
Abstract
Mesenchymal stem cells (MSC) are effective in treating myocardial infarction (MI) and previous reports demonstrated that hypoxia improves MSC self-renewal and therapeutics. Considering that hypoxia-inducible factor-1 alpha (HIF-1α) is a master regulator of the adaptative response to hypoxia, we hypothesized that HIF-1α overexpression in MSC could mimic some of the mechanisms triggered by hypoxia and increase their therapeutic potential without hypoxia stimulation. Transduction of MSC with HIF-1α lentivirus vectors (MSC-HIF) resulted in increased cell adhesion and migration, and activation of target genes coding for paracrine factors. When MSC-HIF were intramyocardially injected in infarcted nude rats, significant improvement was found (after treatment of infarcted rats with MSC-HIF) in terms of cardiac function, angiogenesis, cardiomyocyte proliferation, and reduction of fibrotic tissue with no induction of cardiac hypertrophy. This finding provides evidences for a crucial role of HIF-1α on MSC biology and suggests the stabilization of HIF-1α as a novel strategy for cellular therapies.
Introduction
M
Mesenchymal stem cells (MSC) have been used over the past years as a novel therapy to treat MI and clinical studies using MSC for cardiac tissue repair have been recently completed (Prometheus;
To improve MSC therapeutics, many strategies have been conducted [3]. Preconditioning of MSC with hypoxia induced the pro-survival Akt pathway and increased their therapeutic potential in a model of hind limb ischemia [4]. Hypoxia-inducible transcription factors (HIFs) have been identified as pivot molecular mediators enabling cell adaptation to hypoxia [5 –10] and these molecules have been correlated with cell survival [11 –13]. Ischemic tissue elevates levels of HIF-1α, which in turn regulate the expression of nearly 200 genes affecting cellular responses to stress [13]. Genes regulated by HIF-1α include vascular endothelial growth factor (VEGF), erythropoietin, angiopoietin, placental growth factor, and platelet-derived growth factor [14,15]. Culture of embryonic stem cells on fetal stromal cells overexpressing HIF-1α improves their self-renewal and pluripotency in comparison with maintenance on fibroblast-based feeders [16]. The intracardiac injection of HIF-1α naked DNA reduced the size of infarction and increased neovascularization in the ischemic heart [17].
HIF-1α overexpression in MSC improved their survival and therapeutic properties [18 –20]. Considering the impact of HIF-1α in the adaptative response to hypoxia we wanted to evaluate the impact of HIF activity on the therapeutic potential of MSC during the treatment of ischemic cardiac diseases.
Materials and Methods
All procedures were approved by the Instituto de Salud Carlos III and institutional ethical and animal care committees.
Cells, culture conditions, and lentiviral labeling
Human bone marrow MSC (n=3; Inbiobank San Sebastian) were cultured following manufacturer's instructions and lentivirally transduced with pWIPI-green fluorescent protein (GFP) or pWIPI-HIF-GFP (
Flow cytometry
Human MSC-GFP or MSC-HIF-GFP (passage 6–10) were analyzed by flow cytometry (Coulter EPICS XL flow cytometer; Beckman Coulter) to determine the percentage of GFP-positive cells after lentiviral transduction. Data acquisition and analyses were performed with Expo32 software (Beckman Coulter). The cells were processed according to standard protocols.
Real-time polymerase chain reaction
Total RNA was prepared as described above. cDNA was synthesized using M-MLV Reverse Trascriptase (Invitrogen;
Western blot analysis
Whole extracts from MSC or MSC-HIF were prepared in ice-cold lyses buffer using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific;
Wound healing assay
Cells (MSC or MSC-HIF) were seeded in basal medium supplemented with 10% fetal bovine serum (FBS) and were grown to confluence onto gelatine-coated plates. The wound was made by scraping a plastic blue tip across the monolayer. Cells were allowed to migrate during 18 h. After cell scratching, 18 h later, cultures were photographed with a camera coupled to a phase contrast microscope. Number of cells migrated through the scratched area were counted in MSC-HIF cultures and normalized versus MSC cultures. The assay was performed in duplicated wells and repeated 3 times.
Adhesion assay
Cells were seeded in basal medium onto cover slides previously treated with 10 μg/cm2 of fibronectin (Sigma-Aldrich), 2 μg/cm2 of laminin (Sigma-Aldrich) and 10 μg/cm2 of collagen (Sigma-Aldrich). After 1 h, adhered cells were fixed with 2% paraformaldehyde (PFA), washed with phosphate-buffered saline (PBS), labeled with 4′,6-diamidino-2-phenylindole (DAPI), and counted. The assay was performed in triplicated wells and repeated thrice with MSC from 2 different donors.
MSC migration assay
To study trophism induced in MSC or MSC-HIF by trophic factors, cells were seeded in basal medium (Dulbecco's modified Eagle's medium with 0.5% FBS) at 10,000 cells/cm2 in the top chamber of an 8 μm-pore migration transwell (BD Falcon;
Animals
Male nude rats weighing 200–250 g (HIH-Foxn1 rnu; Charles River Laboratories, Inc.) were used. Initial number of animals included in the study was 70. Mortality in all groups due to surgical procedures was about 30%.
MI and cell transplantation
Permanent ligation of the left coronary artery was performed as previously described [21]. Immediately after left descendent artery (LAD) ligation, rats were transplanted intramyocardically (saline, 2×105 MSC cells or 2×105 MSC-HIF per animal in 3 injections of 5 μL volume at 3 points of the infarct border zone with a Hamilton syringe).
5-Ethynyl-2′-deoxyuridine treatment and analysis of proliferating cells
After saline or cell transplantation, a group of animals was daily given 0.5 mL i.p. injection of 5-ethynyl-2′-deoxyuridine (EdU) (50 mg/kg b.w., i.p.) for 2 weeks. EdU labeled cells in heart tissue were identified using an anti-EdU antibody (Abcam). Proliferation index was calculated as a percentage of EdU labeled nuclei per total of nuclei identified by DAPI staining. Identification of proliferating cardiomyocytes was performed by double staining with anti-EdU antibodies and anti-Troponin I, respectively (Chemicon International). About 10,000 cells were counted per animal.
Functional assessment by echocardiography
Transthoracic echocardiography was performed in rats under inhalatory anesthesia (Sevorane) using an echocardiographic system (General Electrics) equipped with a 10-MHz linear-array transducer as previously reported [21]. Measurements were taken at baseline and post-transplantation (4 weeks). Left ventricular (LV) dimensions in end diastole (LVDd) and end systole (LVDs), anterior and posterior wall (AW and PW) thickness in diastole and systole, end-diastolic area (EDA), and end-systolic area (ESA) were measured. Fractional area change (FAC) was calculated as [(EDA – ESA)/EDA]×100. Fractional shortening (FS) was calculated as [(LVDd−LVDs)/LVDd]×100. Changes in AW were calculated as (AWs−AWd/AWd)×100.
Immunohistochemistry
Four weeks post-implantation, animals were euthanized, the hearts removed, washed with PBS, and fixed in 2% PFA. Heart tissue sections were prepared for immunohistochemistry as previously reported [22].
Vascular density analysis
Immunohistochemical detection of vessels was performed with anti-rat RECA (Chemicon International). Vessels were counted in 10 fields in the peri-infarct zone at 200× and referred as number of vessels per unit area (mm2) using a light microscope and the Image Proplus 7.1 software.
Morphometry
The infarct zone in left ventricles was measured in 8–12 transverse sections of 7 μm (1 slice each 200 μm of tissue) from apex to base fixed with 2% PFA and stained with Masson's trichrome. The fibrotic zone was determined by computer planimetry (Image Proplus 7.1 software). Infarct size was expressed as percentage of total LV area and as a mean of all slices from each heart.
Cross-sectional cardiomyocyte area was measured in the border infarct zone of 3 heart sections from each animal stained with anti-laminin antibody (anti-laminin antibody; Abcam). Myocytes with defined sarcolemmal borders were selected for cross-sectional area measurements. Cardiomyocytes with different sizes (0–1,100 μ2) were measured in 4 animals of each group (around 1,500 cells per animal in saline group and 3,000 cells in MSC or MSC-HIF group) as previously described [23]. Briefly, area of cardiomyocytes was measured by computer planimetry (Image Proplus 7.1 software) and the number of cardiomyocytes ranking from 0–100 μm, 100–200 μm, 200–300 μm, 300–400 μm, 400–500 μm, 500–600 μm, 600–700 μm, 700–800 μm, 800–900 μm, 900–1,000 μm, and 1,000–1,100 μm were calculated in each animal. Median and first and third quartile of small size cardiomyocytes (0–200 μm) in each group were represented in box- and whisker plots using the Graphpad Prism 5 software.
Number of cardiomyocytes of a given size range were expressed as percentage of total cell count for each animal. Distribution frequency of cardiomyocyte cross-sectional areas in saline, MSC, and MSC-HIF groups, were represented in intervals of 100 μm size with overlapping Gaussian curves using the Graphpad Prism 5 software.
Statistical analysis
Data are expressed as mean±standard error of the mean or median (interquartile range). Comparisons between control and experimental groups were done with the Wilcoxon W test or the Mann–Whitney U test, as appropriate. Analyses were conducted with SPSS and GraphPad Prism 5 software. Differences were considered statistically significant at P<0.05 with a 95% confidence interval.
Results
HIF-1α overexpression induces changes in MSC gene expression and cell signaling
MSC were transduced with lentiviral vectors either pWIPI-GFP (named as MSC) or pWIPI-HIF-GFP (named as MSC-HIF). After infection, 90%–95% of cultured cells were GFP positive (Fig. 1B). HIF-1α mRNA levels were elevated 4.88±0.57-fold (Fig. 1B) and MSC-HIF cultures expressed HIF-1α protein in normoxia (Fig. 1D).
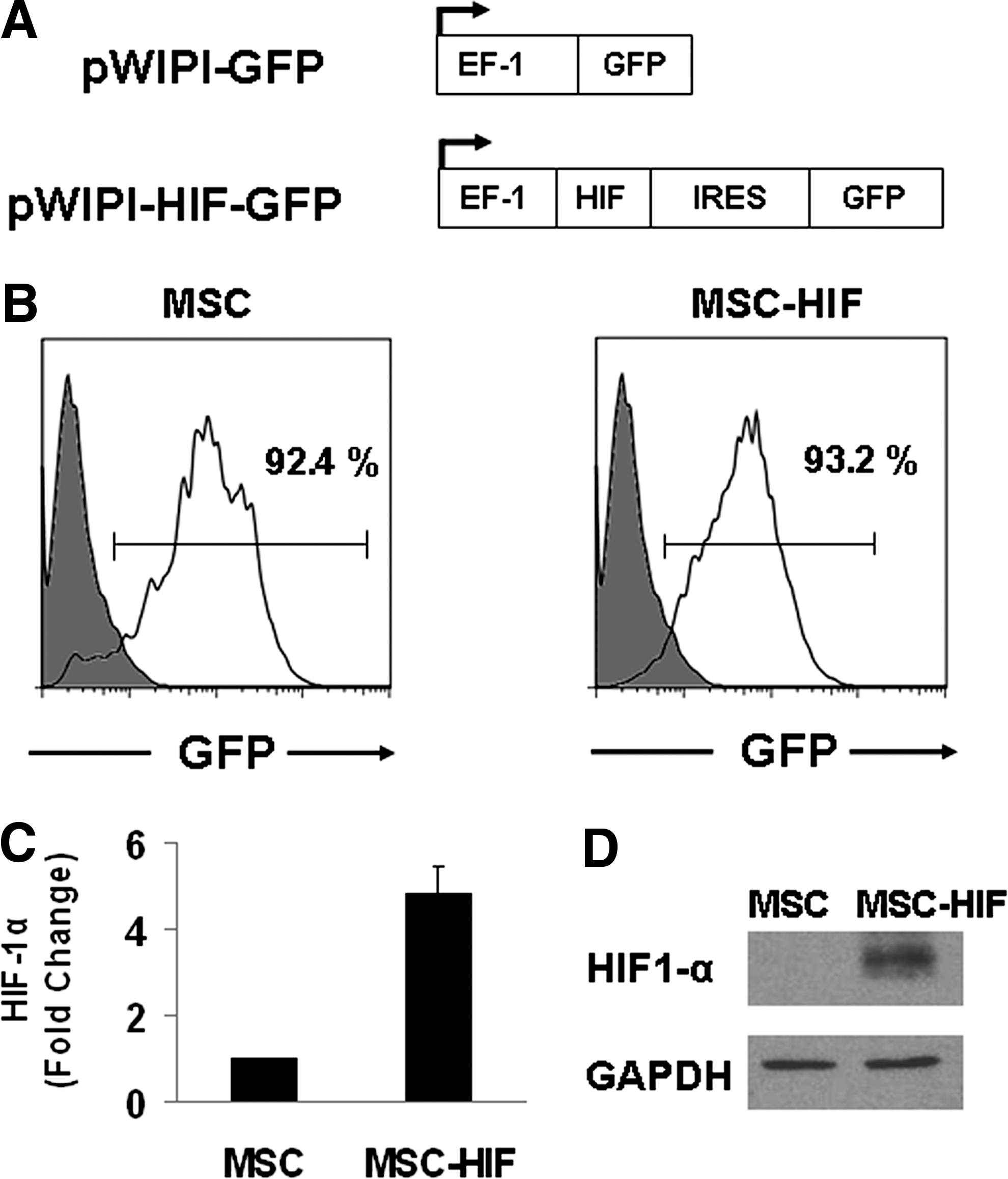
Characterization of transduced MSC cultures.
Based on microarray analysis (article in preparation), we investigated the upregulation of different genes in MSC-HIF. Over 30 HIF target genes analyzed, we detected increased expression in 11 by RT-PCR (Figs. 2A and 3A; Table 1). These genes are implicated in angiogenesis (ANGPT1, APLN, CD3D, CTGF, and CYR61), cell survival (BNIP3, BNIP3L, and PIK3C3), extracellular matrix remodeling [fibroblast growth factor 2, transforming growth factor, beta 2 (TGFβ2)], and chemotaxis (c-MET). Notably, TGFβ2 was upregulated by 7.31±1.25-fold, c-MET was upregulated by 3.88±0.22-fold, and ANGPT1 was upregulated by 2.11±0.21-fold. To investigate if non-hypoxic HIF stabilization had an impact in PI3K/Akt and MAPK pathways immunoblot analysis was carried out (Fig. 2B). Levels of phosphorylated Akt were increased (11.12±1.2 in MSC-HIF vs. 5.54±0.8 in MSC; P<0.05). pMAPK38 (0.87±0.15 in MSC-HIF vs. 0.56±0.1 in MSC; P<0.5) and pMAPKJNK (8.31±1.18 in MSC-HIF vs. 2.07±0.41 in MSC; P<0.5) were also elevated indicating the positive effect of HIF stabilization in MSC proliferation and cell survival.
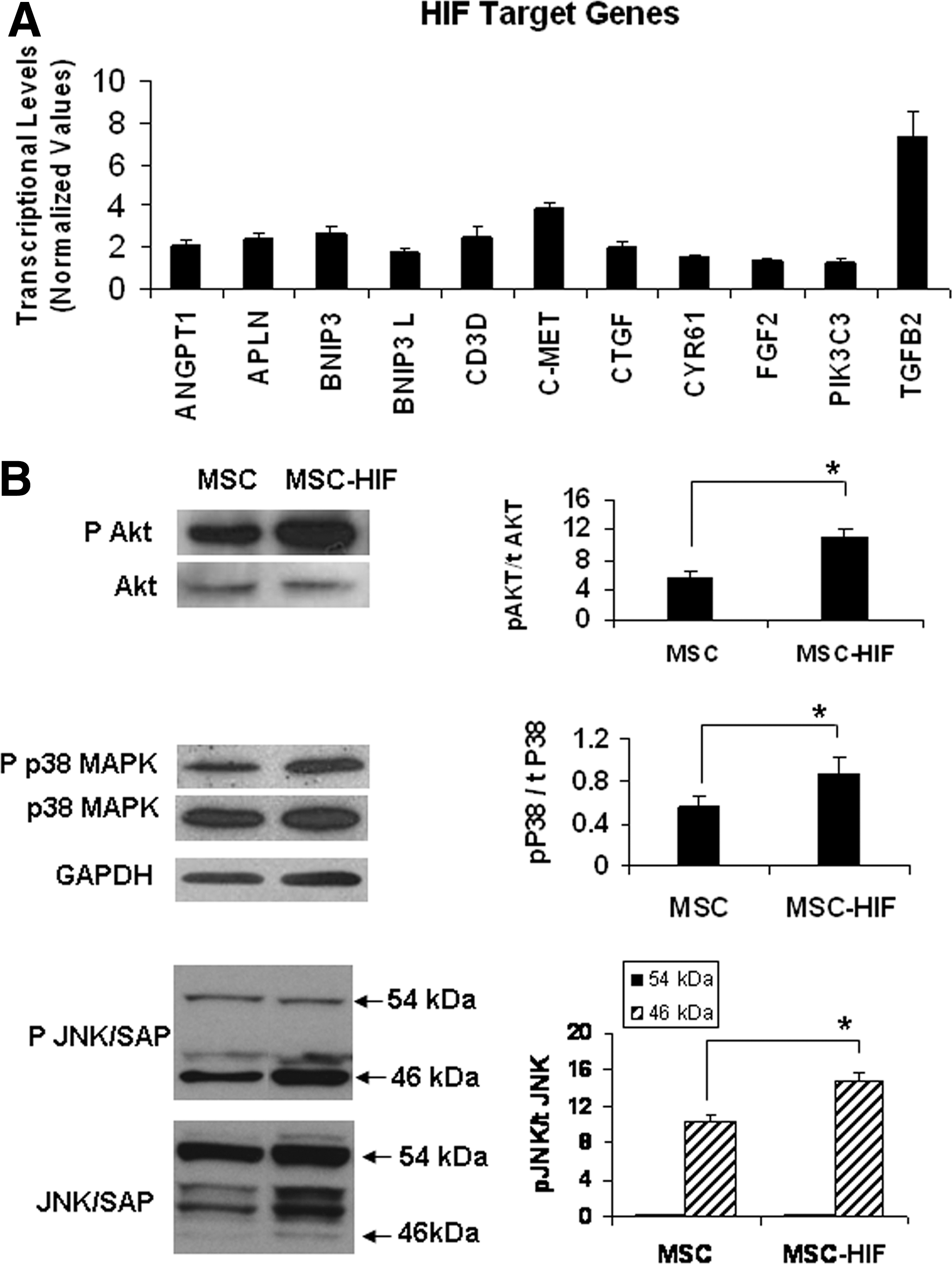
Transduction of MSC with HIF-1α vectors induce biological responses.

HIF-1α expression induces adhesion and migration of MSC.
All values are mean±SEM. AWd, LVd, PWd, AWs, LVs, and PWs are expressed in mm, whereas EDA and ESA are expressed in mm2. FS, FAC, and AWT are expressed as percentage.
MSC, mesenchymal stem cells; HIF, hypoxia-inducible factor; ANOVA, analysis of variance; AWd, anterior wall diastole thickness; AWs, anterior wall systole thickness; AWT, anterior wall thickening; EDA, end-diastolic area; ESA, end-systolic area; FAC, fractional area change; FS, fractional shortening; LVd, left ventricular diastole internal dimension; LVs, left ventricular systole internal dimension; MI, myocardial infarction; PWd, posterior wall diastole thickness; PWs, posterior wall systole thickness; SEM, standard error of the mean.
HIF induces expression of adhesion molecules and promotes migration in response to trophic signals
We next assayed the influence of HIF in MSC cell adhesion and migration (Fig. 3). Quantitative PCR showed increase in fold change expression of several genes implicated in cell adhesion (AMIGO 24.8±0.38-fold, NCAM2 1.27±0.12-fold, ITGA2 2.87±0.12-fold, SDC1 4.44±0.15-fold, and SPARC 2.03±10-fold) and extracellular matrix production (COLXII 3.59±0.69-fold; Fig. 3A). In vitro experiments showed that HIF-1α overexpression promoted cell spreading in a wound healing assay with 2.09±0.5-fold increase at 18 h (P<0.01; Fig. 3B). Cell adhesion to collagen (1.31±0.02-fold; P<0.05) and laminin (1.24±0.09-fold; P<0.05) was also increased in MSC-HIF versus MSC (Fig. 3C). To further investigate the role of HIF-1α in MSC biology, cells were allowed to migrate toward trophic factors in a transwell culture system (Fig. 3D). Migration toward IL-6 (1.93±0.12 in MSC-HIF vs. 1.5±0.18 in MSC; P<0.05), HGF (2±0.25 in MSC-HIF vs. 1.12±0.12 in MSC; P<0.01), and SDF (1.7±0.23 in MSC-HIF vs. 1.17±0.02 in MSC; P<0.01) was significantly elevated. Consistent with results shown in Fig. 3A and B, we observed stronger changes in cytoskeletal architecture and actin reorganization detected by paxillin and phaloidin stainings in MSC-HIF versus MSC when cells were cultured under chemotactic stimulus of SDF (Fig. 3E). These results indicate that HIF-1α induces morphologic/structural changes in MSC that in turn improve cell motility and response to trophism.
MSC-HIF improve cardiac function in a rat model of MI
To test if HIF-1α expression improved therapeutic properties of MSC during cardiac repair, MSC-HIF were transplanted in nude rats after induction of MI. Based on our previous observations [22] and to minimize inflammatory infiltrates due to stem cell transplantation, cell dose was adjusted to 200,000 cells/animal. Thus, saline solution, MSC or MSC-HIF were transplanted into the myocardium of rats shortly after induction of MI by LAD ligation. Four weeks after transplantation, cardiac function parameters were measured to assess the degree of functional recovery achieved by the presence of MSC or MSC-HIF in the infarct border zone. MSC-HIF group displayed a significant recovery of systolic function as calculated by percentage of FAC (33.16%±1.51% in saline group, 38.25%±2.03% in MSC, and 44.01%±1.14% in MSC-HIF; P<0.0001 in MSC-HIF vs. saline and P<0.05 in MSC-HIF vs. MSC in saline group) and FS (23.23%±0.79% in saline group, 24.96%±1.48% in MSC and 30.15%±0.90%, in MSC-HIF; P<0.0001 in MSC-HIF vs. MSC). Beneficial effects were also observed on anterior wall thickening (AWT) at this time point (AWT: 22.64%±0.76% in saline group, 24.73%±1.59% in MSC, and 26.74%±0.92% in MSC-HIF; P<0.01 in MSC-HIF vs. saline; Fig. 4; Table 1).
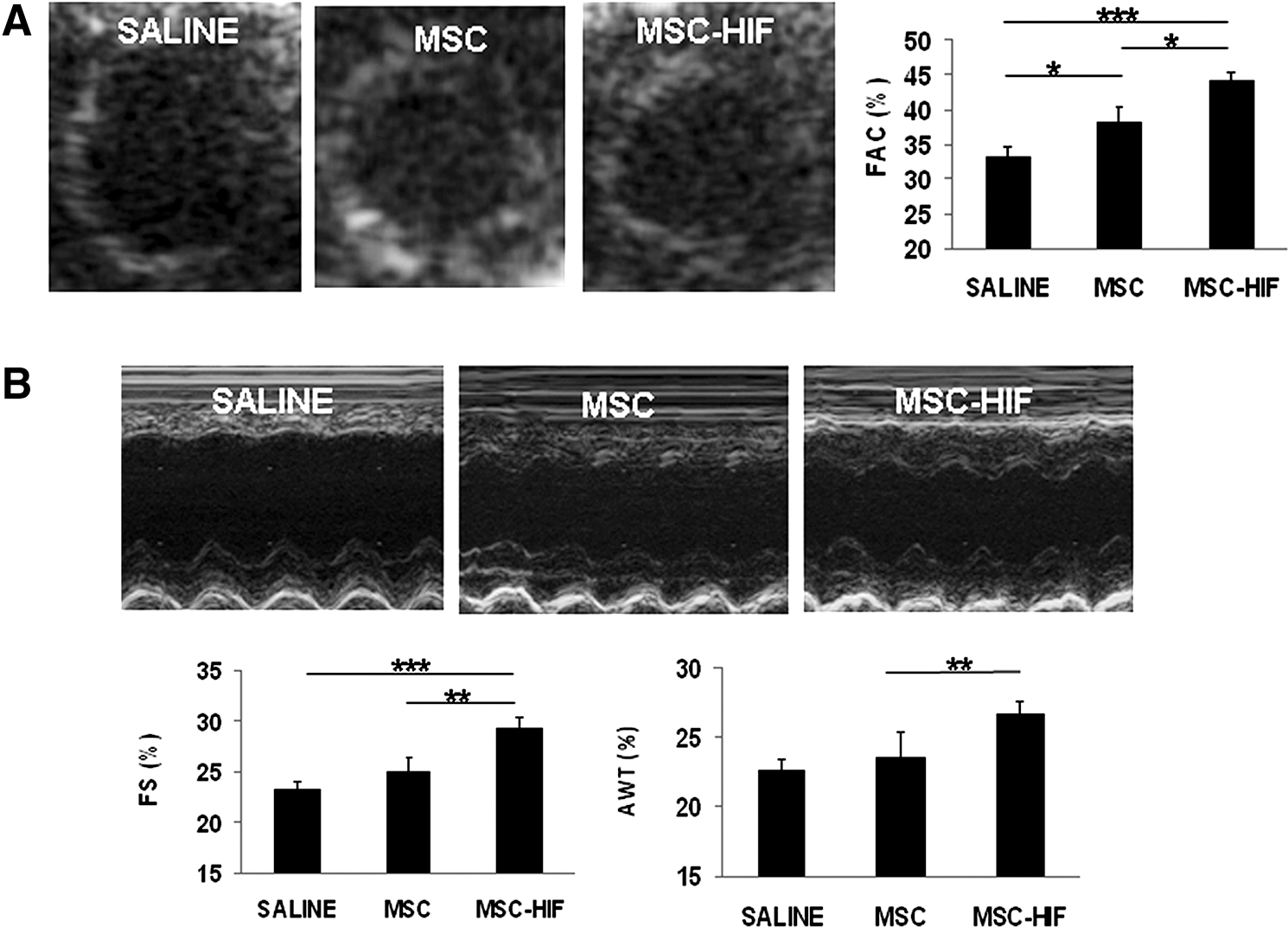
Improvement of LV function in MSC-HIF-treated animals.
MSC-HIF reduce fibrotic scar tissue and induce angiogenesis
Treatment with MSC-HIF significantly reduced the area of fibrous scar tissue (Fig. 5A); percentage of scar tissue was 29.91%±1.72% in saline group (n=8), 18.12%±0.93 in MSC group (n=6), and 12.73%±1.04% in MSC-HIF group (n=11) (P<0.00001 in MSC-HIF vs. saline, P<0.0001 in MSC vs. saline, and P<0.01 in MSC-HIF vs. MSC). Measurement of angiogenesis at the same time point showed that both MSC and MSC-HIF transplantation increased the number of vessels at the peri-infarct (Fig. 5B). Number of capillaries per mm2 was 518±81 in saline, 810±53 in MSC, and 1,252±80 in MSC-HIF (P<0.0001 in MSC-HIF vs. saline, P<0.01 in MSC-HIF vs. MSC and in MSC vs. saline).
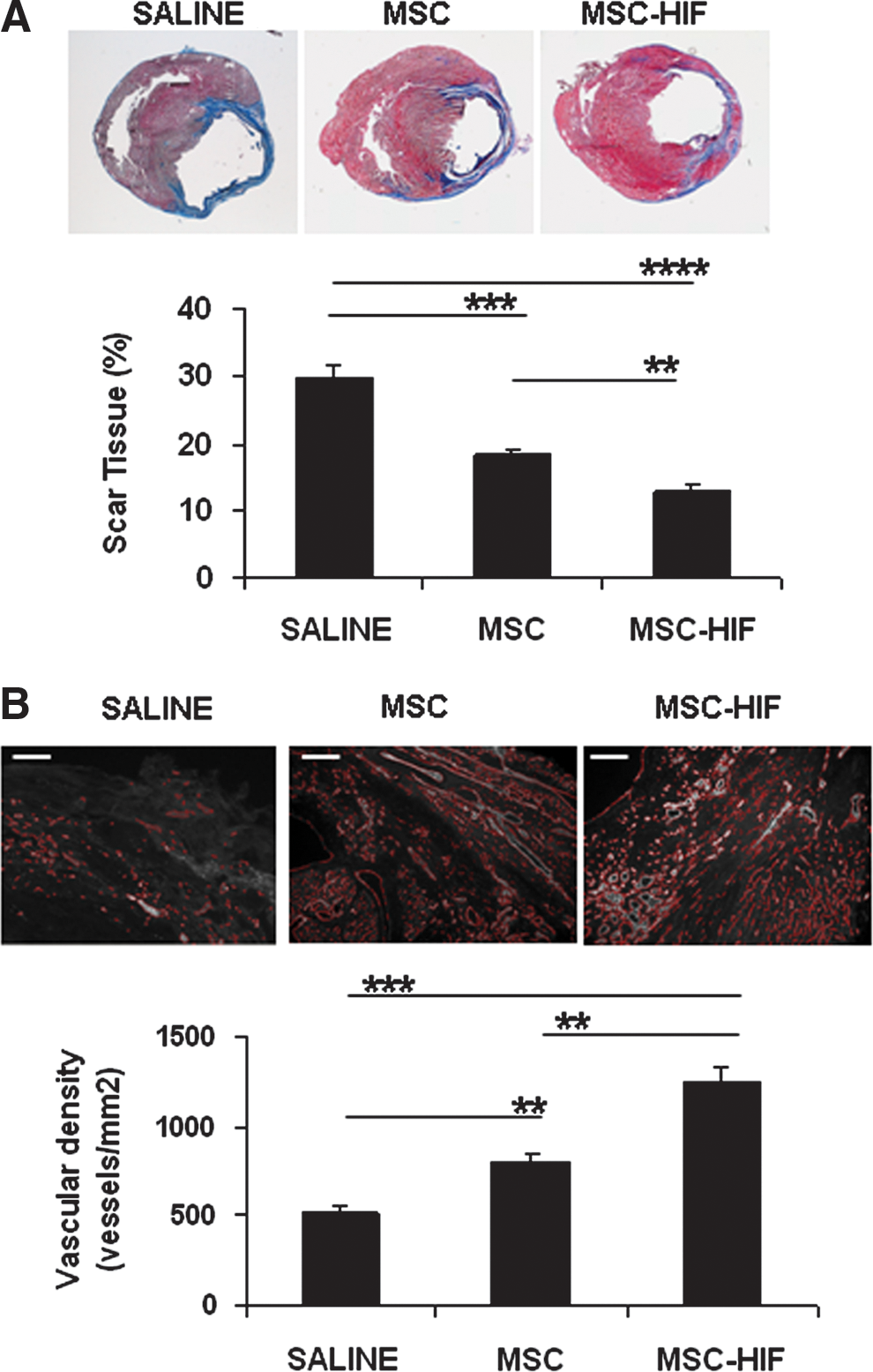
Effect of MSC-HIF transplantation on infarct size and angiogenesis.
MSC-HIF treatment promotes myocardial tissue regeneration
Next, we analyzed the number of proliferating cells in the border zone of EdU-treated animals (n=6 in each group). After 2 weeks, animals treated daily with EdU were sacrificed and the infarcted area was subjected to histological studies. At the doses analyzed, we did not detect engrafted cells in cell-treated animals. However, cell transplantation increased proliferation in heart tissue (Fig. 6A). Proliferation Index was 17.81%±2.86% in saline, 26.82%±3.05% in MSC, and 22.44%±1.22% in MSC-HIF; P<0.05 in MSC and MSC-HIF vs. saline). Double staining Edu/Troponin I revealed the presence of proliferating cardiomyocytes. Cardiomyocyte proliferation index was 2.32%±0.34% in saline group (n=5), 2.86%±0.18% in MSC group (n=6), and 4.39%±0.61% in MSC-HIF group (n=5); (P<0.05 in MSC vs. saline and P<0.01 in MSC-HIF vs. saline). These results point to a paracrine effect as a mechanism induced by cell transplantation.

MSC-HIF-treated animal showed increase in cardiomyocyte proliferation with no hypertrophy of heart tissue.
Finally, we wanted to evaluate if cardiac hypertrophy contributed to the increase in myocardial tissue in animals transplanted with MSC or MSC-HIF. For this purpose, cardiomyocyte area in the border zone of the infarct was calculated from heart sections stained with anti-laminin and anti-troponin I antibodies (Fig. 6B). Small size cardiomyocytes (less than 200 μm2) were more abundant in cell-treated animals (16.21±2.47 in saline, 24.34±7.28 in MSC, and 27.13±4.45 in MSC-HIF; P<0.05 in MSC-HIF vs. saline) (Fig. 6C). However, when analyzing cardiomyocyte global sizes, we observed that relative to saline-treated animals, in both MSC and MSC-HIF groups the distribution of myocyte cross-sectional area was shifted to the left with no significant differences in cardiomyocyte area, indicating the absence of cardiac hyperthrophy.
Discussion
HIF-1α plays different roles in cardiac tissue that have been extensively studied [7]. While accumulation of HIF-1α in cardiomyocytes lead to reduced contractility in the adult and deterioration of ventricular function [24,25], overexpression of these transcription factor attenuated cardiac dysfunction following MI probably due to an increase in blood perfusion and glycolysis and reduction of apoptosis [12,13,26]. In this context, stabilization of HIF-1α in inducible polylhydroxylases (PHD) knockout mice (Phd2flox/flox mice) lead to an HIF-dependent tissue protection in case of MI, with increased capillary density and preserved cardiac structure and function [27]. The authors reported that the success of the approach was to avoid HIF stabilization during heart development, preventing the activation of detrimental pathways, while modulating the expression of PHD2 in the adult. Considering the dangerous potential of HIF-1α stabilization in cardiac tissue, we wanted to potentiate HIF-induced therapeutic mechanisms, without modulation of HIF levels within the heart, using cell therapy.
MSC are an attractive approach to repair ischemic tissues and hypoxia is considered an important stimulus to potentiate their therapeutic mechanisms [28]. In a previous study, we reported that MSC reduced scar tissue formation and increased neovascularization more efficiently than other adult stem cell types [22]. The MSC-dependent paracrine mechanisms observed in vivo seemed to be triggered by hypoxia, since cells injected in the viable myocardium at the border zone often migrated to the ischemic fibrotic tissue, indicating not only a homing to the site of injury but also the ability of these cells to survive in hypoxic environments. Indeed, physiological niches of bone marrow stem cells display reduced oxygen levels and this condition is essential to maintain their self-renewal capacity and pluripotency [29].
Following this rationale, we wanted to investigate the effect of a constitutive form of HIF-1α in MSC biology and therapeutics. This new strategy could be important to: (1) manipulate MSC in normoxia to mimic some of the effects induced by hypoxia, (2) identify the mechanisms by which MSC increase their survival and paracrine effects, and (3) determine HIF target molecules in MSC that could contribute to hypoxia-induced MSC phenotype.
MSC-HIF were analyzed by western blot and RT-PCR to determine HIF-1α expression levels at mRNA and protein level. Analysis of the main signaling pathways activated in MSC-HIF revealed that HIF-1α accumulation activated PI3K/Akt. These results are consistent with previous reports showing that HIF stabilization promotes AKT phosphorylation in MSC [18]. Moreover, it has been suggested that PI3K/Akt are linked to the phosphorylation of the MAPK and SAPK/JNK pathways in MSC and other cell types, which could explain the increase in phosphorylation of p38MAPK and SAPK/JNK kinases in MSC-HIF. Since constitutive expression of HIF-1α induced Akt and SAP/JNK phosphorylation in MSC-HIF (Fig. 2) we hypothesize a possible autocrine loop among these molecules in MSC. Other mechanisms like downregulation of E2A-p21 by HIF-TWIST axes has also been reported to preserve MSC self-renewal capacity and to reduce senescence [28].
We next investigated the impact of HIF-1α on MSC gene expression. Gain-of-function studies in MSC showed that HIF-1α coordinately regulated the expression of genes encoding proteins and growth factors implicated in wound repair, angiogenesis, and extracellular matrix production, most of them previously described as HIF targets [30 –35]. Proangiogenic factors including angiopoietin (ANGPT1), apelin (APLN), and the matricellular protein CYR61 (CNN1) were upregulated in MSC-HIF. Expression of ANGPT1 and APLN1 is modulated by hypoxia and HIF-1α in cardiac myocytes and both promote beneficial effects on cardiac function and blood pressure [32,34]. Similarly, the proangiogenic factor CYR61 (CNN1) has been implicated in fibroblast senescence as a mechanism for controlling fibrogenesis in wound healing [36]. Strikingly, we do not observe upregulation of VEGF mRNA levels in MSC-HIF. Similar results were observed at the protein level in a cytokine assay of MSC-HIF/MSC culture supernatants (not shown). A possible explanation is that MSC express high levels of VEGF in normoxia that make difficult the detection of changes in VEGF levels between MSC-HIF and MSC.
TGFβ and connective tissue factor (CTGF) are also HIF target genes. Several studies suggested that TGFβ may be crucial for stem cell-based myocardial repair by repression of inflammatory gene synthesis mediating resolution of the inflammatory infiltrate [37,38]. In addition, TGFβ increases cell adhesion and regulates matrix metalloproteinases required for MSC migration through extracellular matrix barriers [39]. In this context, CTGF is a downstream mediator of both TGFβ signaling [40] and G-coupled protein receptor 30 (GPER), and recently its activation in HL-1 cardiomyocytes have been linked to stabilization of HIF-1α and associated to anti-apoptotic mechanisms [41,42].
In addition, expression of integrins and genes related to connective tissue synthesis, and cytosqueleton reorganization were upregulated in MSC-HIF, that in turn improved wound healing and MSC locomotory behavior in functional studies of chemotaxis (Fig. 3).
BNIP3/BNIP3L are 2 HIF targets genes that also were upregulated in MSC-HIF. The activation of these molecules may seem contradictory with the rest of survival signals triggered by HIF-1α overexpression, since they belong to the intrinsic mitochondria-linked death pathway. However, the alternative splicing of BNIP3 in ventricular myocytes during hypoxia has been recently described as a novel defense mechanism [43]. Thus, a similar mechanism cannot be discarded in MSC-HIF.
Of particular relevance was the increased migration toward SFD and HGF. These 2 molecules are implicated in the CXCR4 and c-Met axis respectively, that not only are activated in response to hypoxia, but also mediate the migration and homing of bone marrow-derived cells in vivo [29,44 –46]. All these biological processes could be responsible for an improved MSC-mediated cardiac healing.
Thus, the major findings of this study are: (1) HIF-1α triggers activation of PI3K/Akt, p38MAPK, and MAPK/JNK signaling pathways, (2) HIF-1α overexpression increases cell adhesion and induces structural changes and cytoskeleton reorganization, (3) HIF-1α increases cellular responses to trophics factors in vivo and in vitro, (4) MSC-HIF showed increased ability to repair cardiac function in an experimental model of MI, in correlation with increased ability to induce angiogenesis and decrease tissue fibrosis, and (5) Intracardiac injection of MSC-HIF induced cardiomyocyte proliferation in the absence of cardiac hyperthrophy.
These results are in accordance with previous studies in models of calvarial defects showing that treatment with MSC overexpressing HIF-1α promote angiogenesis and osteogenesis [20]. These kinds of approaches, together with others based on direct stabilization of HIF-1α in ischemic tissues, has led to phase I and II clinical studies in patients with severe pheripheral arterial disease [47].
Only 2 PHD inhibitors, FG-2216 and FG-4592, have progressed to clinical setting (
A limitation of our approach is that we have examined the action of HIF-1α in normoxia in vitro, so we cannot discard additional mechanisms in hypoxic culture. However, the observed in vivo effects are likely to be more accurate and to recapitulate the effects of HIF-1α in hypoxic environments resulting from cardiac ischemia.
Overall, this study demonstrates that HIF-1α expression in MSC potentiates therapeutic mechanisms and adds new information about the repair processes induced by MSC.
Footnotes
Acknowledgments
This work was supported in part by grants from the Instituto de Salud Carlos III for the Regenerative Medicine Program of Valencian Community to Centro de Investigación Principe Felipe, KUTXA founding and from the FIS (PI07/784, CP08/80 and PI10/00743). P.S. acknowledges support from Miguel Servet and RETICS programs (Instituto de Salud Carlos III). We thank Dr. R. Muñoz-Chapuli for critical discussion. We also thank Dr. Ortiz de Landazuri for the gift of HIF-1α plasmid, M. Sirerol for cloning assays, E. Nuñez and L. Pardo for immunological studies, M.P. Rubio for western blot analysis, and the service of confocal microscopy at CIPF for technical assistance.
Author Disclosure Statement
All authors have reported that they have no relationships relevant to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.