
Abstract
The domestic pig is an important large animal model for preclinical testing of novel cell therapies. Recently, we produced pluripotency reporter pigs in which the Oct4 promoter drives expression of the enhanced green fluorescent protein (EGFP). Here, we reprogrammed Oct4-EGFP fibroblasts employing the nonviral Sleeping Beauty transposon system to deliver the reprogramming factors Oct4, Sox2, Klf4, and cMyc. Successful reprogramming to a pluripotent state was indicated by changes in cell morphology and reactivation of the Oct4-EGFP reporter. The transposon-reprogrammed induced pluripotent stem (iPS) cells showed long-term proliferation in vitro over >40 passages, expressed transcription factors typical of embryonic stem cells, including OCT4, NANOG, SOX2, REX1, ESRRB, DPPA5, and UTF1 and surface markers of pluripotency, including SSEA-1 and TRA-1-60. In vitro differentiation resulted in derivatives of the 3 germ layers. Upon injection of putative iPS cells under the skin of immunodeficient mice, we observed teratomas in 3 of 6 cases. These results form the basis for in-depth studies toward the derivation of porcine iPS cells, which hold great promise for preclinical testing of novel cell therapies in the pig model.
Introduction
However, the use of retro- or lentiviral vectors for reprogramming prohibits the use of these cells for therapeutic applications in human patients, attributed to risks of insertional mutagenesis and potential oncogene activation [10]. Reprogramming methods, which avoid integration of the vectors into the host genome have been assessed in murine and human cells, including nonintegrating adenoviruses [11,12], episomal DNAs [13 –15], or substitution of reprogramming factors by small molecules [16,17].
A limitation of murine models for preclinical assessments of potential cell therapies is the short life span, small size, and the high level of inbreeding in this species. The pig is an attractive large animal model for preclinical testing of safety and efficacy of iPS-based cell therapies. Porcine organs are largely similar in size and physiology to their human counterparts rendering the domestic pig a suitable model in transplantation, immunology, and surgery [18 –23]. The availability of pluripotent porcine cells would be invaluable for preclinical testing of novel cell therapies. Despite numerous attempts, the derivation of authentic pluripotent porcine cells has been met with limited success [24]. Only recently, putative porcine iPS cells fulfilling several criteria of true pluripotent cells were reported after reprogramming fibroblasts with viral vectors [25 –28]. Notably, in one study, chimeric offspring were reported [28]. After breeding of chimeric pigs, 2 out of 43 F1 offspring carried some of the reprogramming factors in their genome, suggesting germ line transmission [29]. However, current data indicate that reprogramming and in vitro culture conditions are deficient for induction and maintenance of pluripotency in porcine iPS cells [24,30 –32]. In contrast to other species, in the pig, the continuous expression of the exogenous transgenic transcription factors is still necessary to maintain the pluripotent phenotype of presumptive porcine iPS cells [25 –28]. Porcine iPS cells in which the exogenous reprogramming factors are silenced and pluripotency is maintained by the endogenous pluripotency factors have not yet been reported.
Here, we have applied the Sleeping Beauty (SB) transposon system for induction of pluripotency in porcine somatic cells. Recently, we have shown that the SB system, employing a hyperactive transposase variant [33], is an efficient nonviral tool for transgenesis in the pig [34,35]. An apparent advantage of SB-catalyzed transgenesis is the preferential integration into transcriptionally permissive genomic loci [36]. In contrast, random transgenesis frequently resulted in silenced transgenes in the pig [37]. Importantly, the SB transposase has a close-to-random insertion profile in mammalian genomes [33]. The majority of SB insertions occurs in intergenic regions, unlike retro- and lentiviral integrations, which favor promoter and exonic regions [10,12,13]. Thus, the SB-catalyzed DNA integration reduces the risk of insertional mutagenesis and represents a rather safe method of gene delivery [33,36].
Here, fibroblasts of Oct4-enhanced green fluorescent protein (EGFP) transgenic pigs were applied to monitor the reprogramming process. The Oct4-EGFP pigs carry the entire 18 kb genomic locus of the murine Oct4, which is one of the best characterized genes involved in pluripotency, controlling expression of an EGFP reporter [38 –40]. In Oct4-EGFP pigs, the EGFP expression was found to be restricted to pluripotent cells of the germ line [41]. Importantly, EGFP expression was re-activated after experimental reprogramming by either somatic cell nuclear transfer (SCNT), fusion of porcine somatic cells with murine embryonic stem (ES) cells, or viral transduction with a set of 4 reprogramming factors [41]. Here, we employed porcine Oct4-EGFP transgenic fibroblasts and a SB transposon-based reprogramming approach to generate pluripotent iPS cells.
Materials and Methods
Plasmid construction
The PB-CAG.OSKM-puΔtk sequences containing the cDNAs of the open reading frames of the 4 Yamanaka factors (murine Oct4, Sox2, Klf4, and cMyc) were removed from piggyBac (PB) vectors described previously [42] by NheI and NotI restriction and cloned into the SpeI and NotI sites of the pT2HB SB vector. The resulting construct contained a cytomegalovirus enhancer, chicken beta-actin hybrid promoter-driven reprogramming cassette (the reprogramming factors were combined into a single open reading frame separated by sequences encoding viral self-cleaving peptide of foot-and mouth-virus (2A) peptides and a puΔtk cassette), flanked by SB inverted terminal repeats (ITRs).
Derivation of Oct4-EGFP transgenic porcine fetal fibroblasts
The Oct4-EGFP transgenic pigs were produced by SCNT and transferred as reconstructed zygotes to synchronized recipients [41]. One pregnant recipient was sacrificed at day 25 of gestation and fetuses were recovered for fibroblast derivation as described [43]. Porcine fetal fibroblasts were cultured in a high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (PAA), 2 mM L-glutamine, 1 mM sodium pyruvate, 1% nonessential amino acids, 0.05 mM β-mercaptoethanol, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells at passage 3 were used for electroporation with transposon plasmids. A Biorad electroporator with square wave function was used for electroporation. For feeder cells, murine embryonic fibroblasts (MEFs) or transformed mouse fibroblast cells with expression of leukemia inhibitory factor (SNL) cells were grown to subconfluency and inactivated with 10 μg/mL mitomycin C (Sigma) followed by thorough washings.
iPS cell generation and cultivation
Presumptive iPS cells were cultured in a human ES cell medium consisting of DMEM/nutrient mixture F-12 supplemented with 20% knockout serum replacement (Millipore), 1 mM L-glutamine, 0.1 mM nonessential amino acids (Gibco), 0.1 mM β-mercaptoethanol (Sigma), 100 U/mL penicillin, 100 μg/mL streptomycin, and 4 ng/mL basic fibroblast growth factor (bFGF) (Peprotech). Presumptive porcine iPS cells were cultured in a humidified atmosphere consisting of 5% CO2 in air at 37°C. Gelatinized plates, or plates seeded with inactivated MEFs or SNLs were used for porcine iPS cell culture. For subpassaging, a mechanical splitting method [27] was employed. In brief, a Pasteur pipette was pulled over a gas burner, in a second step, the tip was bended by 45–60° over a burner. The bended Pasteur pipette was used to scrape the surface of iPS culture dishes. Typically, small aggregates of cells were released; then a fresh iPS medium was added and cell aggregates were subpassed (1:2 to 1:4 ratio) to gelatinized culture dishes. For gelatinization, the intended culture dishes were wetted with sterile 1% gelatin in phosphate-buffered saline (PBS) and allowed to dry immediately before subpassaging. Alternatively, porcine iPS cells were passaged on feeder cells seeded the day before.
Fluorescence microscopy
For fluorescence microscopy, images were made with an Olympus BX 60 (Olympus) fluorescence microscope equipped with a 12-bit digital camera (Olympus DP 71).
Transmission electron microscopy
Putative iPS cell colonies from passage 16 were mechanically harvested for ultrastructural evaluation. Specimens were fixed in 3% glutaraldehyde in 0.1 M Na phosphate buffer (pH 7.3) at room temperature for 1 h, and then transferred and stored in 0.1 M Na phosphate buffer at 4°C. Samples were embedded in Bacto-agar (Difco Laboratories), dehydrated, and embedded in Epon TAAB 812 Embedding resin (VWR), and processed for semithin and ultrathin sections as previously described [44]. Semithin sections were observed using brightfield microscopy (Leica Microsystems) and ultrathin sections were observed under a transmission electron microscope (CM100; Philips).
In vitro differentiation assays
iPS cells were trypsinized and resuspended in the regular ES cell medium (without bFGF) for generation of embryoid bodies (EBs). To induce EB formation, the hanging-drop method was used and drops of 20 μL containing 600 cells were pipetted onto the lids of 10-cm cell culture dishes and incubated at 37°C for 3 days. EBs were washed off the plate with PBS and transferred to 10-cm dishes containing the ES cell medium in which they were incubated for 3 additional days before harvesting.
For ectodermal differentiation [45], iPS cells underwent 3 weeks of coculture in dishes containing murine stromal cells (MS5; obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen) on gelatin-coated glass coverslips in the serum replacement medium containing DMEM, 15% knockout serum replacement, and 2 mM L-glutamine (all from Invitrogen). Immunocytochemistry for TUJ1 (neuron-specific-β-III-tubulin), SOX2, NESTIN, and GFAP was carried out upon appearance of neuron-like structures. For meso- and endodermal differentiation, iPS cells were detached from the feeder layer by collagenase IV (0.2%) (Invitrogen) dispersed into small clumps and cultured in the differentiation medium (80% Iscove's modified Dulbecco's medium [IMDM] supplemented with 20% fetal calf serum, 1 mM L-glutamine, 0.1 mM β-mercaptoethanol, and 1% nonessential amino acids) in ultralow attachment plates (Corning) for 7 days. Subsequently, EBs were plated onto 0.1% gelatin-coated tissue culture dishes and cultured for further 14 days.
Immunocytochemistry
Cell cultures were grown on gelatinized coverslips, washed with PBS, fixed in 80% methanol, and stored at −20°C. For antibody staining, the coverslips were washed 3 times with PBS/0.01% Triton-X100, and were then incubated with the first antibody in PBS/0.01%Triton-X100. The following antibodies were used: mouse anti-Oct4 (sc-5279, 1:200; Santa Cruz), mouse anti-Sox2 (H-65, 1:200; Santa Cruz), mouse anti-Nanog (H-155, 1:200; Santa Cruz) or goat anti-Nanog (1:50; R&D Systems), mouse anti-cytokeratin 19 [TROMA-III, 1:250; Developmental Studies Hybridoma Bank(DSHB)), mouse anti-vimentin (AMF17b, 1:500; DSHB), murine anti-desmin (1:20; Progen Biotechnik), and goat anti-Sox17 (1:200; R&D Systems). The presence of the molecules was detected with a goat anti-mouse immunoglobulin G (IgG) (H+L) Alexa555 conjugated secondary antibody (A 21424, 1:2,000; Invitrogen Molecular Probes) or an anti-goat IgG Alexa 555 conjugated secondary antibody, respectively. In addition, anti-SSEA-1 (1:200) and anti-TRA-1-60 (1:250) antibodies (both from Millipore) were used in combination with an anti IgM secondary antibody conjugated with Alexa 555. Fibroblasts were used as controls, and samples without the first or without the secondary antibody were run in parallel.
Alkaline phosphatase staining
Cells were fixed with 4% formaldehyde, washed with Tris-buffered saline (with 0.1% Tween-20), and stained with an alkaline phosphatase (AP) staining solution [46].
Quantitative real-time polymerase chain reaction
Total RNA was prepared using a TriReagent (Ambion) from Oct4-EGFP fibroblasts (OG2-PFF) and different passages of iPS cells according to the manufacturer's instructions. Isolated total RNA from cell samples was treated with RNase-free DNase (1 U/μg RNA) (Epicentre Biotechnologies) and 0.5 μg was used for cDNA synthesis. Reverse transcription (RT) was performed in a 20 μL volume consisting of 2 μL of 10× RT buffer (Invitrogen), 2 μL of 50 mM MgCl2 (Invitrogen), 2 μL of 10 mM deoxy-triphosphatnucleotides solution (Bioline), 1 μL (20 U) of RNAsin (Applied Biosystems), 1 μL (50 U) of moloney murine leukemia virus (MMLV) reverse transcriptase (Applied Biosystems), and 1 μL hexamers (50 μM) (Applied Biosystems). The samples were incubated at 25°C for 10 min for primer annealing, and then incubated at 42°C for 1 h. Finally, the samples were heated to 95°C for 5 min. The cDNA was diluted 1:5 and 2 μL (10 ng) were used for Real-Time polymerase chain reaction (PCR) amplification. Real-Time PCR was performed in 96-well optical reaction plates (Applied Biosystems). The PCR mix in each well included 10 μL of 2× Power SYBR_Green PCR Master Mix (Applied Biosystems), 6.4 μL dH2O, 0.8 μL each of the forward and reverse primers (5 μM) (Table 1), 2 μL of cDNA in a final reaction volume of 20 μL. The PCR reaction was carried out in an ABI 7500 Fast Real-Time System (Applied Biosystems) using the following program; activation of the Taq Polymerase for 10 min at 95°C followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The specificity of the PCR product was confirmed by dissociation curve analysis and size detection by agarose gel electrophoresis. Data generated by the Sequence Detection Software 1.4 were transferred to Microsoft Excel for analysis. For normalization, the housekeeping genes GAPDH and EEF1A1 were amplified along with the pluripotency-related genes. Normalization factors were calculated with the excel-based software genorm (
PCR, polymerase chain reaction.
Western blotting
Total protein isolation was performed by cell lysis in RIPA buffer on ice for 30 min. Equal amounts of the denatured protein (10 μg) were separated by 12% sodium dodecyl sulfate–polyacrylamide gels. Proteins were transferred onto polyvinylidenfluoride membranes (Hybond-P; GE-Healthcare) using a semidry blotting technique. Membranes were blocked with Tris-buffered saline/0.01% Triton-X100 (TBST) containing 5% dry milk powder for 1 h. Membranes were incubated with the primary antibody anti-Oct4 [Santa Cruz, sc-5279 (1:500)] in TBST with 5% dry milk at room temperature for 1 hour. Membranes were rinsed with TBST and incubated with a horseradish peroxidase-coupled secondary anti-mouse Fab antibody (1:10,000; Sigma) at room temperature for 1 h. Protein detection was performed with an enhanced chemoluminescence reagent (ECL plus; GE Healthcare) and the FusionFX station (VilberLourmat). Transfer efficiency was controlled by E7 (tubulin; DSHB) blotting.
Teratoma formation
To assess the tumorigenic potential of the porcine iPS cells, 1×106 cells were subcutaneously injected into the flank of CD-1 nude mice. The founder animals were obtained from Charles River, and then maintained in the Institute's mouse house according to approved protocols. Mice were analyzed for tumor formation twice a week, and sacrificed 6–12 weeks after treatment. Tumors were fixed in 4% formaldehyde and cryosections were stained with hematoxylin/eosin for histological analyses.
RNA preparation and microarray analysis
Total RNA was extracted using the TRIzol Reagent (Invitrogen). After RNA isolation, a DNase I digest was performed. A second cleanup of isolated RNA was done by extraction with PhOH/CHCl3/Isoamyl and Isoprop/NaOAc precipitation. The RNA was checked for quantity, purity, and integrity of the 18S and 28S ribosomal bands by capillary electrophoresis using the Agilent 2100 bioanalyzer (Agilent Techologies). Only samples with comparable RNA integrity numbers >8 were selected for microarray analysis.
Microarrays were done using the Low RNA Input linear Amplification Kit Plus, One Color protocol (Agilent Technologies, Inc., 2007; Cat. No.: 5188-5339) and the Agilent RNA Spike-In Kit for One color (Agilent Technologies, Inc., 2007; Cat. No.: 5188-5282), according to the manufacturer's standard protocol. Global gene expression analysis was applied using the porcine 4×44 K design array from Agilent Technologies (G2519F; V2: 026440). A total of 200 ng total RNA was used as a starting material to prepare cDNA. The cDNA synthesis and in vitro transcription were performed according to the manufacturer's recommendation. Quantity and efficiency of the labeled amplified cRNA were determined using the NanoDrop ND-1000 UV-VIS spectrophotometer version 3.2.1. The hybridizations were performed for 17 h at 10 rpm and 65°C in a hybridization oven (Agilent). Washing and staining of the arrays were done according to the manufacturer's recommendation. Cy3 intensities were detected by one-color scanning using an Agilent DNA microarray scanner (G2505B) at 5-μm resolution. Scanned image files were visually inspected for artefacts, and then analyzed.
Intensity data were extracted using Agilent's Feature Extraction software (version 9.5.3.1). All chips passed the quality control and were analyzed using the Limma package of Bioconductor. The data reported in this article were generated in compliance with the minimum information about a microarray experiment (MIAME) guidelines.
The microarray data analysis consisted of the following steps: (1) between-array normalization, (2) global clustering and principal components analysis (PCA), (3) fitting the data to a linear model, (4) detection of differential gene expression, and (5) over-representation analysis of differentially expressed genes. Quantile-normalization was applied to the log2-transformed intensity values as a method for between-array normalization to ensure that the intensities had similar distributions across arrays.
P values were obtained from the moderate t-statistic and corrected for multiple testing with the Benjamini–Hochberg method. The P value adjustment guarantees a smaller number of false positive findings by controlling the false discovery rate.
Results
Transposon mediated reprogramming of Oct4-GFP transgenic porcine fibroblasts
Porcine fibroblasts isolated from Oct4-EGFP transgenic fetuses (passage 3) were co-electroporated with a SB transposon carrying a multigene cassette consisting of the cDNAs for Oct4, Sox2, Klf4, and cMyc, separated by self-cleaving 2A peptide sequences (Fig. 1) and a helper plasmid expressing the hyperactive variant of Sleeping Beauty transposase (SB100X). Jumping genes, such as the class II transposon SB, are mobilized via binding of the transposase to the ITRs, excision of the ITR flanked region and insertion of this sequence into a chromosome [48]. Thereby, a single (monomeric) copy of the ITR flanked transposon is integrated into a genomic locus of the host organism.

Reprogramming porcine fibroblasts with Sleeping Beauty (SB) transposon vectors.
About 10–14 days after transposon electroporation, colonies of cells became visible in the porcine cell cultures (Fig. 1B, C). Mock treatments without plasmid DNA, or only one plasmid of the nonautonomous transposon, resulted in no or very few colonies (Table 2). On day 14, the serum-supplemented culture medium was replaced by the serum-free medium, supplemented with the knockout serum replacement and bFGF, and cells were passaged by mechanical splitting to gelatinized plates. Starting from day 20 after electroporation, first EGFP-positive cells were detected (Fig. 2), which were unevenly distributed in the culture dishes and formed loose colonies (Fig. 2A–C). This observation was confirmed after staining with AP (Fig. 2D, E). After proliferation for >40 passages, the porcine putative iPS cells grew in sheet-like colonies with expression of the Oct4-EGFP transgene. The ratio of EGFP-positive cells varied between 1% and 5% of all cells.
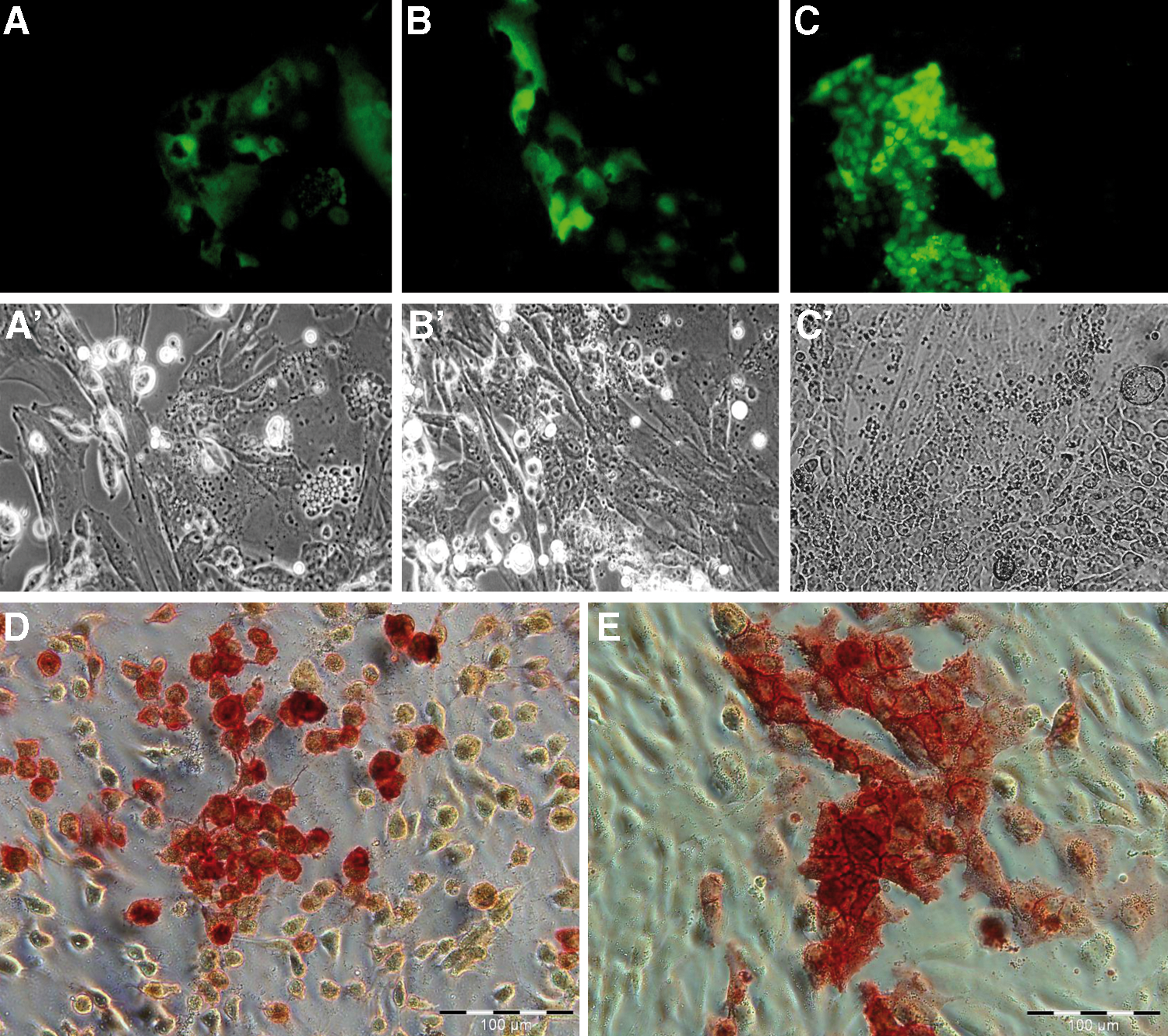
Reactivation of pluripotency reporter Oct4-EGFP.
(n=2); n.a., not applicable; CMV, cytomegalovirus immediate early promoter; SB100X, hyperactive variant of Sleeping Beauty transposase; pOSKM, transposon plasmid carrying the reprogramming factor sequences.
Expression profiling of putative iPS cells
Global gene expression profiling by microarray hybridization revealed a total of 2,166 upregulated and 2,321 downregulated transcripts in the presumptive iPS cells compared to the originating fibroblasts. Putative porcine iPS cells were remarkably stable over different passages (Fig. 1E) and showed expression of stemness-associated genes, such as LIN28, ZFP42 (REX1), SALL4, and TERT. Real-time RT-PCR of selected genes confirmed upregulation of endogenous OCT4, SOX2, UTF1, SALL4, ESSRB, REX1, DPPA5 and TERT (Supplementary Fig. S1; Supplementary Data are available online at
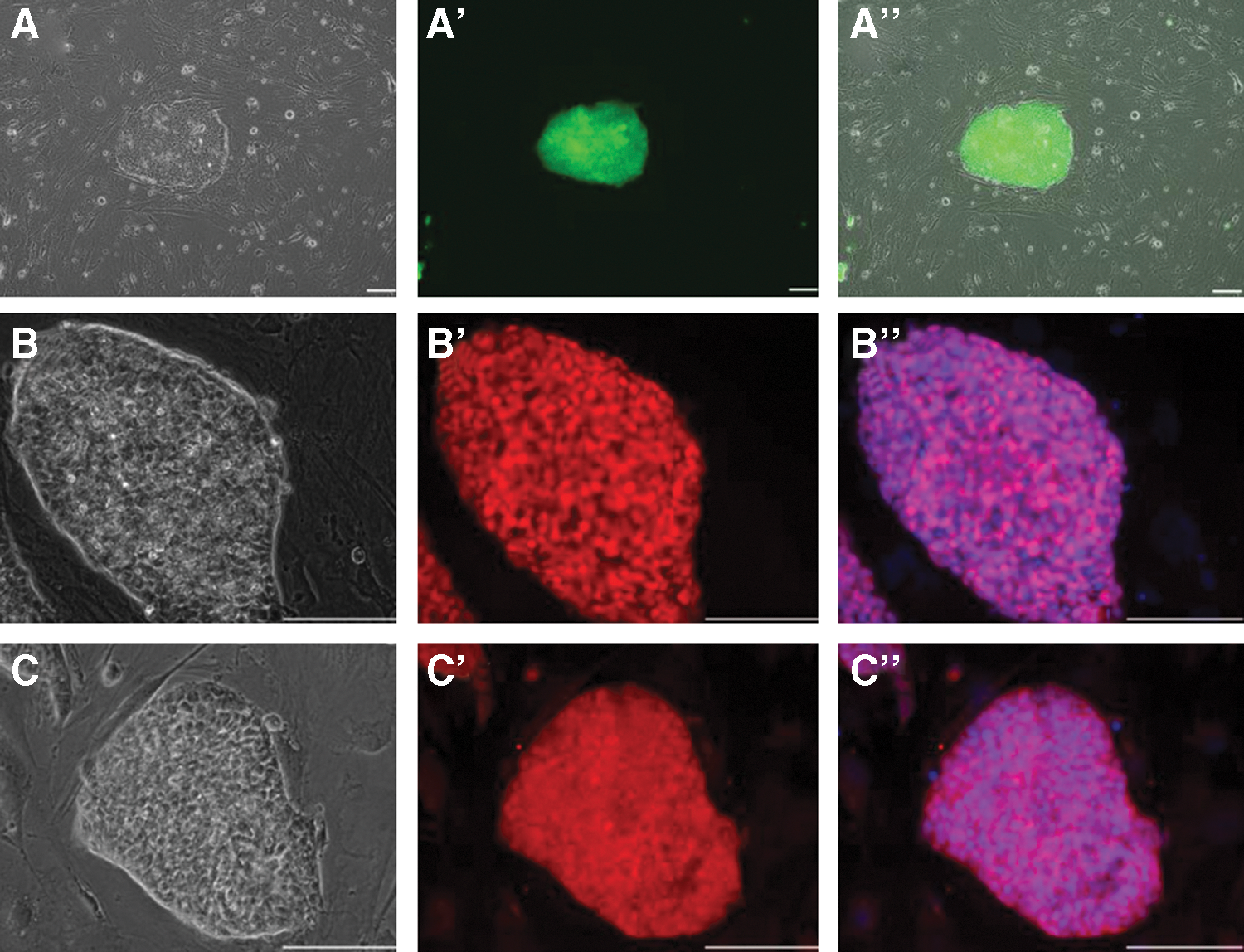
Expression of pluripotency reporter, OCT4 and SOX2 in porcine iPS.
The mRNA expression of the exogenous (transposon-based) multicistronic transcript was determined by species-specific real-time RT-PCR in feeder free-grown porcine iPS cells (Supplementary Fig. S2). Western blotting confirmed the presence of the OCT4 protein in the transposon-treated cells (Supplementary Fig. S2). However, the primary antibody cannot discriminate between the transposon-encoded transcription factor (Oct4) and the porcine OCT4 homolog. In addition, expression of endogenous and exogenous stemness genes were determined by real-time RT-PCR for porcine iPS cells cultured on MEFs (Supplementary Fig. S3). Expression of the surface markers, SSEA1 and TRA-1-60, was verified by indirect immunofluorescence (Supplementary Fig. S4).
Transmission electron microscopy was performed on colonies harvested at passage 16. The analysis revealed that the porcine iPS colonies were compact and composed of cells of uniform morphology (Fig. 4A). The individual cells presented a large, centrally located nucleus rich in euchromatin, typically with a single large nucleolus (Fig. 4B). The cytoplasm only rarely contained organelles, including mitochondria, rough endoplasmic reticulum, and Golgi complexes. Cells in the periphery of the colonies were in some cases connected to each other by tight junctions (Fig. 4C). Apoptotic bodies and cells undergoing apoptosis were occasionally observed (Fig. 4A, D).
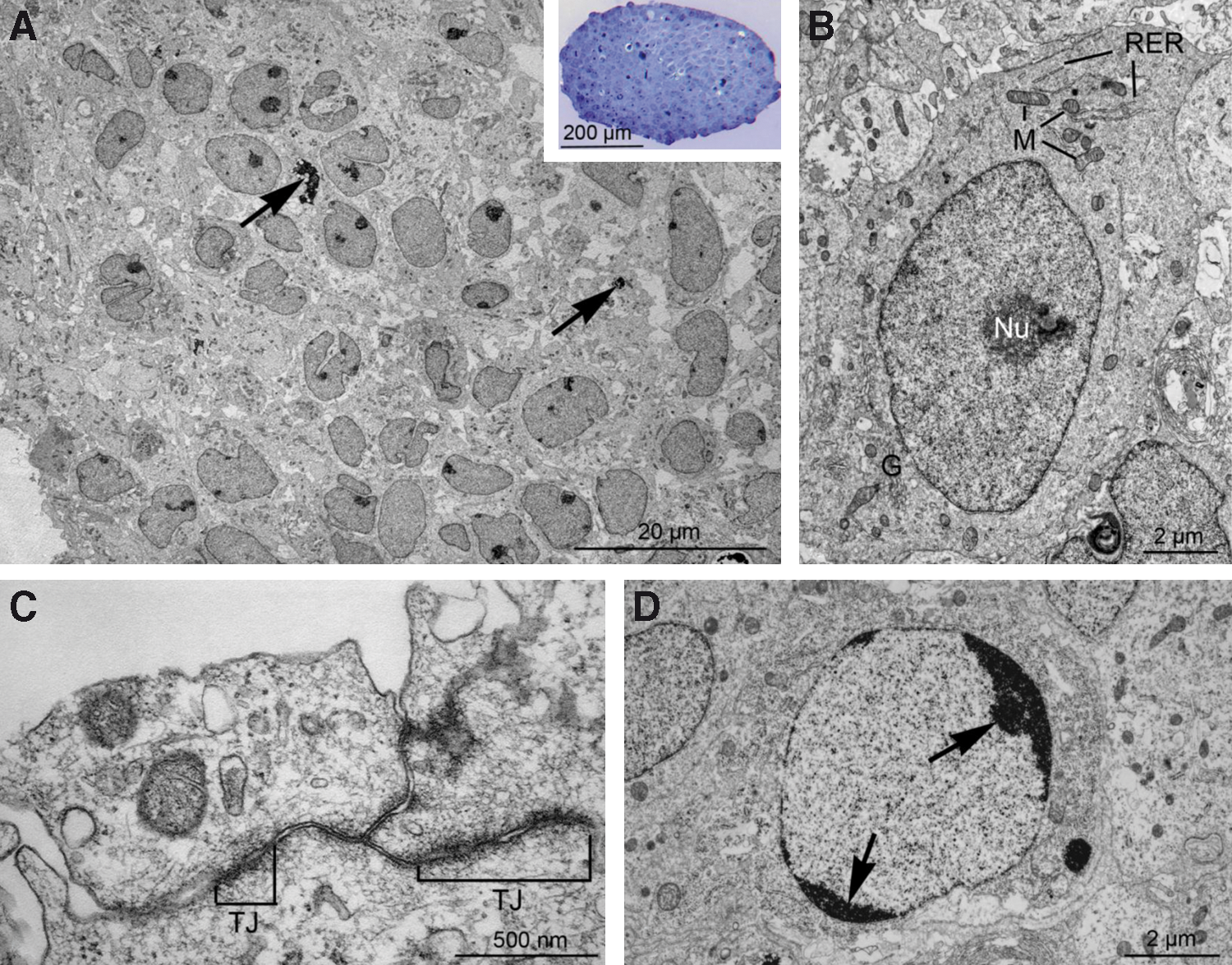
Transposon-derived porcine iPS colonies and cells display typical ultrastructural features of pluripotent stem cells.
Differentiation in vitro and in vivo
In vitro differentiation experiments toward the neuronal lineage [45] resulted in rosette formation and cells, which stained positive for the neuronal progenitor marker NESTIN, SOX2, and the axonal marker TUJ1, all representing ectodermal cell derivatives (Fig. 5). No specific staining for GFAP was found (Fig. 5). Putative mesodermal cells could be identified with antibodies specific for vimentin and desmin; cells of the endoderm were detected with monoclonal antibodies against the endoderm-specific cytokeratin 19 and SOX17 (Fig. 6 and Supplementary Fig. S5). Histochemical staining with oil red and alizarin indicated differentiation toward adipocytes and cartilage cells (Supplementary Fig. S6). In all cases, differentiated cells lost their EGFP fluorescence.
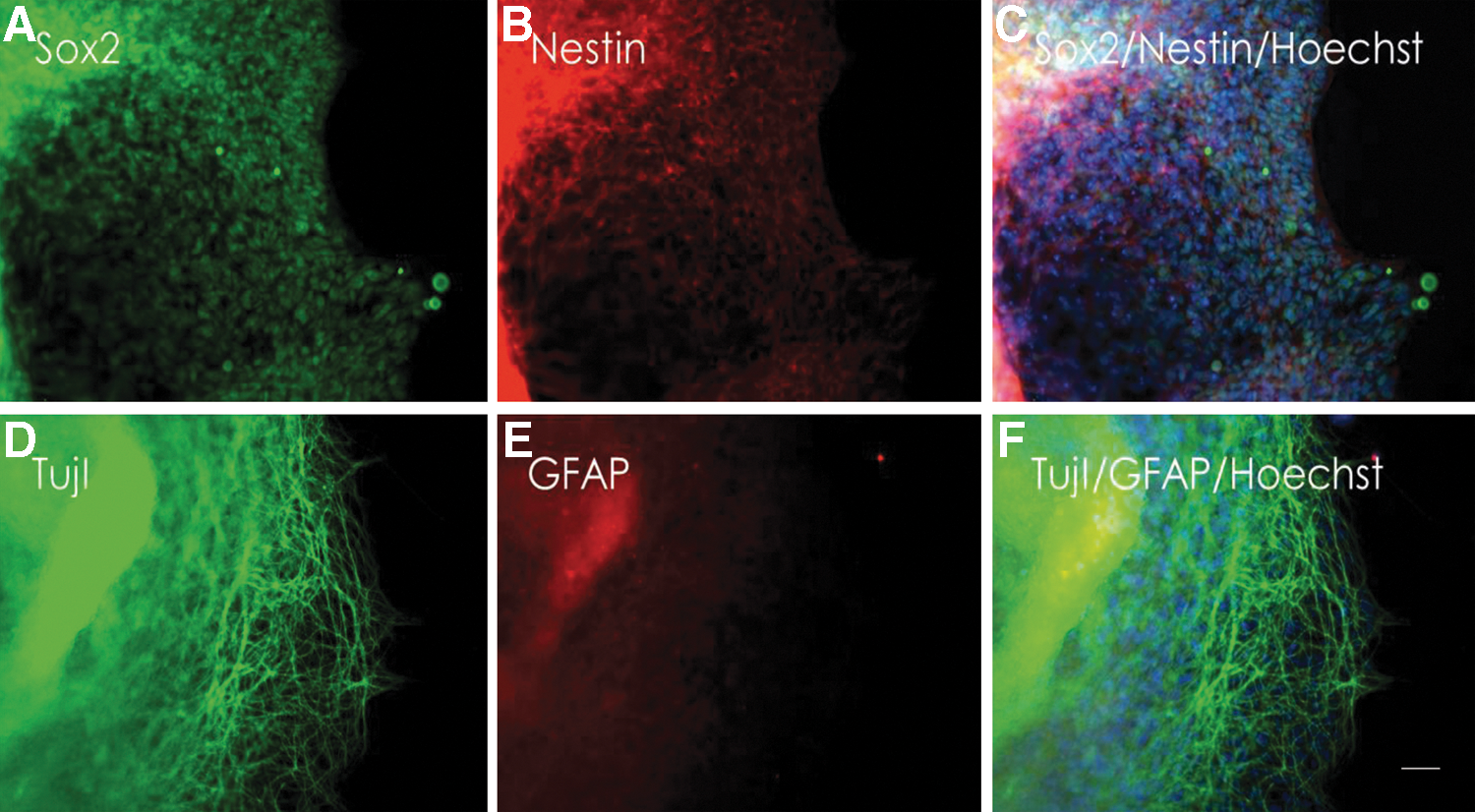
Directed neuronal differentiation of porcine iPS cells.
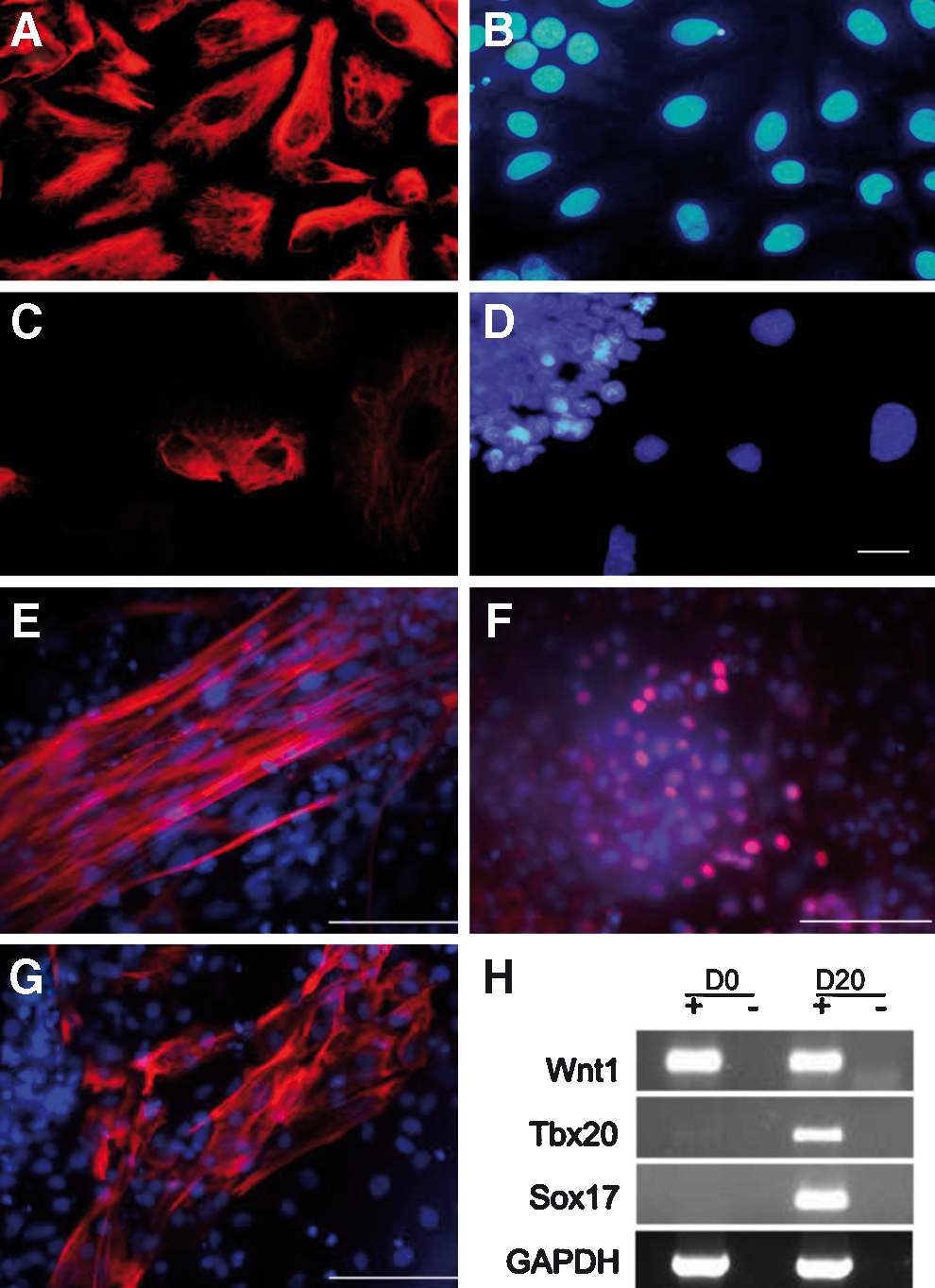
Expression of mesodermal and endodermal markers.
To test their tumorigenic potential, 1×106 putative porcine iPS cells were injected under the skin of 6 immunodeficient nude mice. In 3 animals, a visible tumor growth was observed 2 weeks later. The animals were sacrificed 6–12 weeks after injection and the tumor tissue was sectioned and stained with hematoxylin/eosin. Evidence for the presence derivatives of the 3 germ layers was found indicating the formation of true teratomas (Fig. 7). The sections contained the skeletal muscle, smooth muscle, neuronal, and gland-like structures. Genotyping with species-specific PCR assays confirmed the porcine origin of the teratomas (Supplementary Fig. S7).
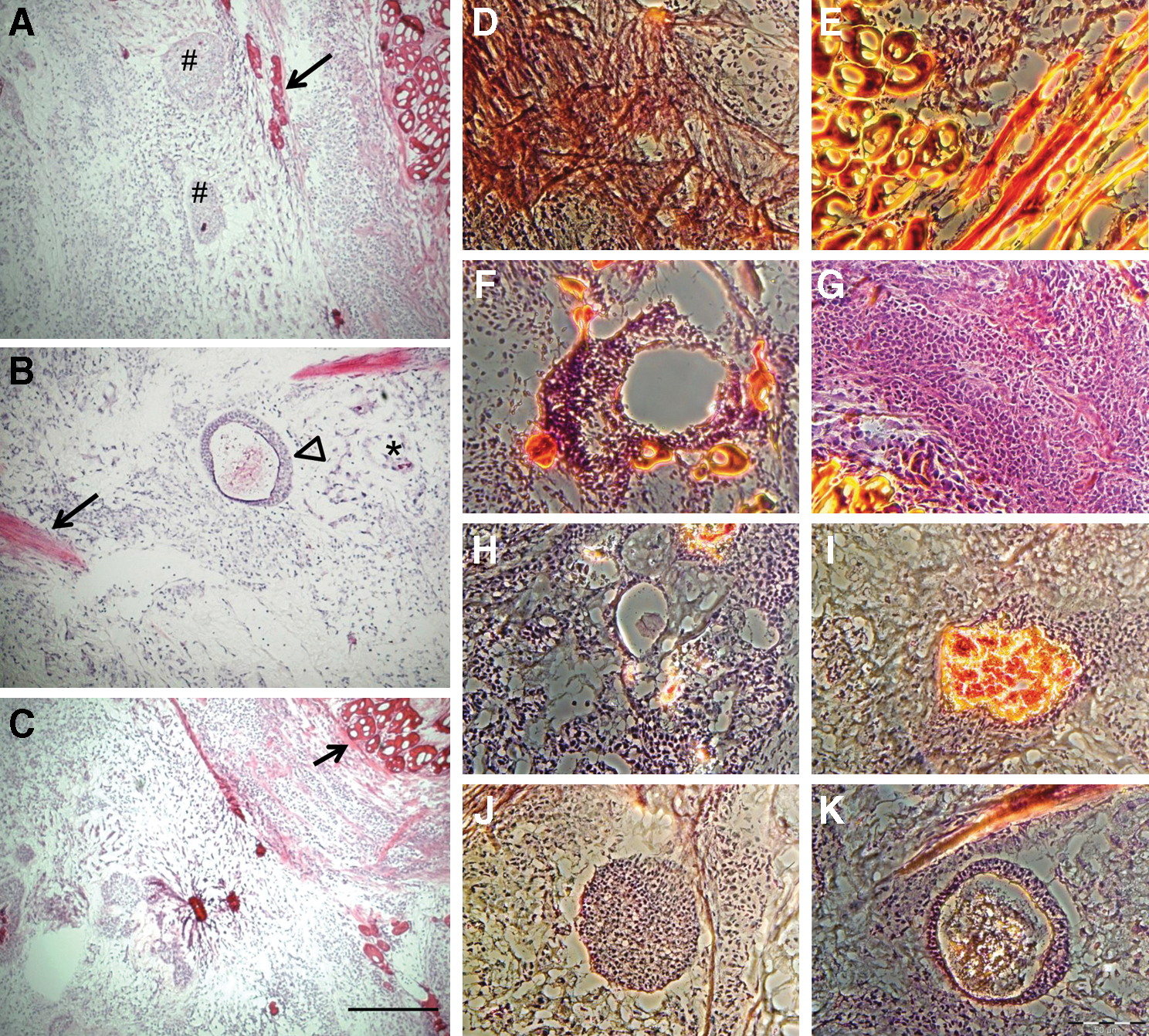
Teratoma formation in nude mice.
Discussion
Here, we used primary fibroblasts derived from Oct4-EGFP transgenic pigs as pluripotency reporter cells and demonstrate their usefulness in reprogramming somatic cells to iPS cells by transposon-mediated delivery of the reprogramming factors. The availability of Oct4-EGFP transgenic pigs allows screening of factors critical for maintenance of pluripotency and to establishing a culture system that is compatible with long-term proliferation of porcine pluripotent cells. Under feeder-free conditions, the porcine iPS cells formed loose colonies. Upon seeding on feeder cells (MEF or SNL), porcine iPS cells grew in typical dense colonies, resembling the morphology of human iPS cells [2,6].
Ultrastructural analysis revealed that the putative iPS cells appeared morphologically uniform and displayed characteristics of typical pluripotent cells, including a large nucleus in a sparse cytoplasm harboring few organelles, a prominent nucleolus, and an abundant amount of euchromatin [49]. At the periphery of the colony, adjacent cells were connected by tight junctions indicating epithelial characteristics; a similar observation has been made in human ES cells [49].
Attempts to derive true porcine iPS cells have met with limited success, although chimerism upon blastocyst complementation with porcine iPS cells [28] and germ line transmission [29] has been reported by one group. Unfortunately, both piglets with germline-transmitted reprogramming factors died around birth [29].
The lack of true porcine ES cells prevents the unequivocal identification of pluripotent cells after reprogramming toward iPS cells in this species. In contrast, the available mouse and human ES cells clearly facilitated the development of protocols for cellular reprogramming in these species [16,50]. In reprogrammed human cells, colonies with phenotypes that resemble true ES cells differed with regard to molecular marker expression and differentiation potential [51]. By analyzing expression of pluripotency markers, the methylation status at the OCT4 and NANOG promoters, and the differentiation potential into teratomas, true iPS cells could be discriminated from cells that were not completely reprogrammed [51]. Mouse reprogramming studies were facilitated by the use of pluripotency reporter gene constructs, driven by either the Fbx15, Oct4, or Nanog promoter, and nascent iPS cells could successfully be identified [1,3,4,13]. Culture conditions for long-term maintenance of pluripotency in murine cells are well defined [52]. Recent findings indicate that long-term maintenance of pluripotency of murine ES and iPS cells depends on the presence of a rare two-cell (2C) stage [53].
In the present study, we used somatic cells from Oct4-EGFP transgenic pigs, and show that this facilitated reprogramming, thus representing a valuable tool for studies into the reprogramming of somatic cells into iPS cells. Experiments have been initiated to excise the reprogramming SB-transposon to generate foot print-free porcine iPS cells. In PB reprogrammed murine iPS, the traceless removal of a reprogramming transposon has been shown [54]. Recently, the suitability of SB-mediated reprogramming of murine fibroblasts to iPS cells has been demonstrated [55].
The transposon-mediated reprogramming reported here has distinct advantages over viral approaches reported previously [25 –28]. Transposon plasmid preparation is straightforward and does not require additional security precautions, as for retro- and lentiviral vectors. The SB transposase catalyzes integration into the host genome in a random manner, without preference for promoter or exonic regions, thus reducing the risk of insertional mutagenesis [54,56,57].
In conclusion, results of the present study demonstrate the usefulness of porcine Oct4-EGFP cells and transposon-mediated reprogramming for the derivation of porcine iPS cells. However, this study also highlights the limitations of current culture systems. Variable cell morphology in different passages and a low ratio of Oct4-EGFP-positive cells indicated that maintenance of pluripotency in reprogrammed porcine cells is not yet fully achieved under the present conditions. Most likely this is due to the culture conditions and culture media, which were adapted from work with human and mouse pluripotent cells. The remarkable differences between human and murine pluripotent cells [58 –61] suggest that porcine iPS require species-specific media and growth factor supplements. A molecular basis for this different responsiveness to culture conditions could be the species-specific expression of regulatory micro-RNAs (miRNA); certain miRNA-clusters are correlated with stemness and ensure fine tuning of post-transcription regulatory networks [62]. Nevertheless, results of this study set the basis for in-depth studies on pluripotency sustaining factors in porcine cells.
Footnotes
Acknowledgments
The authors are grateful for the expert technical support of Stephanie Holler and Nicole Cleve. We thank Dr. S. Galge (MHH) for support with teratoma analysis. Gratefully, Prof. R. Michael Roberts (University of Missouri) gifted lentivirus reprogrammed porcine iPS, described in [![]() ]. The authors thank Dr. S. Petkov and Dr. T.R. Talluri for critical reading of the manuscript. Financial support by the Bundesministerium für Bildung und Forschung (iPSILAM and ReGene consortia), the EU projects EU FP7 PartnErS (PIAP-GA-2008-218205) and PluriSys (HEALTH-2007-B-223485) and InduStem, the Danish National Advanced Technology Foundation, and the Danish Council for Independent Research, Technology and Production Science is gratefully acknowledged.
]. The authors thank Dr. S. Petkov and Dr. T.R. Talluri for critical reading of the manuscript. Financial support by the Bundesministerium für Bildung und Forschung (iPSILAM and ReGene consortia), the EU projects EU FP7 PartnErS (PIAP-GA-2008-218205) and PluriSys (HEALTH-2007-B-223485) and InduStem, the Danish National Advanced Technology Foundation, and the Danish Council for Independent Research, Technology and Production Science is gratefully acknowledged.
Ethics Statement
Animals were maintained and treated according to German laws for animal welfare and for genetically modified organisms.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.