
Abstract
Parathyroid hormone (PTH) anabolic osteoporosis therapy is intrinsically limited by unknown mechanisms. We previously showed that disabling the transcription factor Nmp4/CIZ in mice expanded this anabolic window while modestly elevating bone resorption. This enhanced bone formation requires a lag period to materialize. Wild-type (WT) and Nmp4-knockout (KO) mice exhibited equivalent PTH-induced increases in bone at 2 weeks of treatment, but by 7 weeks, the null mice showed more new bone. At 3-week treatment, serum osteocalcin, a bone formation marker, peaked in WT mice, but continued to increase in null mice. To determine if 3 weeks is the time when the addition of new bone diverges and to investigate its cellular basis, we treated 10-week-old null and WT animals with human PTH (1–34) (30 μg/kg/day) or vehicle before analyzing femoral trabecular architecture and bone marrow (BM) and peripheral blood phenotypic cell profiles. PTH-treated Nmp4-KO mice gained over 2-fold more femoral trabecular bone than WT by 3 weeks. There was no difference between genotypes in BM cellularity or profiles of several blood elements. However, the KO mice exhibited a significant elevation in CFU-F cells, CFU-FAlk Phos+ cells (osteoprogenitors), and a higher percentage of CFU-FAlk Phos+ cells/CFU-F cells consistent with an increase in CD45−/CD146+/CD105+/nestin+ mesenchymal stem cell frequency. Null BM exhibited a 2-fold enhancement in CD8+ T cells known to support osteoprogenitor differentiation and a 1.6-fold increase in CFU-GM colonies (osteoclast progenitors). We propose that Nmp4/CIZ limits the PTH anabolic window by restricting the number of BM stem, progenitor, and blood cells that support anabolic bone remodeling.
Introduction
A
While the mechanisms regulating the extent of the PTH anabolic window are unknown, we demonstrated that disabling the transcription factor nuclear matrix protein 4/cas-interacting zinc-finger protein (Nmp4/CIZ) in mice significantly extends and augments PTH bone-forming capacity; treatment of wild-type (WT) and Nmp4- knockout (KO) mice with intermittent PTH for 7 weeks resulted in significant increases in serum osteocalcin, a marker for bone formation, but these serum profiles as a function of time were strikingly different [7,8]. In the WT mice, serum osteocalcin peaked at 3 weeks of treatment and returned to baseline by 7 weeks of hormone administration [7]. However, in the null mice, this PTH-induced surge in serum osteocalcin exceeded that observed in the WT mice and was still climbing at the end of the 7-week treatment regimen [7]. Consistent with this sustained serum osteocalcin surge, at the end of the 7-week treatment period, the null mice had gained significantly more femoral, vertebral, and tibial trabecular bone than WT mice while maintaining robust increases in cortical bone [7,8]. These enhanced increases in cancellous bone in the Nmp4-KO skeleton all showed significant treatment×genotype interactions, thus demonstrating that Nmp4/CIZ suppresses PTH-stimulated anabolism [7,8].
When in the PTH treatment regimen does bone formation in the Nmp4-KO mice eclipse WT growth, and what sustains this extended and enhanced anabolic activity? The WT and Nmp4-KO mice exhibited equivalent PTH-induced increases in trabecular bone during the first 2 weeks of treatment; however, at this treatment point, femoral mRNA profiles revealed a transient enhanced increase in PTH-stimulated c-fos and Fra-2 expression in the null mice as well as an elevated expression of Nfatc1 in these animals [7]. Although these transcription factors mediate numerous functions within the context of bone, this is consistent with an enhanced PTH-induced increase in mesenchymal stem cell self-renewal and/or recruitment of null osteoblasts and osteoclasts into the anabolic window [9 –13]. Interestingly, the untreated Nmp4-KO mice had a modest, but significantly elevated, bone mineral density and bone mineral content as compared to WT animals [8] despite modestly elevated levels of serum C-terminal telopeptides of type I collagen (CTX), a marker for bone resorption [7]. The Nmp4-KO bone marrow (BM) yielded ∼1.8-fold more osteoclasts in vitro as compared to WT marrow, and the null osteoclasts were significantly more active than their WT counterparts [7]. Therefore, bone formation was exceeding resorption, but how this occurred was not clear (eg, osteoblast–osteoclast coupling [14] and/or intrinsic differences in stem and progenitor pools that support bone formation or resorption).
To address whether the enhanced PTH-stimulated addition of trabecular bone in the Nmp4-KO mice is coincident with the initial surge in the serum osteocalcin, and to determine the cellular basis of this sustained enhanced anabolic activity, we treated WT and Nmp4-KO female mice with intermittent PTH for 3 weeks before harvesting femurs, femoral BM, and peripheral blood (PBL). Our data reveal that the Nmp4-KO mice show significantly enhanced PTH-stimulated addition of trabecular bone at 3 weeks of hormone treatment, and that Nmp4 has a profound regulatory role in BM population dynamics. Disabling this transcription factor results in alterations in stem, progenitor, and blood cell populations that accommodate the prolongation of the PTH anabolic window while maintaining bone remodeling. These data reveal novel aspects of how the PTH anabolic window is regulated and have implications for a novel adjuvant osteoporosis therapy.
Materials and Methods
Mice
Nmp4-KO mice, backcrossed onto a C57BL/6J background for 6–7 generations, [7,8], and their WT littermates were used for these studies. Our local Institutional Animal Care and Use Committee approved all experiments and procedures involving the production and use of the mice described in this investigation.
PTH treatment
Before initiating hormone treatment, 8-week-old female WT and Nmp4-KO mice were given 100 μL sterile saline by subcutaneous (sc) injection, once daily to habituate them to handling. At 10 weeks of age, animals were sorted into 4 treatment groups based on equivalent mean-group–body weight. These 4 groups included (1) vehicle-treated WT; (2) PTH-treated WT; (3) vehicle-treated Nmp4-KO, and (4) PTH-treated Nmp4-KO mice. Experimental animals were injected sc with human PTH 1–34 [hPTH(1–34), Bachem Bioscience, Inc.] at 30 μg/kg/day, daily or vehicle control (0.2% BSA/0.1% 1.0 mN HCl in saline; Abbott Laboratory) for 3 weeks. There was no significant difference in initial and final mean-group-body weights between genotypes. In a separate experiment, the BM of untreated female WT and Nmp4-KO mice (13 weeks of age) was harvested to compare the multipotent mesenchymal stem cell (CD45−/CD146+/CD105+/nestin+) frequency.
CFU-FAlkPhos+ assay [15]
BM was flushed from femurs; single-cell suspensions were prepared, and cells were seeded into 6-well plates at an initial density of 1×106 cells/well. Each culture well contained 2 mL of complete α-MEM supplemented with 100 IU/mL penicillin, 100 μg/mL streptomycin, 25 μg/mL amphotericin, 2 mM
Flow cytometry
Whole BM was isolated by flushing the femurs of experimental mice with α-MEM supplemented with 10% FBS. PBL was collected from the mice by cardiac puncture. The red blood cells (RBCs) were lysed with an RBC lysis buffer (Qiagen) before the PBL and BM were processed for flow cytometric analysis. All antibodies for flow cytometry were purchased from BD Biosciences. Stained samples were analyzed on an FACS Caliber (BD Biosciences), and results were quantified using FlowJo Version 8.8.6 software (Tree Star, Inc.).
Clonogenic assays
Colony-forming units (CFU-Cs) were assayed as previously described [16]. Briefly, 2.5×104 BM mononucleated cells or 25 μL PBL was seeded onto a 35-mm gridded dish containing methylcellulose and murine stem cell factor (100 ng/mL), murine granulocyte–macrophage colony-stimulating factor (10 ng/mL), murine interleukin 3 (IL3, 5 ng/mL), murine recombinant macrophage–colony stimulating factor (10 ng/mL), and human erythropoietin (4 U/mL) for 7 days at 37°C in a 5% CO2 incubator. Colonies were scored using an inverted light microscope. All cytokines were purchased from PeproTech.
Hemavet analysis
PBL was collected from the WT and Nmp4-KO mice and processed for blood cell enumeration using the Hemavet 950 FS according to the manufacturer's instructions (Drew Scientific).
Micro computed tomography
We previously reported no differences in the femur length between Nmp4-KO and WT mice at 8 and 17 weeks of age [8]. Therefore, after euthanasia, a 2.6-mm span (∼5 mm3 of medullary space) of the distal femoral metaphysis was scanned in 70% ethanol on a desktop microcomputed tomography (μCT) (μCT 35; Scanco Medical AG) at 10-μm resolution using 55-kVp tube potential and 400-ms integration time to measure trabecular three-dimensional (3D) morphometric properties as previously described [17]. From the 3D constructs, trabecular bone volume per total volume (BV/TV, %), connectivity density (Conn.D, mm−3), structure model index (SMI), trabecular number (Tb.N, mm−1), trabecular thickness (Tb.Th, mm), and spacing (Tb.Sp, mm) were calculated using Scanco software.
Statistical analysis
The program JMP version 7.0.1 (SAS Institute) was used to process all statistical evaluations. We employed a 2-way analysis of variance (ANOVA) for the PTH studies using genotype and treatment as the independent variables. If a genotype×treatment interaction was indicated, the data were analyzed by a Tukey honestly significant difference (HSD) post hoc test to determine significant differences between the experimental groups. Statistical significance was set at P≤0.01 to guard against type I errors. A separate experiment was conducted using a distinct group of our experimental mice for the purpose of comparing the frequency of multipotent mesenchymal stem cells (CD45−/CD146+/CD105+/nestin+) in untreated female WT and Nmp4-KO mice. These data were analyzed with a 2-sample t-test, assuming unequal variances and statistical significance was set at P≤0.05. The numbers of mice per treatment group are indicated in the appropriate Figures and Tables.
Results
Nmp4-KO mice exhibited an enhanced increase in femoral trabecular bone after 3 weeks of treatment
To determine if the divergence between the WT and Nmp4-KO mice in serum osteocalcin levels at 3 weeks is coincident with the beginning of the enhanced addition of trabecular bone in the null animals observed at 7 weeks [7], we sorted WT and Nmp4-KO mice into 4 treatment groups and harvested the femurs for μCT analysis as described in the Materials and Methods section. Although the WT and null mice had previously shown equivalent PTH-induced increases in trabecular bone at 2 weeks of treatment [7], in the present study, the null mice exhibited significantly augmented PTH-stimulated increase in femoral trabecular bone as compared to their WT littermates at 3 weeks (Fig. 1). The Nmp4-null mice showed a more robust PTH-stimulated increase in BV/TV compared to the WT animals during the first 3 weeks of treatment (Fig. 1A). The KO mice added ∼2.3-fold more bone than their WT littermates in response to PTH (Fig. 1A). The 2-way ANOVA indicated a strong genotype×treatment interaction, and the Tukey HSD post hoc determined that there was no difference in BV/TV between the vehicle-treated WT and KO animals (Fig. 1A). While PTH treatment increased connectivity parameters (Conn.D, mm−3) for both genotypes, a significantly greater enhancement was observed in Nmp4-KO mice compared to WT mice (Fig. 1B). Again, there was no difference between the vehicle-treated WT and null mice. The SMI was used to evaluate the PTH-induced alteration in femoral trabecular morphology. This parameter measures changes from a rod-like to a plate-like form, and the lower the value, the more plate-like the shape, which is indicative of an increase in bone strength [18]. PTH treatment of both WT and Nmp4-KO mice resulted in a significant transition to a more plate-like morphology, which was more pronounced in the null mice with a significant genotype×treatment effect (Fig. 1C). Interestingly, the SMI value for the femoral bone of the KO mice treated with vehicle was statistically equivalent to that of the WT mice treated with PTH (Fig. 1C). While PTH treatment significantly increased trabecular thickness (Tb.Th, mm, Fig. 1D) in null mice, treatment did not alter trabecular thickness in WT mice. Finally, PTH equivalently increased the trabecular number (Tb.N, mm−1, Fig. 1E) and decreased spacing (Tb.Sp, mm, Fig. 1F) in both genotypes.

Disabling Nmp4 enhanced PTH-induced increases in femoral cancellous bone after 3 weeks of treatment. Microcomputed tomography-acquired femoral trabecular architecture, including
BM cellularity, spleen weight, and the profiles of most blood elements did not differ between the WT and Nmp4-KO mice
To address whether there are differences between the Nmp4-null and WT mice in the BM or PBL cellular profiles supportive of the observed enhanced PTH-induced addition of trabecular bone, we obtained immunophenotypic, clonogenic, and hematological profiles at 3 weeks of treatment (Tables 1 –3). The spleen weight measured, as a% of total body weight, did not differ with the genotype, but did modestly increase with PTH treatment in both WT and null mice (Table 1). The profiles of blood elements between the Nmp4-KO and WT mice were unremarkable. We observed no differences between any of the treatment groups in the BM and PBL profiles of the RBCs, WBCs, platelets, neutrophils, lymphocytes, eosinophils, monocytes, B-cell lineages, CD4+ T cells, or the Lin(-)Sca-1(+)c-Kit(+) (LSK) cells (Tables 1 and 2). Finally, there were no differences between WT and Nmp4-KO mice in CFU-C, CFU-G, CFU-GEMM, and CFU-M cells (Table 3). PTH treatment had no impact on any of these parameters (Tables 1 –3).
WT and null mice were treated with intermittent PTH or vehicle for 3 weeks (number of mice/experimental group=9–14). A 2-factor ANOVA was used to evaluate the impact of genotype and treatment on the individual parameters. Statistical significance was set at P<0.01 to guard against type I errors. Percent (%) spleen weight is the weight of the organ divided by the total body weight.
WT, wild type; VEH, vehicle; PTH, parathyroid hormone; KO, knockout; ANOVA, analysis of variance; EO, eosinophils; LY, lymphocytes; MO, monocytes; NE, neutrophils; PLT, platelets; RBC, red blood cell; WBC, white blood cells.
Mice were treated with PTH or vehicle for 3 weeks (number of mice/experimental group=11–14). Statistical significance was set at P<0.0l.
LSK, lin-/Scal+/c-Kit+; BM, bone marrow; PBL, peripheral blood.
WT and null mice were treated with intermittent PTH or vehicle for 3 weeks (number of mice/experimental group=10–14). A 2-factor ANOVA was used to evaluate the impact of genotype and treatment on the individual parameters. Statistical significance was set at P<0.01 to guard against type I errors.
Nmp4-KO BM yielded more multipotent MSCs (CD146+/nestin+), CFU-FAlkPhos+, CFU-GM, and CD8+ T cells than WT BM
To determine if the source of this augmented bone formation in Nmp4-null mice is derived, in part, from an expanded pool of osteoprogenitors, we obtained BM from our experimental groups for analysis of CFU-FAlkPhos+ colonies as described in the Materials and Methods section. We recovered ∼4-fold more CFU-FAlkPhos+ colonies from the null mice than the WT animals (Fig. 2A). The total number of CFU-F colonies was significantly elevated in the Nmp4-KO cultures (Fig. 2B), and the percentage of CFU-FAlkPhos+/total CFU-F colonies was significantly increased in the cultures from the Nmp4-null BM as compared to the WT BM (Fig. 2C). There was a trend toward increased yield of CFU-F and CFU-FAlkPhos+cells with PTH treatment in both genotypes, but this was not significant. Therefore, we next addressed whether the frequency of the self-renewing multipotent mesenchymal stem cell (CD45−/CD146+/CD105+/nestin+), the precursor of CFU-F-derived lineages, including osteoprogenitors, is elevated in untreated Nmp4-KO mice. Indeed, we observed a nearly 4-fold increase in this cell phenotype in the null BM (Fig. 2D).
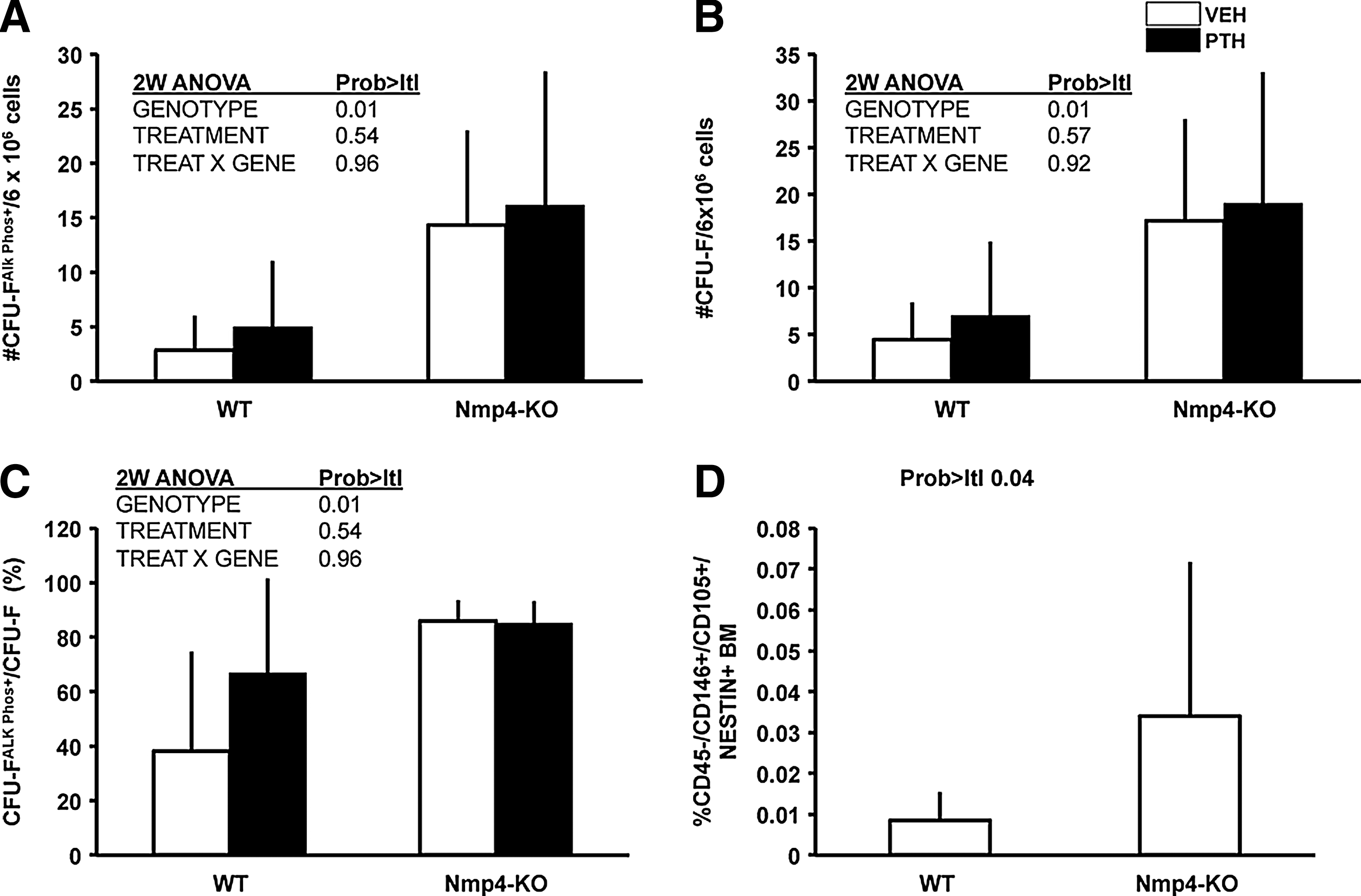
Nmp4-KO BM yielded more osteogenic stem and progenitor cells irrespective of treatment.
Nmp4 has no significant influence on the percentage of CD4+ T cells in the BM or PBL (Table 2), but recent studies have demonstrated that CD8+ T cells play an obligatory role in the PTH anabolic response via their release of the glycoprotein Wnt10b, a potent agonist for osteoprogenitors [19,20]. Indeed, the present data show that the percentage of CD8+ T cells in the null BM was 2-fold greater than that observed in the WT BM (Fig. 3A), but there was no difference in the percent CD8+ T cells in the PBL between the genotypes (Fig. 3B). Additionally, PTH treatment had no effect on the size of this population of cells in either the BM or PBL.
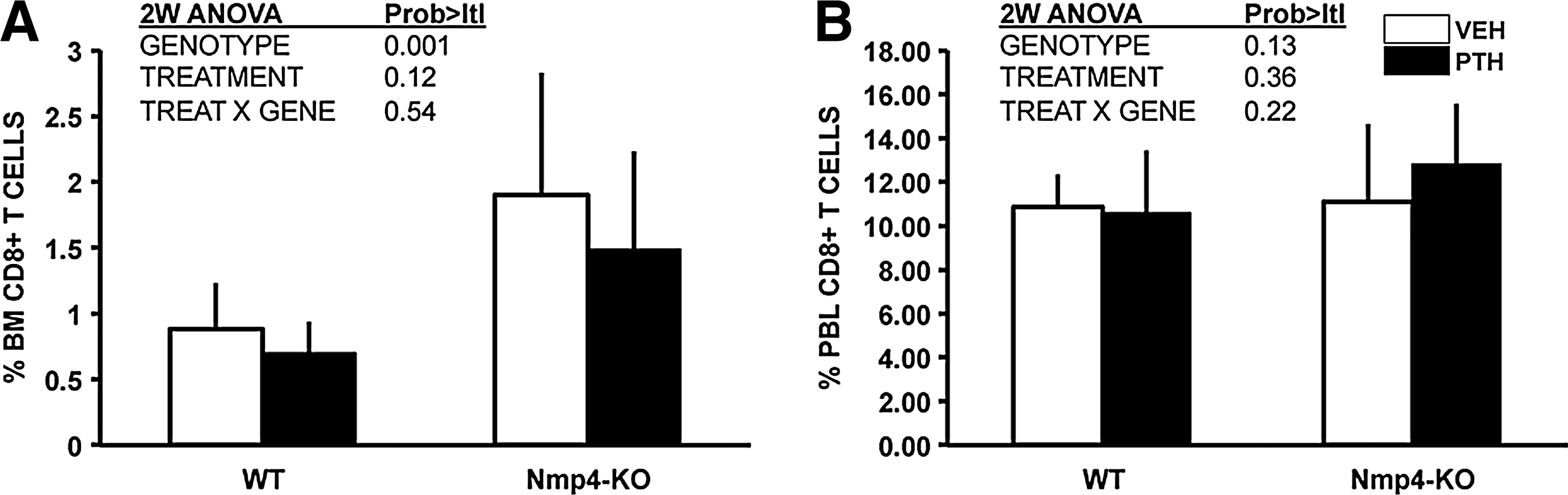
Nmp4-KO BM harbored more CD8+ T cells than WT BM irrespective of treatment.
Next, to address whether the observed modest elevation in bone resorption in the null mice and the enhanced number of osteoclasts derived from their BM [7] is due, in part, to an increase in osteoclast progenitors, we evaluated the number of CFU-GM cells from our treatment groups. Indeed, the Nmp4-null mice exhibited a modest (∼1.6-fold), but significant, increase in CFU-GM cells as compared to their WT littermates (Fig. 4). PTH had no effect on the number of these cells (Fig. 4).

More CFU-GM cells were obtained from Nmp4-KO mice than WT mice, irrespective of treatment. Intermittent hPTH (1–34) 30 μg/kg/day or vehicle was administered for 3 weeks as described in the Materials and Methods section (average±SD, number of mice/experimental group=10–14; statistical differences were determined using a 2-way ANOVA).
Discussion
A significant drawback to the use of PTH as an osteoporosis drug is that its anabolic potency declines within a relatively short period of time, which is particularly problematic in treating a chronic degenerative disease [21]. The cellular and molecular mechanisms underlying this closing of the PTH anabolic window are unknown. We have recently determined that deleting the transcription factor Nmp4/CIZ from mice significantly extends the PTH anabolic window and results in enhanced trabecular bone formation without compromising hormone-stimulated gains in cortical bone [7,8].
An intriguing aspect of the Nmp4-KO mouse response to anabolic doses of PTH is that the enhanced addition of trabecular bone requires a lag period to materialize [7]. Previously, we reported that both WT and null mice exhibited equivalent PTH-stimulated increases in trabecular bone during the first 2 weeks of a 7-week treatment. In this study, we compared hormone-induced increases in femoral cancellous bone after 3 weeks of treatment and indeed observed that the Nmp4-KO mice exhibited a >2-fold increase in PTH-induced accrual of femoral trabecular bone formation as compared to their WT littermates. This enhanced response to PTH was manifested in an augmented increase in BV/TV, trabecular connectivity (Conn D), and trabecular thickness (Tb.Th). Additionally, PTH had a greater impact on the SMI in the null mice. A decrease in SMI indicates a change in cancellous architecture from a rod-like to a plate-like morphology and is a result of alterations in modeling and remodeling [18,22,23]. This suggests that PTH-stimulated increases in bone strength are enhanced in the Nmp4-KO mice, although this must be confirmed by biomechanical testing.
Our data indicate that deleting Nmp4/CIZ establishes a BM microenvironment that is primed for anabolic signals. We observed no differences in femur cellularity, % spleen weight, or in the profiles of the vast majority of blood elements; however, there was a striking difference in the number of osteoprogenitor cells as evaluated by the clonogenic CFU-FAlkphos+ assay. The Nmp4-null BM yielded 4-fold more of these colonies than did the WT BM. In an earlier study, Noda and colleagues observed that BM cultures from null mice yielded about 3-fold more mineralized nodules than WT mice [24], which is equivalent to measuring CFU–osteoblast colonies [25]. In the present study, we also determined that the total number of CFU-F colonies obtained from the null mice was significantly elevated as was the% CFU-FAlkphos+/total CFU-F. These data together suggest that Nmp4 suppresses the frequency of CFU-F cells and impedes commitment to the osteogenic lineage. This is consistent with the elevated number of CD45−/CD146+/CD105+/nestin+ cells obtained in the Nmp4-KO mice. These cells are self-renewing multipotent mesenchymal stem cells and contain all the bone marrow colony-forming-unit fibroblastic colony activity [26,27]. PTH did not significantly impact the number of CFU-FAlkPhos+ colonies recovered from the BM of either genotype, although there was a trend toward modestly elevating the frequency of these cells. A variety of studies have shown conflicting stimulatory and inhibitory effects of PTH on osteoprogenitor proliferation [28 –30]; however, the prevailing view is that intermittent PTH recruits osteoprogenitors into the osteoblast differentiation pathway and enhances their survival instead of increasing the size of this progenitor pool [31, and references therein]. It is the accumulation of repeated new waves of osteoprogenitors with enhanced osteogenic potential that mediates the PTH-stimulated increase in bone mass [28,32]. This may also explain that the observed lag period before the enhanced PTH-induced bone formation phase is initiated in the Nmp4-null mice. If indeed the anabolic effect of intermittent PTH is the result of consecutive waves of committed osteoblast differentiation accumulated from each PTH exposure, in which hormone only acts on the BM early osteoprogenitor cells [28], then the rate of PTH osteoprogenitor recruitment would be equivalent in both the WT and KO mice, but the WT osteoprogenitor pool would be depleted before the KO population. This is consistent with the observed equivalent addition of bone during the first 2 weeks of treatment, but the divergence in both serum osteocalcin and bone formation in the null mice at 3 weeks [7 and the present work]. Finally, the Nmp4/CIZ-KO osteoblast exhibits a modest, but significant, enhanced response to numerous anabolic stimuli, including PTH, BMP2, and mechanical loading [24,33 –35]; therefore, an expanded population of such cells is certainly consistent with the augmented skeletal bone mineral density and bone mineral content of the null animals.
The expanded Nmp4-KO osteoprogenitor pool may be supported by the 2-fold increase in BM CD8+ T cells as compared to the WT mice. CD8+ T cells express the PTH receptor PTHR1 and support intermittent hormone anabolic activity via their secretion of the glycoprotein Wnt10b, a potent agonist of osteoblast activity [19,20]. PTH-induced bone formation was significantly reduced in T-cell-deficient mice and in these mice reconstituted with Wnt10b—/—
T cells [20]. Interestingly, we observed no difference in the level of CD8+ T cells in the PBL, suggesting that the recruitment and/or the retention of these cells is enhanced in the null BM microenvironment. BM CD8+ T cells consist chiefly (∼50%) of CCR7+
A second provocative aspect of the Nmp4-KO skeletal phenotype is that the baseline bone mineral density and bone mineral content are slightly increased despite a modest elevation of bone resorption [7]. While the increase in osteoclast number may be attributed to coupling (eg, increased osteoblast support of an increase in osteoclastogenesis [14 and references therein]), the present data suggest that this reflects intrinsic differences in osteoclast progenitor populations. We observed a modest (1.6-fold), but statistically significant, increase in CFU-GM cells in the null mice as compared to their WT counterparts. Although CFU-C cells were elevated in the Nmp4-KO mice, this only approached significance, and there was no difference in the levels of CFU-M cells between the genotypes. The precise lineage of the osteoclast and its relationship to other hematopoietic cells is controversial; however, there are a number of studies supporting the hypothesis that the osteoclast lineage branches to terminal differentiation via the CFU-GM cells before further passage toward the monocyte/macrophage lineage [39,40].
The present data suggest that the heightened bone anabolism and modestly elevated bone resorption in the global Nmp4-KO mouse are derived, in part, from a unique confluence of BM stem, progenitor, and blood cells. The null BM harbors an expanded pool of MSCs (CD146+/nestin+), osteoprogenitors, and CD8+ T cells, which together supply the osteoblasts necessary for the observed augmented bone-forming activity, even in the presence of elevated bone resorption driven by the modestly enlarged CFU-GM pool (1.6-fold) that contributes to the osteoclasts. This may support an environment of enhanced anabolic remodeling.
Our data do not directly relate the differences in cellular composition observed in the Nmp4-KO mice to enhanced PTH-stimulated increases in trabecular architecture. To address this issue, a combination of genetic-, drug-, and transplantation-based approaches will be required, because all of these methods have strengths and drawbacks, yet their intersection reveals complementary aspects of the phenomenon under study. However, the previously observed heightened PTH-responsiveness and osteogenic capacity of Nmp4-KO BMSCs and osteoblasts in culture [24,33,35] and the enhanced number of the progenitors of these cells (present study) likely make a substantial contribution to the extended anabolic window. Additionally, Nmp4/CIZ deficiency augmented newly formed trabecular bone mass after femoral BM ablation as compared to WT mice [24], confirming the enhanced osteogenic capacity of the reconstituted KO BM. It is certainly tenable that multiple stem/progenitor types are necessary for maintaining an open PTH anabolic window, and that one transcription factor has significant direct and/or indirect control over these populations was unexpected despite the fact that Nmp4/CIZ is expressed in multiple cell and tissue types [41]. Nmp4/CIZ has been proposed as a potential target for osteoporosis therapy, [42] and the present data further develop this idea, suggesting that disabling Nmp4/CIZ may provide an adjuvant therapy for extending PTH clinical efficacy by expanding the stem/progenitor populations sustaining its anabolic action.
Footnotes
Acknowledgments
This work was supported by grants from the Leukemia & Lymphoma Society (6234-12, FCY), Department of Defense (NF100087, FCY), and from NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), contract grant number DK053796 (JPB).
Author Disclosure Statement
No competing financial interests exist.