
Abstract
The characterization of adipose-derived stromal/stem cells (ASCs) remains difficult due to the lack of a definitive and unique cellular marker. Therefore, a combination of markers is necessary to identify the cells. No comprehensive analysis of the immunophenotype of expanded plastic adherent ASCs has been published. Therefore, the aim of this study was to characterize the general phenotype of cultured ASCs and to further analyze cellular subsets. ASCs were isolated from lipoaspirates from patients undergoing cosmetic liposuction and cultured in standard cell culture. A comprehensive phenotype characterization was done with the BD Lyoplate™ Human Cell Surface Marker Screening Panel containing 242 antibodies and isotype controls. Cultured ASCs not only showed the characteristic expression profile of mesenchymal stem cells (MSCs), but also revealed donor-specific variability in the expression of 49 other markers. We further detected markers with a scattering in the fluorescence intensity, indicating subpopulations with different expression profiles. Therefore, a multi-color flow cytometric analysis was done after staining the cells with direct-labeled antibodies against CD73, CD90, CD105, and either CD34, CD140b, CD200, CD201, or CD36 to verify the selected subpopulations of ASCs. We detected no CD34-CD36 double-positive population, but CD34+-CD36− and CD34—CD36+ subpopulations, both of which are positive for the 3 main MSC markers, CD73, CD90, and CD105. All other detected subpopulations also co-expressed the 3 main MSC markers, and therefore fulfill the minimal phenotypic criteria for the definition of cultured MSCs. Our study demonstrates the first comprehensive phenotypic characterization of ASCs and clearly highlights donor-specific variability in ASC preparations.
Introduction
M
Adipose tissue contains a large number of multipotent cells. These so-called adipose-derived stromal/stem cells (ASCs) possess the advantages of a minimally invasive harvesting procedure from adipose tissue and a high cell yield compared with bone marrow (reviewed in [6]). It has been described that stem and progenitor cells in the uncultured stroma-vascular fraction (SVF) from adipose tissue usually amount to up to 3% of the whole cells, and this is 2,500-fold more than the frequency of stem cells in bone marrow [7]. Others have also described that adipose tissue provides a large numbers of stem cells compared with bone marrow. A bone marrow transplant contains approximately 6×106 nucleated cells per mL [8], of which only 0.001%–0.01% are stem cells [9]. In comparison, the number of SVF cells that can be isolated from subcutaneous liposuction aspirates is approximately 0.5–2.0×106 cells per gram of adipose tissue [8,10 –13], whereby the percentages of stem cells range from 1 to 10% [12,14,15], most likely depending on the donor and tissue-harvesting site. Approximately 0.5×104 to 2×105 stem cells can be isolated per gram of adipose tissue, varying between patients. Nevertheless, MSC preparations, especially at the beginning of the culture period, are heterogeneous cell cultures comprising a subset of stem cells (or different subsets of stem cells) and more differentiated (progenitor) cells [6].
To date, there has been no comprehensive analysis of the immunophenotype of expanded plastic-adherent ASCs. Therefore, the aim of this study was to characterize the overall phenotype of cultured ASCs, the variability of this phenotype between cell isolation of different donors and to further analyze subsets/subpopulations by multi-color measurement.
Materials and Methods
Isolation and culture of cells
Human adipose-derived stromal/stem cells (ASCs) were isolated from lipoaspirates from patients undergoing cosmetic liposuction, as described earlier [10,16]. Our study was approved by the ethics committee of the clinic of the Goethe University, Frankfurt. Liposuction aspirates were obtained from 16 subjects undergoing plastic surgical procedures. Aspirated tissue was digested at 37°C with 0.075% collagenase I (230 U/mg; CellSystems) and continuously agitated for 45 min. The stromal-vascular fraction was separated from the remaining fibrous material and the floating adipocytes by centrifugation at 300 g. The cells sedimented were washed with phosphate-buffered saline (PBS) and filtered through a 100 μm pore filter (Millipore). Erythrocyte contamination was reduced by density gradient centrifugation with Biocoll (Biochrom). High contamination with erythrocytes was found to markedly decrease cell adherence and proliferation. A preceding density gradient separation provided a better yield of adherent cells than treatment with an erythrocyte lysing buffer. Finally, the cells were plated for initial cell culture, and cultured at 37°C in an atmosphere of 5% CO2 in humid air. We used Dulbecco's modified Eagle's medium (DMEM; Sigma) with a physiologic glucose concentration (100 mg/dL) supplemented with 10% fetal calf serum (FCS; PAA) as the culture medium. The medium was replaced every 3 days. Subconfluent cells were passaged by trypsinization. The adipogenic and osteogenic differentiation potential of cultured ASCs was proved by specific media, as previously described [17]. Primary isolated and cultured ASCs were characterized in detail in earlier studies [16,18 –20].
The ASCs used in all experiments (Lyoplate and multi-color measurements) were isolated from 13 different female donors (age 40.0±11.2, body mass index 26.0±5.5).
Bone marrow-derived mesenchymal stem cells (BM-MSCs) were used as control cells for the multi-color measurements. BM-MSCs were isolated by plastic adherence. 4.2×106 of bone marrow mononuclear cells were resuspended in 6 mL of fetal bovine serum containing medium and plated in a 25 cm2 tissue culture flask. The cells were cultured in an incubator at 37°C with 5% CO2 and 95% humidity. After 72 h, the medium containing nonadherent cells was removed and replaced with fresh medium. The adherent spindle-like cells were further cultured for 10–14 days until the cells reached about 70%–80% confluence as evaluated by microscopy. During this time, the medium was changed every 3 days. Subconfluent cells were passaged by trypsinization and plated at a density of 2×103 MSCs/cm2. BM-MSCs were expanded until passage 3 and then used for the phenotypic analysis.
Cell surface marker screening by Lyoplate technology
The BD Lyoplate™ Human Cell Surface Marker Screening Panel (BD Biosciences) was used to characterize cultured ASCs. The kit contains 242 purified monoclonal antibodies to cell surface markers, and also isotype controls for assessing unspecific backgrounds. The assay was processed as described by the manufacturer, with minor modifications. ASCs (n=5, passage 2–4) were detached from cell culture plastic by accutase and processed. 50,000 cells were resuspended in 100 μL buffer per 96 well and were stained in each well with the specific primary antibody (10 μL) for 25 min on ice. Then, the cells were washed and stained with the secondary antibody (50 μL) for 25 min on ice. Finally, the cells were resuspended in 75 μL buffer and measured (10,000 cells per well) in a BD FACSArray™ cytometer.
We used the automatic evaluation of the FACSArray cytometer. Nevertheless, the background fluorescence was set manually for each plate. Data are presented as a heat map generated by BD FACSDiva™ software and heat map representation in Excel 2007 (
Verification of selected markers by multi-color flow cytometry
Multi-color flow cytometric analysis was carried out to verify selected markers of ASCs (ASC subsets). ASCs (n=7–9, passage 2–4) were detached by accutase and stained with antibodies against direct-labeled CD73 (PE-Cy7), CD90 (APC), CD105 (PerCP), and either CD34-PE, CD140b-PE, CD200-PE, CD201-PE, or CD36-FITC. The analyses were 4-color measurements (CD73/90/105 and a PE-mAb), except for CD34-PE/CD36-FITC (5-color). Briefly, 1.5×105 ASCs were stained for 15 min in the dark (without fixation), washed twice, and the labeled cells were analyzed using an FACSCanto II flow cytometer (BD Biosciences). All experiments included negative controls without antibodies or with corresponding isotype controls. The flow cytometer was set using isotype controls. Cells were gated by forward and sideward scatter to eliminate debris.
Bio-imaging
ASCs were cultured in 96-well plates (BD Imaging plate) to 70% confluency. Live cells were stained and processed as recommended by the manufacturer. Briefly, the cells were rinsed thrice with PBS and then stained with the primary antibodies for 35 min on ice. Next, the cells were washed twice and stained with the secondary antibodies for 25 min in the dark. The cells were washed and fixed with 4% para-formaldehyde for 10 min in the dark. Finally, the nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) for 5 min in the dark. The plates were stored at 4°C in the dark until imaging. Images were made by an automated confocal fluorescence microscope (BD PathwayHT, 20× objective) and colored/merged by the Attovision software (BD Bioscience).
Statistical analysis
Analysis was performed using GraphPad Prism 5.0 (Graph-Pad Software). Data are presented as median and range. The Mann–Whitney rank sum test was used for statistical analysis. P-values of < 0.05 were considered significant.
Results
Phenotype analysis by Lyoplate measurements
Cultured ASCs from 5 different donors were evaluated by the Lyoplate technology for their surface expression of 242 markers. Our study presents the first comprehensive phenotypic characterization of cultured ASCs. ASCs used for the Lyoplate assay were in passage 2–4, and the median culturing time was 45 days (range 35–50). First, we titrated the recommended required quantity of ASCs per well. We used 5×104 ASCs per well, though the user will only need approximately 12.7 million cells for the whole Lyoplate assay. This is 10 times less than that recommended by the supplier (from 5×105 to 1×106 per well).
Flow cytometric data were analyzed by automatic evaluation of the FACSArray cytometer after gating the debris. The background fluorescence was set manually for each plate and compared with isotype controls (both were negative). Then, the expression of every antigen was evaluated by setting a marker and analyzing percent positive cells in the histograms. The results are shown in a heat map (Fig. 1) and by representative flow histograms (Fig. 2A). Characteristic markers are expressed (CD13, CD29, CD73, CD90, CD105, and CD166), and the cells lack the expression of a panel of uncharacteristic markers (CD11b, CD14, CD19, CD31, CD45, and HLA-DR) (Fig. 2A). Our results are, therefore, consistent with the minimal phenotypic criteria for the definition of cultured MSCs proposed by the International Society for Cellular Therapy [5]. In addition, we also show bio-imaging of a characteristic marker (CD44) after processing by the Lyoplate technology (Fig. 2B).

Heat map of cell surface marker expression by adipose-derived stromal/stem cells (ASCs). The BD Lyoplate™ Human Cell Surface Marker Screening Panel was used to characterize cultured ASCs. The kit contains 242 purified monoclonal antibodies to cell surface markers and isotype controls. 50.000 ASCs were resuspended in 100 μL buffer, plated into a 96 well, stained, and measured in an FACSArray cytometer. The median of percent-positive ASCs (n=5, passage 2–4) is presented. For mAb-clones and abbreviations, see
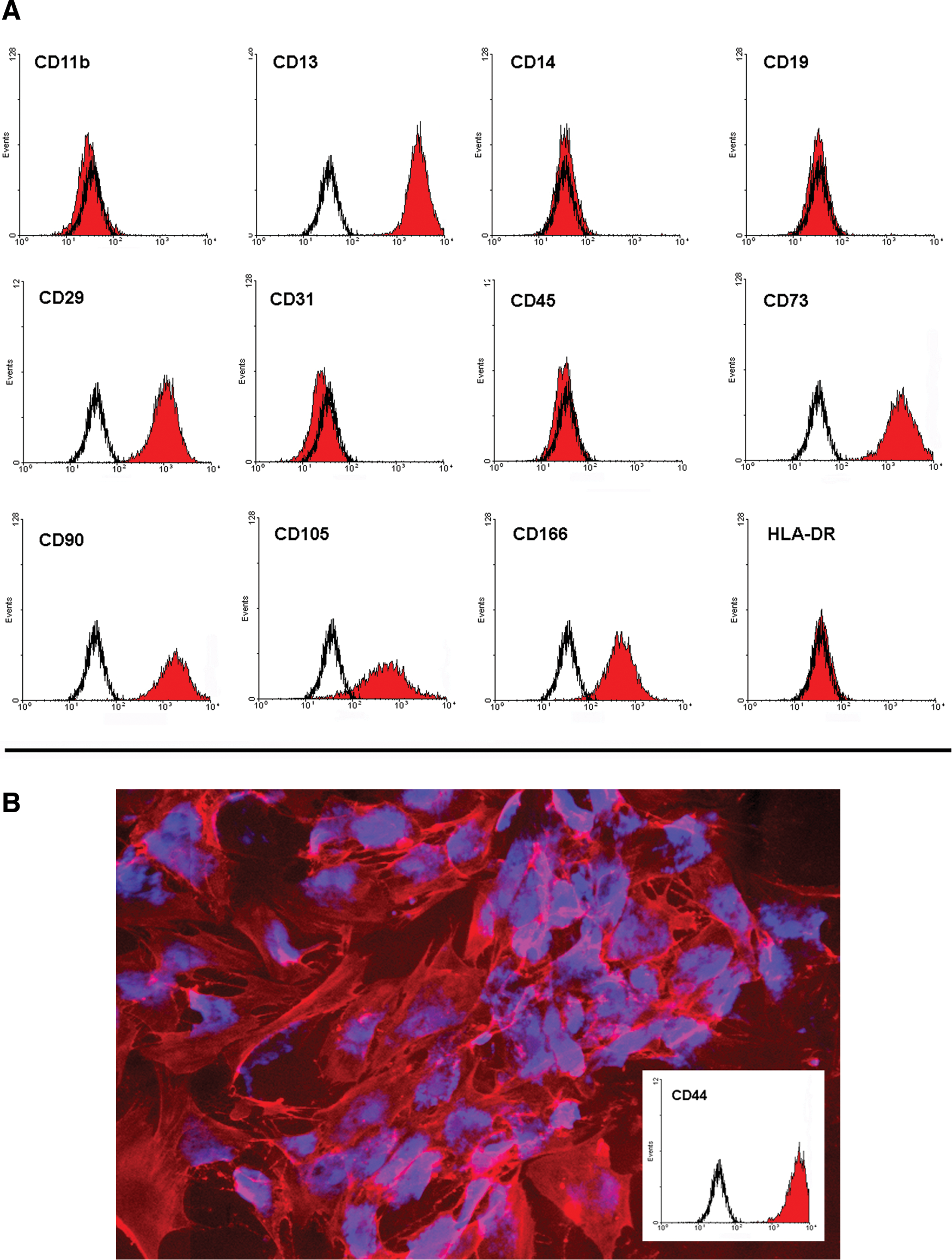
Expression analysis of typical cell surface markers on ASCs.
We further assessed the histograms of all markers for scattering in the fluorescence intensity, indicating subpopulations with different expression profiles. We detected markers with a broad scattering of the fluorescence, such as CD200 (Fig. 3B) and CD201 (Fig. 3C). Furthermore, the data revealed donor-specific differences in marker expression accompanied by a high variability in the expression profile between the donors, for example, CD36 (Fig. 3D) and CD34 (Fig. 3E). The isolated cell population from different donors differed highly in the expression profile (no expression, scattering of the fluorescence intensity, positive), as shown by the expression of CD36 and CD34. Altogether, 49 markers of the 242 markers researched showed a high variability in the expression of a specific antigen. The criteria for this variability were that the (a) expression of the marker showed a broad range between the 5 donors, and (b) cells of at least 2 out of 5 donors express the marker (>5% positive cells). These markers are summarized in Table 1.

Representative expression of specific markers.
The table shows antigens whose expression pattern between the different donors showed a broad range (as shown in Figs. 3D and E). We used the following criteria: Expression of the marker showed a broad range between the 5 donors and cells of at least 2 out of 5 donors express the marker with more than 5% positive cells. The median and range of the percentage of positive adipose-derived stromal/stem cells generated from 5 donors is shown.
Multi-color flow cytometry
Subsequently, we performed multi-color flow cytometry to analyze the expression of selected antigens from cultured ASCs in greater detail, especially with regard to subpopulation composition. ASCs in passage 2–4 were generated from 9 different donors, whereby the median culturing time was 44 days (range 27–62). The gating strategy is demonstrated in Fig. 4A and B. All measurements confirmed the results from the Lyoplate analyses, whereas median values differed from the Lyoplate results. We show specific subsets that are positive for CD34, CD36, CD140b, CD200, or CD201 (Fig. 4). CD140b (PDGFR-b) was used as a positive control, because the expression of this antigen is generally described for MSCs/ASCs.

Characterization of ASC subsets by multi-color flow cytometry.
We performed 5 color measurements in the case of CD34-PE/CD36-FITC (Fig. 4B). We detected no CD34+-CD36+ double-positive population, but CD34+-CD36− and CD34—CD36+ subpopulations, both of which are also positive for CD73, CD90, and CD105 (Fig. 4C and D). Gating of all subsets revealed that all populations are CD73+-CD90+-CD105+ (data for CD140b, CD200, and CD201 not shown) Therefore, all subsets fulfill the minimal phenotypic criteria described for MSCs [5]. Furthermore, we were only able to detect CD34+ cells with a PE-labeled antibody. If we labeled the same cells with an FITC-labeled anti-CD34 antibody (from the same clone), we were not able to detect these cells in our multi-color cytometric analyses (data not shown).
In contrast to ASCs, BM-MSCs showed no CD34+ or CD36+ subsets, and only a small portion of CD200+ and CD201+ cells (Fig. 5). This difference in the antigen expression between ASCs and BM-MSCs was significant in the cases of CD34, CD36, and CD201 (Fig. 5).
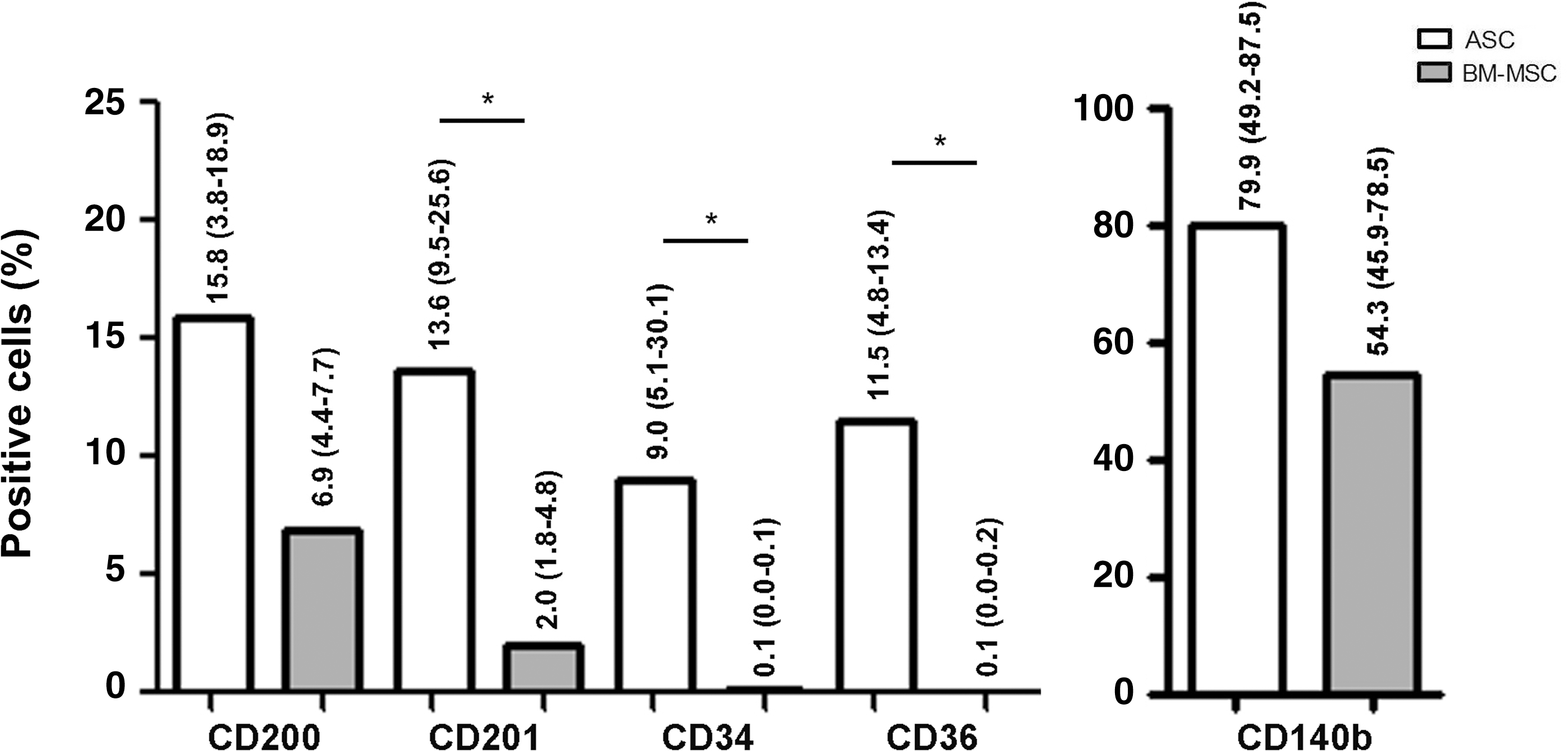
Differences in the antigen expression between ASCs and bone marrow-derived mesenchymal stem cells (BM-MSCs). Median (and range) of percentage of positive cells is shown (analysis/gating corresponding to Fig. 4B). In both, ASCs and BM-MSCs, all subsets are CD73+CD90+CD105+ (data not shown). Significant differences in marker expression between ASCs and BM-MSCs are marked [*P<0.05, ASC (n=7–9), BM-MSC (n=3)].
Discussion
Over the last decade, different studies have characterized some surface markers of ASCs and compared freshly isolated ASCs with cultured in early and later passages [10,14,21 –23]. Unfortunately, no unique single marker for ASCs has been found so far. Therefore, the combination of markers is necessary to identify cultured ASCs. Primary ASCs at the beginning of the culture period do not uniformly express all surface proteins that are supposed to be characteristic for ASCs [14,21,24]. The expression profile of ASCs changes during culture time.
ASCs in passage 2 or 3 are morphologically a homogeneous population of fibroblastoid cells. These cells uniformly express the characteristic MSC markers: CD29, CD44, CD73, CD90, CD105, and CD166, and lack the expression of CD11b, CD14, CD31, and HLA-DR. To date, no comprehensive and thorough surface expression profile of cultured ASCs in the early passage has been reported. In this study, therefore, we characterized the overall phenotype of cultured ASCs, assessed the donor-dependent variations of the ASC phenotype, and further analyzed subsets/subpopulations of ASCs by multi-color flow cytometry.
The Lyoplate analysis clearly demonstrated that ASCs express the main characteristic markers. However, the expression of some markers varied from donor to donor. Since all donors were of the same gender, we assume that this variable marker expression may be influenced by the age of donors, body mass index, ethnicity, and their medical history (e.g., preexisting diseases, nicotine, or alcohol abuse). The variability between ASC isolation from different donors or different liposuction sides has been reported in many studies, but in none of these has the phenotypic variations between cultured ASCs isolated from different donors been described. It has been shown, for example, that the body mass index is negatively correlated to the number of stromal cells per gram and their differentiation capacity [25]. The liposuction procedure may differ between different clinics, the liposuction (or biopsy) side is different, and the time lapse until the isolation procedure starts differs between the laboratories [6]. It has also been reported that the liposuction side, liposuction procedure, age, or body mass index play an important role in the yield of ASCs and their frequency and growth capacity [12,25 –29], but it is not clear whether this favors different subsets in cultured ASCs. All these variables may affect the composition of the isolated initial cells and the following cultured population(s), but it is extremely difficult, if not impossible, to standardize these variables [6].
ASCs, similar to MSCs in general, are selected by their adherence to the cell culture plastic. This generally used isolation technique might be selected for a heterogeneous primary cell population. The cells of this fraction are characterized early during primary culture by a slightly heterogeneous morphology indicating different stem and precursor cell subpopulations, and (maybe) more differentiated cells (dedifferentiated endothelial cells, smooth muscle cells, and pericytes). The heterogeneity of MSC isolations, in general, has been discussed in many publications [30 –32]. Nevertheless, when analyzing the adherent population by flow cytometry, no macrophages, endothelial cells, lymphocytes, or granulocytes seem to remain [16]. The presence of endothelial cells, for example, is not detectable [16]. However, endothelial cells in culture are extremely susceptible to culture conditions, such as supplements and particularly shear stress, and, therefore, may dedifferentiate or trigger apoptosis under static culture conditions [33].
Variations in the ASC expression pattern reported by different laboratories may be due to donor-dependent differences, differences in the liposuction side or in the isolation and culture methods. Regardless of the reason for this, it creates difficulties in reproducing and comparing the results of different laboratories. The expression of most characteristic markers is consistently found to be expressed by cultured ASCs, and others are consistently not found to be expressed, but the expression of others is described in a very contrary manner between different laboratories (summarized in [34]). Some authors reported the expression of antigens such as CD34, CD54, CD107, or CD146 to be expressed on cultured ASCs, while others could not observe any expression of these antigens. The expression of (some) markers is described to be dependent on the culture conditions. In this case, it is also important to note that differences in marker expression may be influenced by factors secreted by accessory cells in the initial passages [35]. Nevertheless, one main critical point seems to be the donor-specific variability in the expression of some markers, as shown overall for 49 of the 242 markers researched in our study. We detected CD34+ cells in ASC cultures in passage 2–4 in our Lyoplate and multi-color analyses, for example, but this CD34 expression was highly variable between the donors. Moreover, it should be mentioned that the verification of CD34 expression in the multi-color measurements was only possible with a PE-labeled antibody, and not with an FITC-labeled antibody from the same clone. This is potentially due to a very low expression of the antigen that needs a bright fluorescent label for the proof of expression. Although CD34 is reckoned as a hematopoietic stem cells-associated surface marker, its expression is also described in vascular endothelial cells (which are also CD31+). In addition, it is described to be expressed by ASCs' subsets, but gets partly lost in later passages depending on the culture conditions, such as plating density and culture medium [14,36,37]. Although the role of CD34 in cell adhesion and hematopoietic stem cell differentiation has been described, the functional implications of CD34 expression in ASCs remain unclear [36,38,39]. CD34 expression in hematopoietic cells decreases with cell maturation, suggesting that CD34 expression indicates immature cell status [38]. The same interpretation has been suggested for ASCs' CD34 expression [36]. The loss of CD34 expression may reflect the differentiation or commitment of ASCs [36]. It has been demonstrated that more than 85% of the stroma vascular fraction which had initially adhered to the culture surface had a CD31−/CD34+/CD45−/CD146− phenotype [24]. Two subpopulations with different phenotypes have been identified within the CD34+ cells (a CD34dim and CD34bright subpopulation) [23]. As already reported, an immunomagnetically isolated CD34+ subpopulation from the stroma-vascular fraction of adipose tissue lacks the expression of the hematopoietic marker CD45, and these cells were not able to give rise to hematopoietic colonies under specific stimuli [40]. Another study compared sorted CD34+ and CD34- subsets of ASCs, and found that CD34+ cells are more proliferative and have a higher ability to form colonies, while CD34− cells have a greater ability to differentiate into adipogenic and osteogenic lineages [36]. The authors speculated that the loss of CD34 expression may be related to the physiological process of commitment and/or differentiation from immature status into specific lineages [36]. Furthermore, our multi-color analyses showed that all CD34+ cells fulfill the minimal phenotypic criteria for MSCs (positive for CD73, CD90, and CD105 expression). This was also the case for all CD36-, CD200-, and CD201-positive ASCs, and for CD200- and CD201-positive BM-MSCs. BM-MSCs were negative for CD34 and CD36 expression, as already described in the literature and contrary to ASCs [41]. We further used the PDGFR-β (CD140b) as a positive control, because its expression by ASCs and BM-MSCs is generally accepted. All CD140b+ ASCs and BM-MSCs in our study also expressed CD73, CD90, and CD105.
In conclusion, our study provides the first comprehensive immunophenotypic analysis of expanded plastic-adherent ASCs and characterizes subsets of ASCs. While the main characteristic antigens for ASCs/MSCs were shown to be expressed, and all isolations meet the criteria of the International Society for Cellular Therapy, the expression of 49 markers was highly variable between different donors. Therefore, the analysis clearly shows donor-specific variability in ASC preparations and highlights the critical antigens. Further studies need to be done to investigate the source of these variable expressions. This can now be done by selecting 1 or 2 of the described antigens and an effort to correlate the expression to the liposuction site or donor-specific varieties. In addition, the study can also be used as a starting point of further studies to characterize specific cellular subsets of ASCs. The existence of these subsets in the primary isolates and changes during the cell culture period should be examined. Furthermore, the influence of different media on the expression of the described specific antigens (and, therefore, on the cellular subsets) should be investigated by upcoming studies, also in terms of producing GMP-compliant ASCs for transplantation.
Footnotes
Acknowledgments
Dr. G. Sattler, Rosenparkklinik Darmstadt, is acknowledged for the supply of lipoaspirates. Parts of this work were supported by the Adolf Messer-Stiftung.
Author Disclosure Statement
The authors report no conflicts of interest in connection with this article.