 1 μL per reaction for qRT-PCR.
For comparison of pluripotency-related gene expression levels between fESCs and normal ESCs, we used 2 previously established normal rabbit ESC lines [20]. The normal ESCs and the fESCs were cultured on feeder layers in ESM supplemented with bFGF for 72 h after passaging. ESC colonies were collected using CTK colony-digestion solution and treated with TRIzol reagent for RNA isolation.
Inhibitor-based determination of the signaling cascades related to pluripotency maintenance in fESCs
Confluent fESCs were passaged onto Matrigel (BD Falcon, Bedford, MA) and cultured in MEF-conditioned rESC-medium (rESM) for 24 h to eliminate feeder cells. The medium was then replaced with rESM containing 8 ng/mL bFGF, 1,000 U/mL LIF (Millipore, Billerica, MA), and 20% MEF-conditioned medium. For inhibition of signal transduction, 1 μM JAK inhibitor I (Merck, Darmstadt, Germany), 1 μM MEK/Erk inhibitor PD0325901 (Stemgent, Cambridge, MA), or 0.5 μM ALK4/5/7 inhibitor A83-01 (Stemgent) was added to the culture medium. After 48 h, cultures were collected and analyzed for the pluripotency marker gene Nanog using qRT-PCR.
Evaluation of multiple differentiation potentials
For in vitro differentiation, fESC colonies were collected with CTK solution, washed once with fresh ESM, and then transferred into suspension culture in 10% FCS–supplemented DMEM (Invitrogen). After 6 days in suspension culture, embryoid bodies (EBs) formed from the fESCs were collected and replated onto gelatin-coated culture dishes. The cells derived from the EBs were evaluated by immunofluorescent staining with specific antibodies following 10 days in culture.
Differentiation properties of the fESCs were evaluated in vivo via teratoma formation assay. Cell suspensions containing 5×106 fESCs were subcutaneously injected into the femora of severe combined immunodeficient (SCID) mice. After 8 weeks from the time of cell injection, teratomas were collected, fixed by Mildform 10N (Wako Pure Chemical Industries) overnight at room temperature, dehydrated, and embedded in paraffin.
Immunohistochemical staining
Immunohistochemical (IHC) staining was performed on 6-μm-thick deparaffinized sections. Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxide. Next, the sections were blocked with Block Ace and incubated with primary antibodies (Table 1) overnight at 4°C. After washing, HRP-conjugated secondary antibodies were applied, and samples were further incubated for 30 min at room temperature. Finally, the slides were visualized by DAB immunostaining using the Liquid DAB+ substrate Chromogen System (DAKO JAPAN, Kyoto, Japan).
Statistical analysis of the data
In experiments comparing gene expression levels between ICMs, scores obtained by ΔΔCt calculations were analyzed by Student's t-test. For comparing gene expression levels between ESC lines, scores obtained by ΔΔCt calculations were analyzed by Tukey–Kramer HSD test or Student's t-test. Both statistical analyses were performed with JMP software version 7.0.2 (SAS Institute, Cary, NC). P-values <0.05 were considered significant.
Results
Production of MII oocytes, blastocysts, and ESCs by in vitro follicle culture, IVM, ICSI, and ICM culture
We obtained 264 MII oocytes from 349 early antral follicles (75.6%). All oocytes included in the preculture follicles were in GV stage when evaluated by nuclear staining with DAPI (Fig. 1D), and these proceeded to MII stage after in vitro culture and IVM (Fig. 1E). After ICSI and embryo culture, 53 embryos reached the blastocyst stage (20.1%, from MII oocytes). These blastocyst embryos were morphologically normal, and some of them hatched at the end of the culture period (Fig. 1G). We isolated ICMs from the blastocysts by immunosurgery. In total, 24 ICMs were collected.
We first examined whether ICMs obtained from follicular-culture-derived embryos (fICMs) possessed the pluripotency necessary for establishment of ESC lines. To confirm pluripotency, we performed real-time PCR analysis for genes related to pluripotency using ICMs collected by immunosurgery. No statistically significant differences were observed for Nanog, Sox2, and Klf4 expression; however, the expression of the pluripotency-related gene POU5f1 was significantly lower and the early differentiation marker GATA6 expression was significantly higher in fICMs (Fig. 2A). To examine the possibility that differential gene expression of POU5f1 might influence the appearance of undifferentiated cells, we performed in vitro culture of ICMs on MEF feeder layers and observed POU5f1 expression levels. After 4 days in culture, ICMs exhibited growth and expansion out of the ICMs. Although no significant morphological differences were observed between the fICMs and the control ICMs, observation of immunofluorescence revealed that POU5f1 was expressed at lower levels in fICMs (Fig. 2B). In control ICMs, many POU5f1-expressing cells appeared at the initial ICM attachment point as well as in the outgrowth regions. In fICMs, on the other hand, only a few POU5f1-expressing cells emerged in the center area of the original ICM cell clump and even fewer were observed in the outgrowth regions. After isolation of the undifferentiated areas from the cultured fICMs, cells were passaged onto fresh feeder layers. These cells gave rise to very heterogeneous colonies consisting of many differentiated cells and a few undifferentiated cells (Fig. 2C). This high heterogeneity continued during early passages at least up to passage 6. However, continued careful cloning enabled establishment of stable undifferentiated cell lines. In total, 3 stable cell lines were established from 24 fICMs by this method.

Analysis of pluripotency-related gene expression in the fICMs.
Evaluation of fESC pluripotency
To evaluate the pluripotency of fESCs, we performed an ALP activity assay, as well as immunofluorescence, RT-PCR, and western blot analyses. All 3 cell lines formed flat colonies and individual cells exhibited prominent nuclei. All 3 cell lines showed high ALP activity and expressed the pluripotent cell markers POU5f1, SSEA1, and SSEA3 (Fig. 3A). RT-PCR analysis and western blot analysis confirmed that the fESCs expressed these markers. Nanog, POU5f1, and Sox2 were detected by RT-PCR (Fig. 3B) and Klf4, POU5f1, and Sox2 were detected by western blot (Fig. 3C). Nanog could not be detected by western blot because there were no specific antibodies available for rabbit Nanog protein. We then compared expression levels of the pluripotent-related genes Nanog, POU5f1, and Sox2 among the fESCs and the 2 normal rabbit ESC lines at passages 20 to 24. The expression levels were the same among the normal rabbit ESCs and the 3 fESC lines (Fig. 4A).

Characterization of the fESCs.
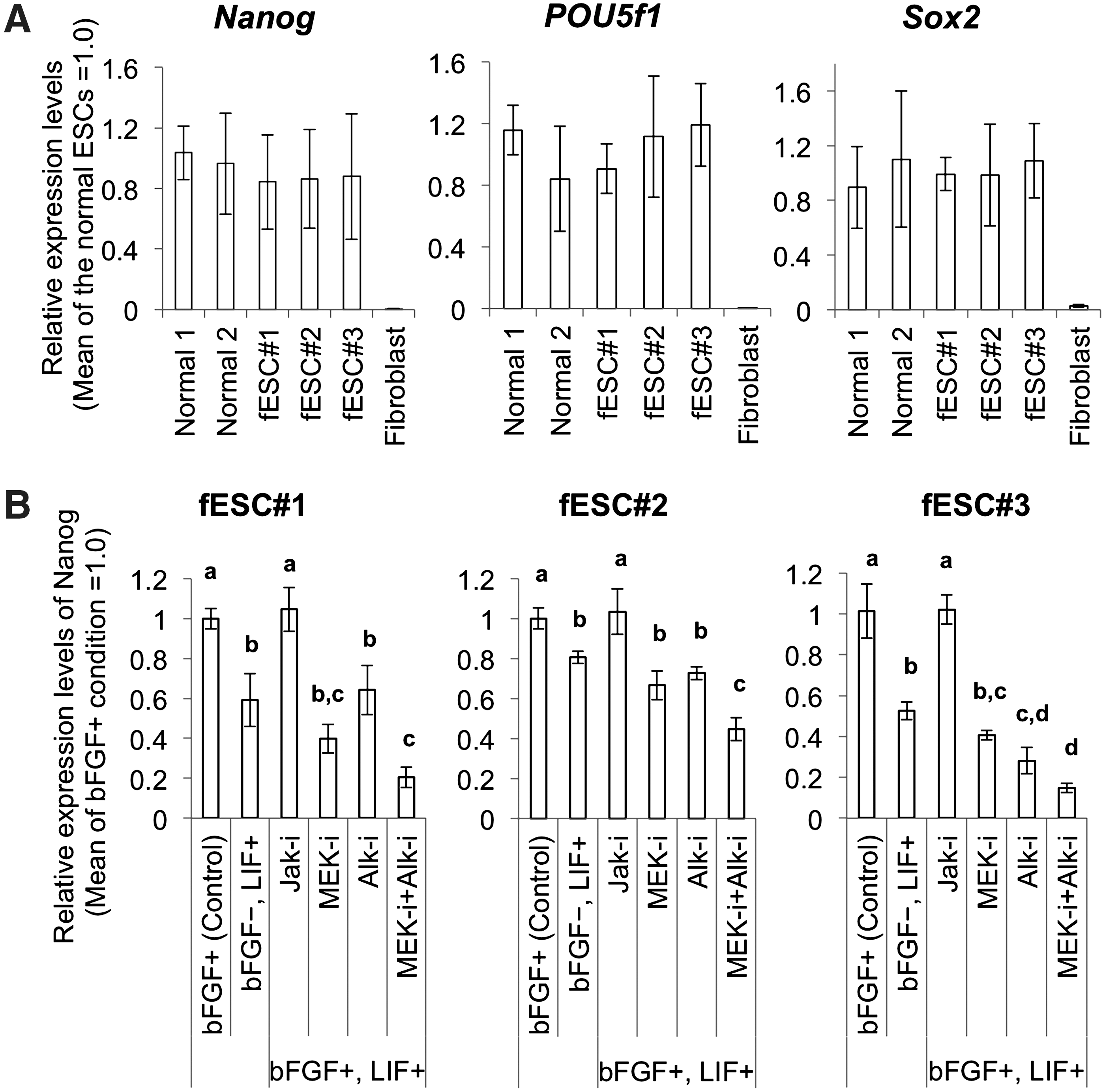
Quantitative real-time polymerase chain reaction gene expression analysis for characterization of fESCs.
Next, we blocked 3 independent signal transductions pathways related to ESC self-renewal to determine which signaling cascades were required to maintain fESC pluripotency. Nanog expression was lower for fESCs cultures treated with LIF, than that for fESCs cultures treated with bFGF. We next cultured fESCs in MEF-conditioned medium containing both bFGF and LIF and 1 of 3 inhibitors: either a JAK inhibitor to block LIF-JAK-Stat3 signaling, a MEK inhibitor to block bFGF-MEK/Erk signaling, or an ALK inhibitor to block Activin-Smad signaling. JAK inhibition did not affect Nanog expression, whereas MEK or ALK inhibition caused a significant decrease in Nanog expression. Combined application of MEK and ALK inhibitors resulted in even lower expression of Nanog. These results demonstrate that our fESCs rely on LIF-independent signaling pathways that involve both bFGF-MEK/Erk signaling and Activin-Smad signaling to maintain their pluripotency, similar to what has been observed for other rabbit ESCs and human ESCs (Fig. 4B).
fESCs possess multiple differentiation potentials both in vitro and in vivo
To analyze the differentiation potential of the fESCs, we performed suspension culture to induce EB formation. After 6 days in suspension culture, the ESCs formed cystic EBs. Ten days after attachment to culture dishes, the EBs differentiated into various types of cells, including fibroblasts, neurons with axons, and beating cardiac cells. Immunofluorescent analysis using anti-TuJ, anti-α-fetoprotein, anti-desmin, and anti-cardiac troponin T antibodies revealed that neural cells, endodermal cells, and cardiac myocytes were formed from each of the 3 fESC lines (Fig. 5). To test this in vivo, we transplanted 2 fESC clones (#1 and #2) into the femurs of SCID mice. In all cases, solid teratoma formation was observed after 8 weeks. Hematoxylin-eosin stain (HE) staining and IHC staining revealed that these teratomas contained various tissues derived from all 3 germ layers, such as epidermal tissues (ectoderm), TuJ-expressing neural tissues (ectoderm), desmin-expressing muscular tissues (mesoderm), and intestinal epithelium (endoderm) (Fig. 6).
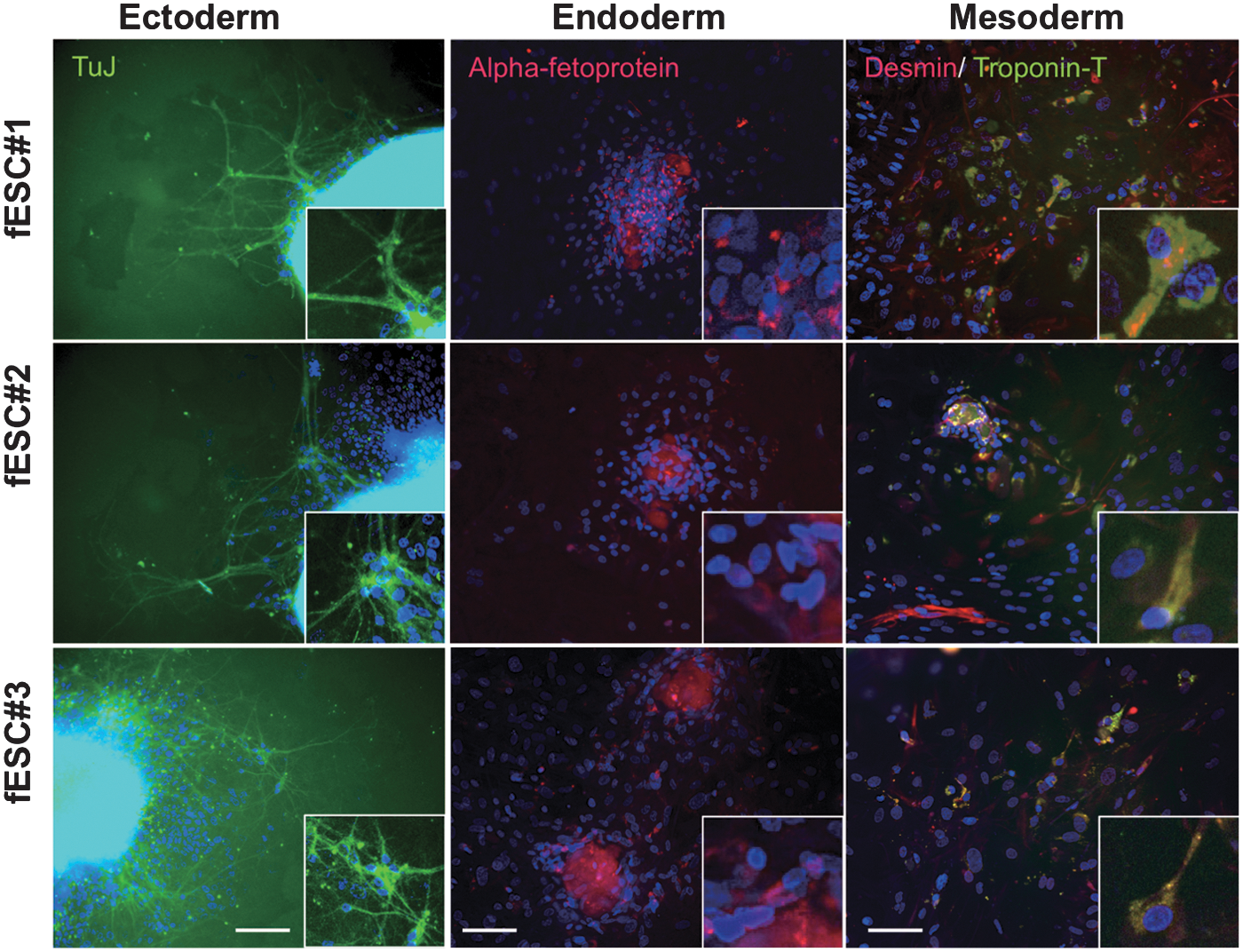
Embryoid body (EB)–mediated in vitro differentiation of fESCs. All panels show images of differentiated cells 10 days after EB attachment. TuJ-expressing neural cells (left), α-fetoprotein-expressing endodermal cells (middle), and Desmin- and cardiac troponin T–expressing cardiac myocyte (right) are shown. Scale bars=200 μm.
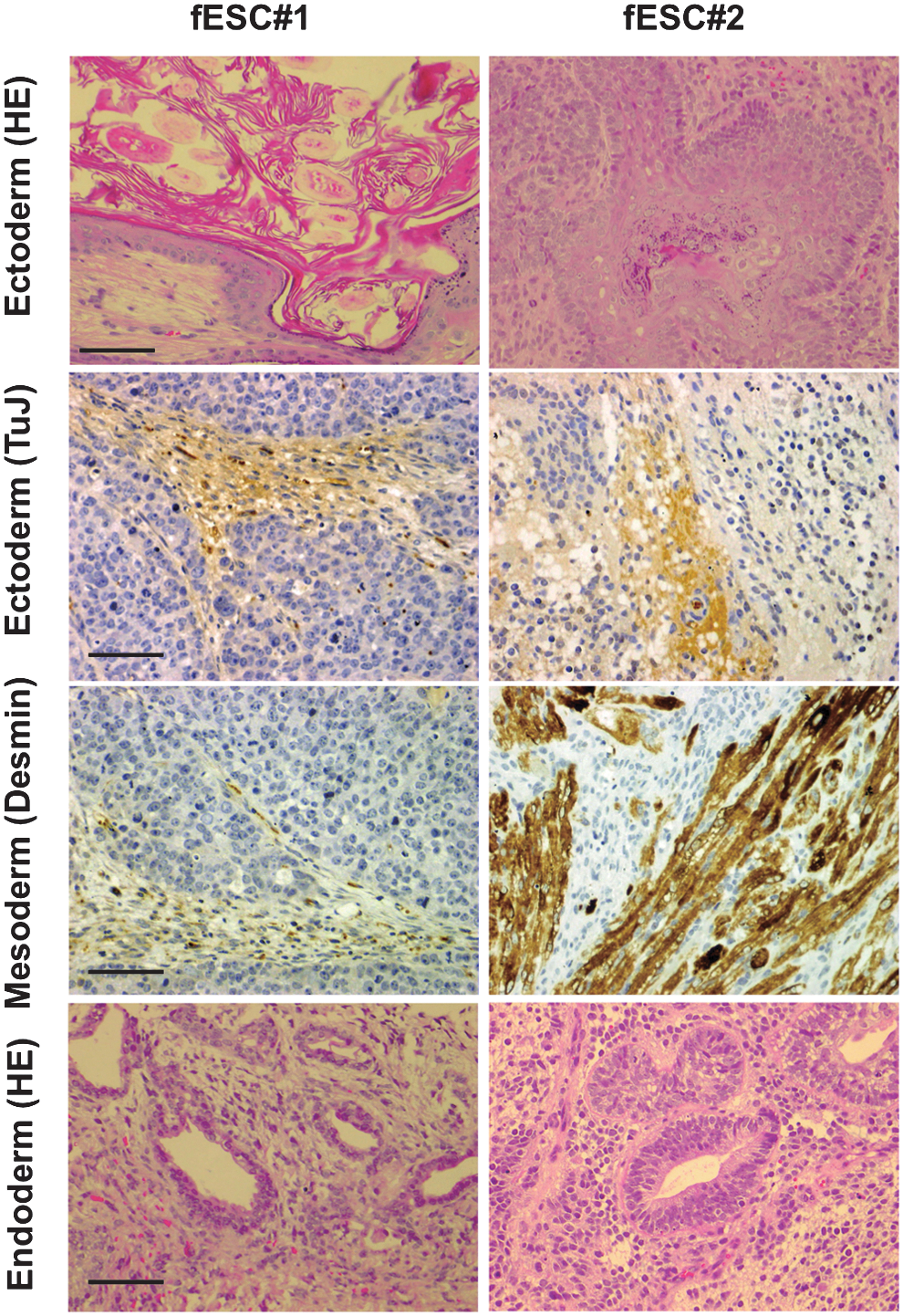
Histological evaluation of teratomas derived from the fESCs (#1 and #2). Images show epidermal tissues, TuJ-expressing neural tissues (immunohistochemistory; IHC), Desmin-expressing muscular tissues (IHC), and intestinal epithelium (HE). Scale bars=200 μm.
Discussion
To provide another valuable source of oocytes for clinical use and research, it is necessary to examine the potency of the ovarian follicle. Here, we demonstrated that it is possible to establish ESC lines from immature ovarian follicles in rabbits. By using postovulated ovaries treated with gonadotropins, we think it may be possible to extrapolate some of our results to humans, where oocytes are typically collected following superovulation, although further studies will be required to verify this assertion. At the end of the study, we established fESCs that met the evaluation criteria for rabbit undifferentiated ESCs; that is, they exhibited ALP activity and expressed SSEA1, SSEA3, and a variety of other pluripotent markers. The fESCs were capable of differentiating into 3 different germ layers both in vitro and in vivo. This is the first report of ESC production from immature follicular oocytes obtained from postovulated ovaries in a nonmouse species. As described earlier, rabbit ESCs seem to possess human-type pluripotent stem cell–like characteristics at least in terms of colony morphology, marker expression, and signal transduction pathways essential to maintain pluripotency. Therefore, the fact that the ESCs established from ovarian follicular oocytes are obtainable from rabbits is important for the stem cell research field.
We recently developed a method for obtaining oocytes that develop to the blastocyst stage from rabbit ovarian follicles via in vitro follicle culture [28]; therefore, we attempted to utilize this system for establishing ESCs. First, we determined the pluripotency of cells in rabbit ICMs following immunosurgical isolation using qRT-PCR for pluripotency-related genes. We found that the expression level of POU5f1 was significantly lower in ICMs from follicular-oocyte-derived embryos. Subsequent experiments showed that this low level of POU5f1 expression was carried over through early passages in vitro for ICMs cultured on MEF feeder layers. The fICMs gave rise to very heterogeneous populations of cells, including many differentiated cells. It should be noted, however, that this phenomenon usually does not occur during establishment of ESC lines, even from control (normal) embryos.
Until now, it has been accepted that POU5f1 (also known as Oct4) expression persists throughout peri-implantation development as a major factor involved in regulating pluripotency [31 –34]. In mouse embryo development, Oct4 expression competes with Cdx2 to determine the fate of cells [ICM or trophectodermal cell (TE) fates]. Reduced Oct4 expression results in enhanced Cdx2 expression, committing cells to the TE fate [31,35]. However, some reports suggest that the contradictive relationship between Oct4 and Cdx2 may be species specific. For example, Harvey et al. monitored expression patterns of Oct4, Nanog, and Cdx2 in blastocysts from cynomolgus monkeys and did not observe a contradictive relationship between Oct4 and Cdx2 [36]. In our study using rabbits, Cdx2 expression was not detected in the fICMs, even though relatively low levels of POU5f1 expression were observed, suggesting that Oct4 and Cdx2 may also not follow a contradictive relationship in rabbits. Recently, Kobolak et al. analyzed expression of rabbit POU5f1 in detail. They reported that POU5f1 is expressed in the TE in addition to the ICM [37]. This expression pattern was similar to other mammalian models, including monkey, porcine, and bovine, although expression of POU5f1 was much higher in the ICM. Interestingly, it has also been demonstrated that if Oct4 is removed from Cdx2-KO mouse ESCs, then the ESCs initiate differentiation [31]. This clearly indicates that Oct4 has important functions besides determination of cell fate between ICM cells or TE cells in combination with Cdx2.
Oct4 is part of the pluripotency-maintaining core circuit with Nanog and Sox2 [33,38]. Nanog and Sox2 are also expressed in the ICM and act as important elements for establishment and control of pluripotency. However, at least 3 facts obscure the indispensability of Sox2 and Nanog in establishment of pluripotency. First, deletion of Sox2 can be partially compensated for by other Sox family members, such as Sox4, Sox11, and Sox15 [32,39,40]. Second, forced expression of Oct4 can rescue the loss of pluripotency in ESCs triggered to differentiate by Sox2 deletion [32]. Finally, pluripotency of mouse ESCs can be maintained even if Nanog is removed [33,41]. From these 3 lines of evidence, Oct4 function can be thought of as the most important factor involved in pluripotency. Thus, it is likely that the POU5f1 (Oct4) depletion observed in our system may directly alter the outgrowth of ICM cells, resulting in heterogeneous cell populations containing many differentiated cells.
Once the undifferentiated cells were isolated by repeated cloning and entered a stable cycle of self-renewal, they expressed pluripotent cell markers at comparable levels to normal rabbit ESCs and could be stably maintained for at least 50 passages. Recently, it was reported that heterogeneous populations of ESCs contain at least 3 types of cells [42,43]. Pluripotent cells, differentiated (or differentiating) cells, and transitory cells can appear in the same culture and the frequency at which they appear can vary according to culture conditions and cell origins. If these findings from mouse ESCs are also applicable to rabbit ESCs, then it is highly possible that the ESC lines we established were the result of multiple selections for undifferentiated cells.
After entering a phase of stable proliferation, no significant differences in gene expression levels were observed for pluripotent cell markers among the fESC lines and the 2 normal rabbit ESC lines. To permit comparison of pluripotency among all 5 cell lines (3 fESCs and 2 rESCs), we maintained the cells on feeder layers. This prevented bias due to differential vulnerability of cell lines to feeder-free culture. All fESC lines showed higher expression levels of the pluripotent marker genes than that of somatic cells, although the variation in expression level was somewhat large. This large variation can be attributed to unavoidable factors, such as contamination with feeder cells or unevenness in quality of the feeder layers. There are 2 possible reasons why the fESCs showed comparable pluripotency gene expression to control ESCs. Either cloning-mediated selection of the ESCs possessing typical ESC characteristic as described previously or alteration of the epigenetic status of the cells may have occurred. In mouse and human ESCs, epigenetic status can be affected by in vitro culture [44]. Surprisingly, Horii et al. reported that epigenetic modification can occur within as few as 5 passages, over which time differences in expression patterns between ESCs established from embryos produced via IVF and in ESCs from in vivo embryos can disappear [45]. In our study, it is possible that in vitro culture of the fESCs altered their epigenetic profile, resulting in expression characteristics that were comparable to control ESCs.
To determine whether the pluripotency of the fESCs was of a primed pluripotent form, we performed signal transduction blocking studies. These studies demonstrated that the fESCs maintained pluripotency through bFGF-MEK/Erk and Activin-Smad signaling mechanisms characteristic of normal rabbit ESCs and other primed-form ESCs. To isolate the effects of inhibitors on fESCs, the fESCs were first transferred into feeder-free conditions. Reactivity of the fESCs to inhibitors was generally consistent, but subtle differences between cell lines were observed. It is possible that the fESC lines possessed a diverse range of characteristics in terms of sensitivity to the presence of cytokines or signal transduction inhibitors, similar to what has been observed for human ESCs, in which the reactivity to Activin varies among ESC lines [46]. Based on the fact that the fESCs showed typical rabbit ESC characteristics, including human-ESC-like pluripotency [19,20], our results provide important evidence to support the argument that ovarian follicles could be a valuable source for ESC generation.
From another viewpoint, our results provide valuable information about the developmental properties of embryos produced from in vitro ovarian follicle culture. No viable newborns have yet been reported from the in vitro culture of immature follicles from rabbits [28] and it is known that ICM pluripotency is a prerequisite for fetus formation. Although limited development to blastocyst stages has been reported for rabbit embryos, the pluripotency of embryonic ICM cells has not been confirmed in immature follicular cultures. In our study, the fact that ESCs could be generated from the ICMs of rabbit embryos derived from in vitro culture of immature follicles provides indirect evidence that the ICMs of embryos produced in follicular culture systems are pluripotent. This finding is important because successful embryo development to the blastocyst stage from ovarian immature follicular culture systems has been reported for species that are of importance to the livestock industry and that can also serve as model systems for reproductive medicine, including bovine [5,6], porcine [7,8], and buffalo [9]. In porcine [47,48] and bovine [49], procedures for obtaining pluripotent ESCs have already been developed. Further, Xu et al. recently succeeded in producing MII oocytes from immature secondary follicles by in vitro culture [50]. Although it may be difficult to obtain developmentally competent oocytes from primates, the techniques for ESC production have been well established in these species [51 –53]. Therefore, application of our technique for deriving ESCs from immature follicles may lead to advancements in ESC research that are both economically and clinically important.
Footnotes
Acknowledgments
This work was supported in part by Grant-in Aid for Young Scientists B (KAKENHI, 23791670) from the Japan Society for the Promotion of Science. We thank Ms. Kanae Shigi, Kinki University Faculty of Medicine, for excellent technical assistance.
Disclosure Statement
The authors have nothing to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.