
Abstract
Mesenchymal stromal cells (MSCs) are a heterogeneous cell population capable of differentiating toward several cell lines in vitro and, possibly, in vivo. Within cultured MSCs, we identified and purified a precursor cell population [mesodermal progenitor cells (MPCs)] retaining robust proliferation potential and ability to differentiate into endothelial or mesenchymal cells. MPC-derived MSCs retain the ability to further differentiate into osteoblasts, cartilage, or fat cells. Here we further characterized MPCs and MSCs by evaluating expression of integrins and adhesion molecules showing their ability to assemble the molecular machinery involved in endothelium adhesion. MPCs were shown to interact with activated and nonactivated endothelium, whereas MSCs exhibited activation of focal adhesion complexes, higher cell motility, and reduced or absent adhesiveness onto endothelial cells, suggesting a matrix remodeling vocation. We also reported a consistent expression of CXCR4 on the MPC cell surface, suggesting that the different phenotypic behavior could be related to specific functions of the cell in each differentiation stage.
Introduction
We previously described a new bone marrow cell population coisolated in autologous serum MSC cultures that we named mesodermal progenitor cells (MPCs) for their ability to differentiate into early-stage MSCs as well as toward endothelial and cardiomyogenic lineages [17]. MPCs have peculiar morphological, phenotypic, and molecular features allowing to discriminate them from MSCs [18]. MPCs firmly adhere to plastic supports, and they are not easily detached by trypsin digestion [19]. Interestingly, MPCs are resting cells expressing pluripotency-associated proteins as SSEA-4, Oct-4, Nanog, and Nestin [20]. Therefore, MPCs appear to be of high interest in view of their potential for clinical applications in regenerative medicine.
Controversies about MSC expression of surface receptors or functional structures to sustain tissue engraftment [21] prompted us to further characterize MPCs that may contaminate MSC cultures to investigate the presence of surface integrins (ITGs) as well as specific adhesion/invasion structures that could play a role in cell homing and cell activation.
Materials and Methods
Primary cell cultures
Bone marrow samples were obtained from six patients (four men/two women, median age: 66 years) undergoing hip replacement, after written consent. To isolate MSCs, bone marrow mononuclear cells (BMMNCs) were standard cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) using hydrophilic tissue culture-treated flasks (TC flasks). In parallel, the BMMNC samples were cultured in DMEM supplemented with 10% pooled human AB serum (PhABS) from male donors only (Lonza) on no-gas-treated hydrophobic plastic flasks as previously described [18], to isolate MPCs. The media were changed every 48 h, and the cultures maintained at 37°C and 5% CO2 for 10–12 days. The cells were detached by TrypLE Select® (Invitrogen) digestion and processed for cytological characterization and RNA extraction.
Further, six samples (three men/three women, median age: 71 years) were processed to grow mixed cultures of MPCs and MSCs. BMMNCs were cultured in DMEM supplemented with 5% FBS and 5% PhABS in TC flasks. After 10–12 days, cells were detached by TripLE Select digestion and processed for flow cytometry. In parallel, mixed cultures were also grown in Lab-Tek® double-chamber slides (Nunc) to obtain fluorescence microscopy specimens.
Cytofluorimetric validation of primary culture samples
The purity of cell preparations was assayed using anti-SSEA-4 Alexa-Fluor-488-conjugated (Biolegend), anti-MSCA-1 PE-conjugated (Miltenyi Biotec), and anti-CD90 PE/Cy5-conjugated (Becton Dickinson) antibodies. Aliquots of cell suspensions were washed in PBS/0.5% BSA/0.01% NaN3 and stained with fluorescent primary antibodies for 30 min at 4°C. The samples were then acquired using FACSCanto II® (Becton Dickinson) and analyzed by Diva Software®. MPC cultures were selected for further analysis when the MSC mesenchymal component (SSEA-4negMSCA-1+CD90bright) was lower than 2%. Similarly, MSC cultures were selected when the MPC component (SSEA-4+MSCA-1negCD90neg) was lower than 2%.
Molecular expression profiling of extracellular matrix and adhesion molecule genes
Total RNA was extracted using an RNeasy Mini Kit (Qiagen GmbH), as indicated by the manufacturer's protocol. On-column DNase I digestion was performed. In detail, 100 ng of RNA samples was retrotranscribed by the QuantiTect Whole Transcriptome Kit (Qiagen), and 50-fold cDNA dilutions were analyzed by quantitative real-time PCR using the iCycler-iQ5 Optical System (Bio-Rad Laboratories) and iQ SYBR Green SuperMix (Bio-Rad).
The expression profile analysis of the extracellular matrix (ECM) and adhesion molecule (AM) genes was performed using the RT2 Profiler™ PCR array kit (SABioscience, Qiagen) according to the manufacturer's instructions. Relative quantitative analysis was carried out by following the 2−ΔΔCt Livak method [22]. The genes were considered differentially expressed when at least 1 log difference was detected. Gene expression was defined as “consistent“ when the relative fold expression was 0.01 or higher, “mild” with values between 0.01 and 0.001, and “not expressed“ with values lower than 0.001.
CXCR4 mRNA expression was investigated using specific primer pairs designed to detect CXCR4 v1 and CXCR4 v2 variants (NCBI Ref. No.: NM_001008540, NM_003467):
CXCR4 v1 Sense 5′-GCTTGCTGAATTGGAAGTGAATG-3′,
CXCR4 v1 Antisense 5′-CCACAATGCCAGTTAAGAAGATGA-3′,
CXCR4 v2 Sense 5′-CAGCAGGTAGCAAAGTGA-3′,
CXCR4 v2 Antisense 5′-TCGGTGTAGTTATCTGAAGTG-3′.
Quantitative PCR was performed as previously described.
Human phosphokinase antibody array
To obtain a mixed-cell preparation of MPCs and MSCs, four bone marrow samples (two men/two women, median age: 69 years) were cultured as described in the culture method section in either PhABS or FBS, and cells detached by TrypLE Select®. Proteome profiling was performed using the Human Phosphokinase Antibody Array kit from R&D Systems according to the manufacturer's instructions. Leica QWin image analysis software (Leica) was used for densitometric evaluations. Data were presented as a net pixel density calculated by subtracting the median gray levels of dots.
Cytofluorimetric analysis of integrin surface expression in mixed cultures
Detached cells from mixed cultures were processed for flow cytometry and stained with anti-CD11a, anti-CD49f, anti-CD104, or anti-CD105 FITC-conjugated antibodies; anti-CD11b or anti-CD11c PE-conjugated antibodies; anti-CD90 or anti-CD184 PE/Cy5-conjugated antibodies; or anti-CD18 APC-conjugated antibodies (all antibodies from Becton Dickinson). Appropriate isotypic controls for each fluorescence channel were used to set quadrants on the dot-plots. To discriminate between MPCs and MSCs, the events were displayed on an SSC vs CD90 dot-plot; the SSClow/highCD90neg events were gated as MPCs, whereas the SSClowCD90bright events were gated as MSCs.
Tricolor immunofluorescence of mixed cultures
Slides were fixed for 15 min in periodate-lysine-paraformaldehyde and subsequently permeabilized by 0.05% Triton X-100 for 30 min. Immunofluorescence was performed using mouse monoclonal antibodies against Integrin-α6, β2, and β4, Gelsolin (Abcam), and Paxillin (Becton Dickinson) and then revealed by Goat anti-mouse SFX kit (Invitrogen), according to the manufacturer's instructions using Alexa-Fluor®-488 anti-mouse IgG. The slides were stained by Phalloidin Alexa-Fluor®-555-conjugated antibody (Invitrogen) for 30 min to reveal F-actin organization, and mounted in Prolong® Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) to detect the nuclei. For colocalization experiments, Integrin-β2 was revealed, and then slides were stained with rabbit monoclonal antibodies against Integrin-αL, αM, or αX (Abcam) and revealed using Goat anti-rabbit IgG (H+L) DyLight™-549-conjugated (KPL). Pictures were taken and combined using a standard fluorescence DMR Leica microscope (Leica) equipped with Leica CW4000 image software (Leica).
Endothelial cell adhesion assay
Human umbilical vein endothelial cells (HUVECs) were obtained after written consent by using a standard method [23]. Briefly, after 30 collagenase digestions, the cell suspensions were washed twice in D-PBS and plated in 75-cm2 fibronectin-coated culture flasks in an EGM-2® medium (Lonza). At confluence, cells were detached by TrypLE Select® digestion and replated in 96-well plates at 50,000 cells/well density. The following day, confluent cultures were activated by adding 50 ng/mL recombinant IL-1β, 50 ng/mL TNF-α (Miltenyi), or a combination of the two. Nonactivated HUVEC wells were also prepared. Ten bone marrow samples (six men/four women, median age 68) were also processed as described in the culture method section to harvest MPCs. To obtain paired MSC samples, 20,000 MPCs/cm2 were differentiated in TC gas-treated T75 flasks using the MesenPRO® medium (Invitrogen) as previously reported [17], and cultures maintained for two passages (P2-MSCs). To perform endothelial cell adhesion assay, cells were detached, and suspensions incubated at 37°C overnight in a fresh culture medium under constant agitation. MPCs and P2-MSCs were fluorescence-labeled by CSFE (Invitrogen) according to the manufacturer, and 100,000 cells/well plated on an HUVEC monolayer. In parallel, control plates with HUVECs only were set. After 4 h in the dark, floating cells were washed out, and fluorescence of adhered cells was quantified using a Victor2 1420 plate reader (Applied Biosystems) equipped with 485-/530-nm excitation/emission filters.
Statistical analysis
Statistical analyses were carried out using The Prism Version 5 package (GraphPad). Numerical data are expressed as means±SEM. All experiments were analyzed by two-tailed Student's t-test for unpaired samples, to determine the P value. Where multiple comparisons were made, one-way ANOVA with Bonferroni post-test was performed.
Results
MPCs and MSCs were unambiguously identified by morphology and phenotype as described previously [17 –20], thus allowing us to perform molecular profiling assays on homogeneous cell-type populations. In particular, two out of six MPC primary cultures whose mesenchymal-contaminating component (SSEA-4negMSCA-1+CD90bright) resulted higher than 2% were excluded from further analysis. The remaining four validated MPC cultures showed monomorphic, rounded, highly rifrangent cells in PhABS and typical spindle-shaped cells in FBS (data not shown).
RT2 Profiler PCR array for ECM and AMs provided quantification of expression for 84 human genes, including matrix proteins, ITGs, metallopeptidases (MMPs), as well as their inhibitors (TIMPs), and other AMs. MSCs expressed 50 out of the 84 genes. In particular, 44 genes were consistently expressed with 26 showing expression over 1 log higher than in MPCs (Fig. 1A). We found that 17 of the MSC-expressed genes were coding for matrix proteins or were directly related to the synthesis of matrix components. Four genes were characterized as MMPs (MMP-2, MMP-3, MMP-11, and MMP-16; Fig. 1B), while the remaining five genes were identified as coding for cytokines CTGF and TGFβ1 or AMs (VCAM1, ITGA1, and ITGA8; Supplementary Fig. S1A; Supplementary Data are available online at

Quantitative molecular expression profiling of ECM and adhesion molecules.
To analyze and characterize the surface integrin profiles, mixed MPC/MSC cultures were used to avoid possible misinterpretations due to different culture conditions. After 10–12 days, mixed cultures showed two morphologically distinct populations: fried egg-shaped MPCs alongside with classic spindle-shaped MSCs (Fig. 2). Cytofluorimetric data confirmed gene expression results for Integrin-α6 (CD49f), αL (CD11a), αM (CD11b), αX (CD11c), and β2 (CD18) showing positive stain exclusively on MPC events (dark green dots in Fig. 2), while Integrin-β4 (CD104) was detected neither in MPCs nor in MSCs (data not shown).
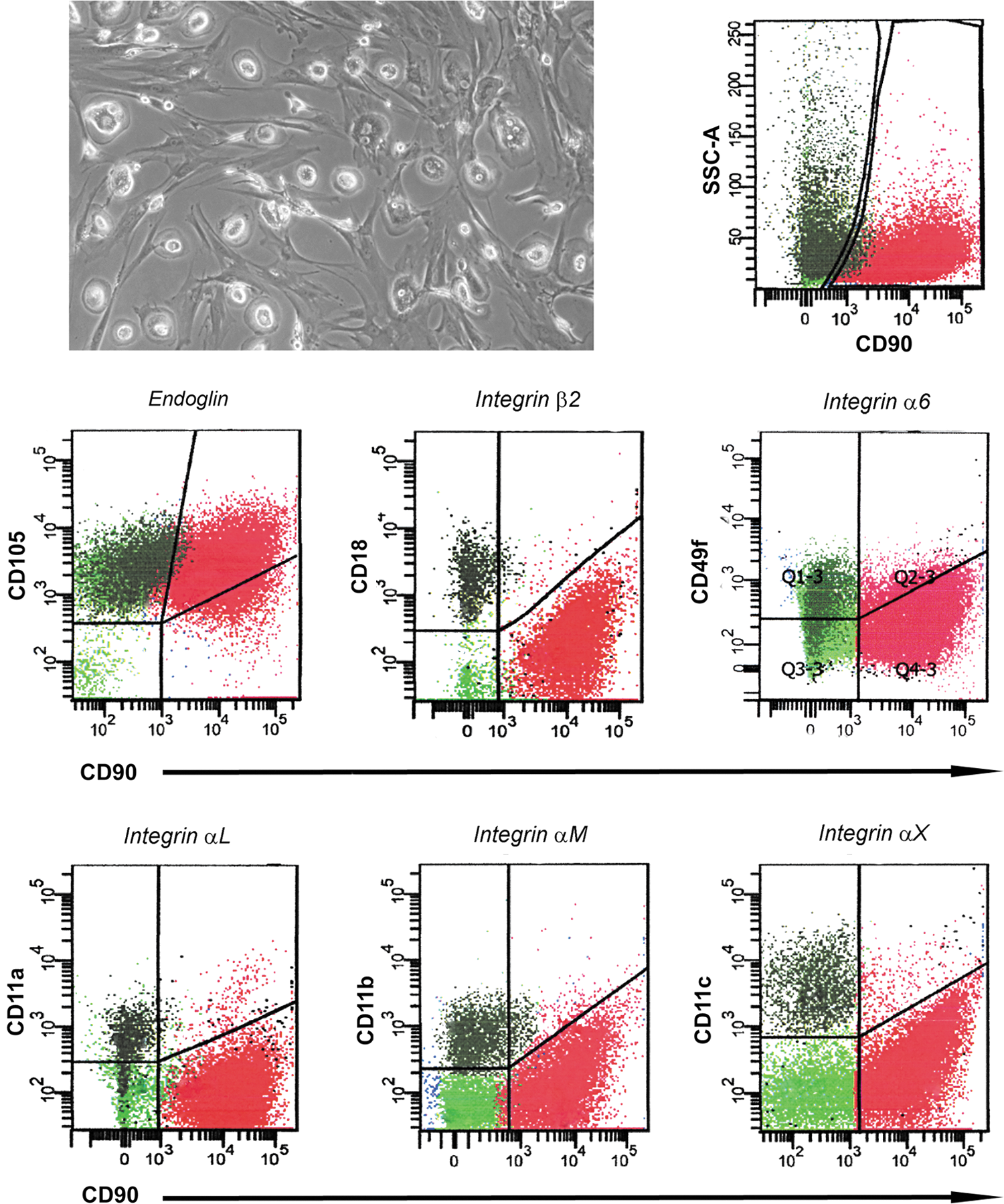
Surface integrin expression in cells from mixed cultures. Culturing bone marrow mononuclear cells on tissue culture-treated flasks with 5% of pooled human AB serum and 5% of fetal bovine serum, MPC/MSC mixed cultures were obtained. Phase-contrast microscopy verified coisolation of the two populations. Flow cytometry revealed SSClow/highCD90neg events (MPCs, dark green dots) alongside with SSClowCD90bright events (MSCs, red dots). MPC-exclusive expression of Integrin-α6 (CD49f), αL (CD11a), αM (CD11b), αX (CD11c), and β2 (CD18) was confirmed.
Immunofluorescence confirmed the different F-actin organization patterns between MPCs and MSCs reported previously. MPCs were characterized by the presence of a high number of F-actin dots, sometimes grouped in rosettes, as revealed by Phalloidin stain (red in Fig. 3). The typical dotted pattern is usually associated to a transient functional adhesion structure called podosome, this including activated rosette structures. Gelsolin bright positive stain (green in Fig. 3) colocalized with F-actin dots (yellow in Fig. 3) and not with other actinic structures as ruffles (Fig. 3A) or filipodia (Fig. 3B), confirming their podosomal nature. Paxillin was not detected on MPCs. On the contrary, MSCs showed the characteristic organization of stress fibres (Fig. 4A) starting from focal adhesion (FA) plaques, as revealed by the Paxillin bright stain (green in Fig. 4A), at the ends of long polymerized chains of F-actin (red in Fig. 4A). Further, MSCs exhibited higher phosphorylation of FA complex kinases Paxillin, FAK, and Src (Fig. 4B) alongside with downstream phosphokinases ERK1/2, MEK1/2, JNK, and c-Jun. These data confirmed the activation of FA signaling in MSCs only. Time-lapse microphotography confirmed FA activation, showing for adherent MSC highly active cell motility with intense ruffling at the periphery (Supplementary Movie S1). Conversely, MPCs showed neither cell motility nor active short-term turnover of filipodia. Expression of Integrin-β2, which appeared very bright in MPCs (green in Fig. 5A), was spatially associated with podosomes surrounding the actinic dotted structures (Fig. 5B) or colocalized in rosettes (yellow in Fig. 5C). Integrin-β2 showed no association with other nonpodosomal actin structures, as ruffles and lamellipodia. Immunofluorescence for Integrin-α chains, exclusively expressed by MPCs, revealed that Integrin-αX colocalized with Integrin-β2 (Supplementary Fig. S2A) surrounding the podosomes (Supplementary Fig. S2B) with Integrin-αL, and Integrin-αM (Supplementary Fig. S3). Integrin-α6 in MPCs was not spatially associated to podosomes (Supplementary Fig. S4).
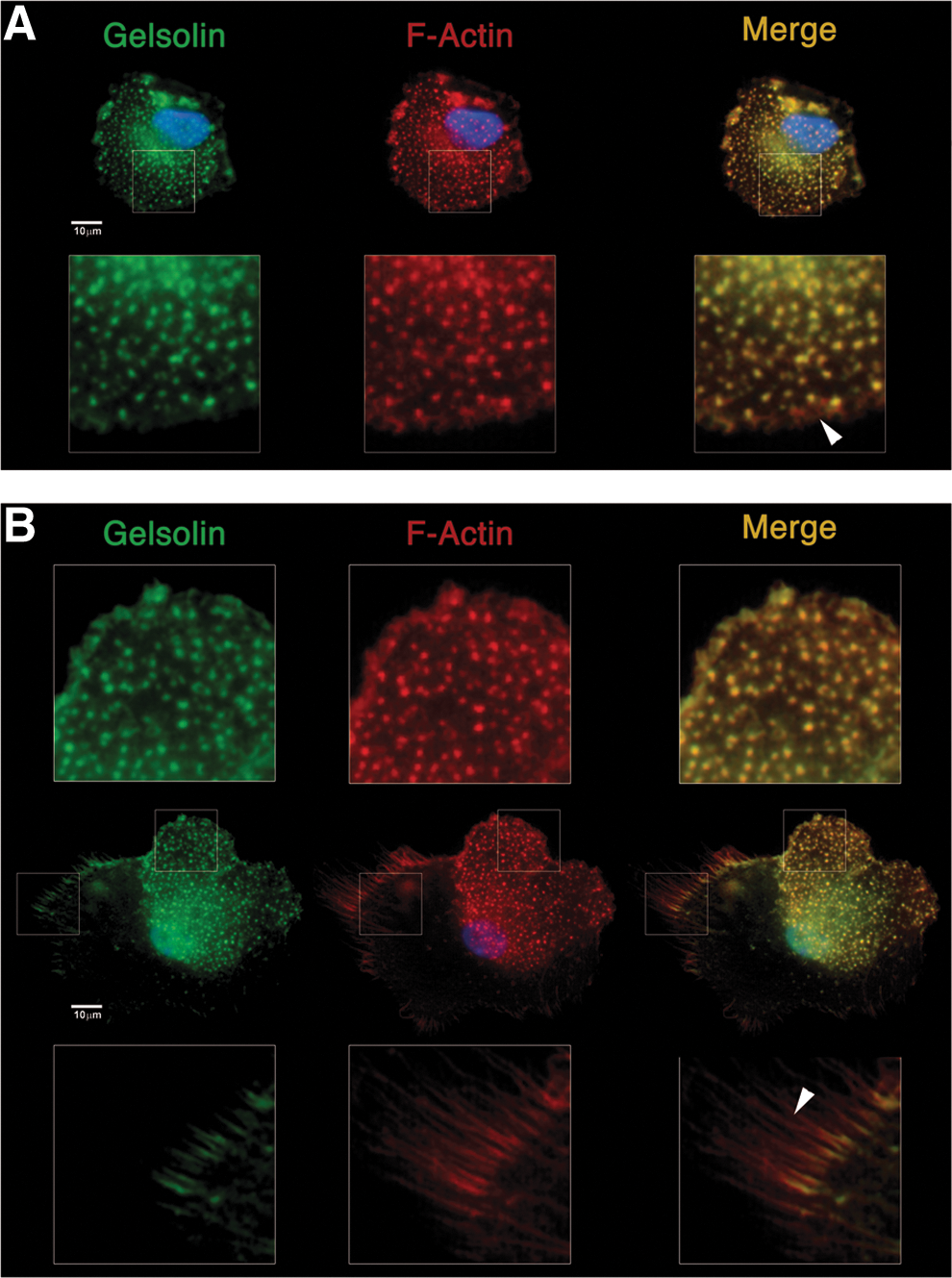
F-actin organization and podosomal structure in MPCs. MPCs showed a characteristic dotted F-actin (red) pattern, typically associated with podosomes. Gelsolin bright stain (green) colocalized with actinic dots (yellow in merged picture) and confirmed their podosomal nature. Structures with different F-actin organization as
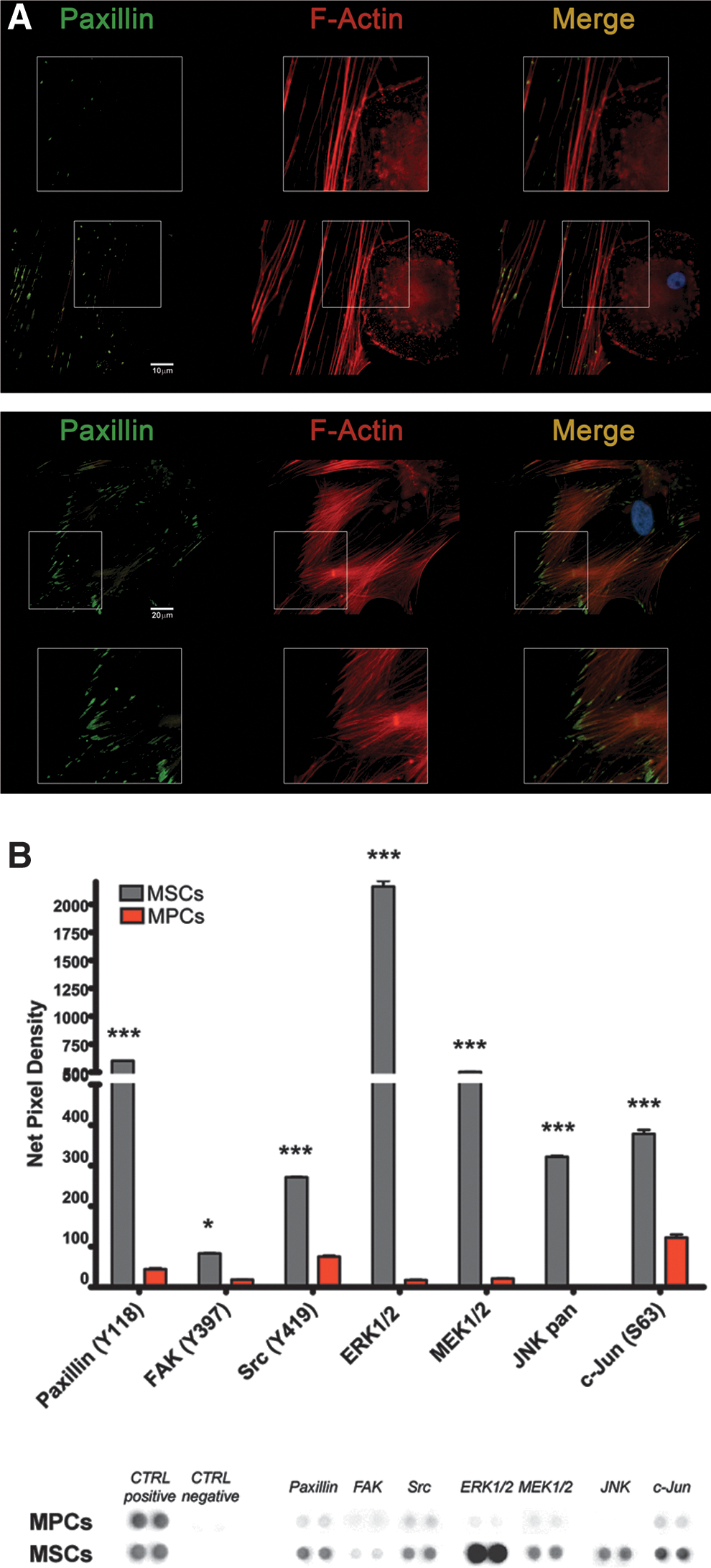
F-actin organization and focal adhesion plaques in MSCs.
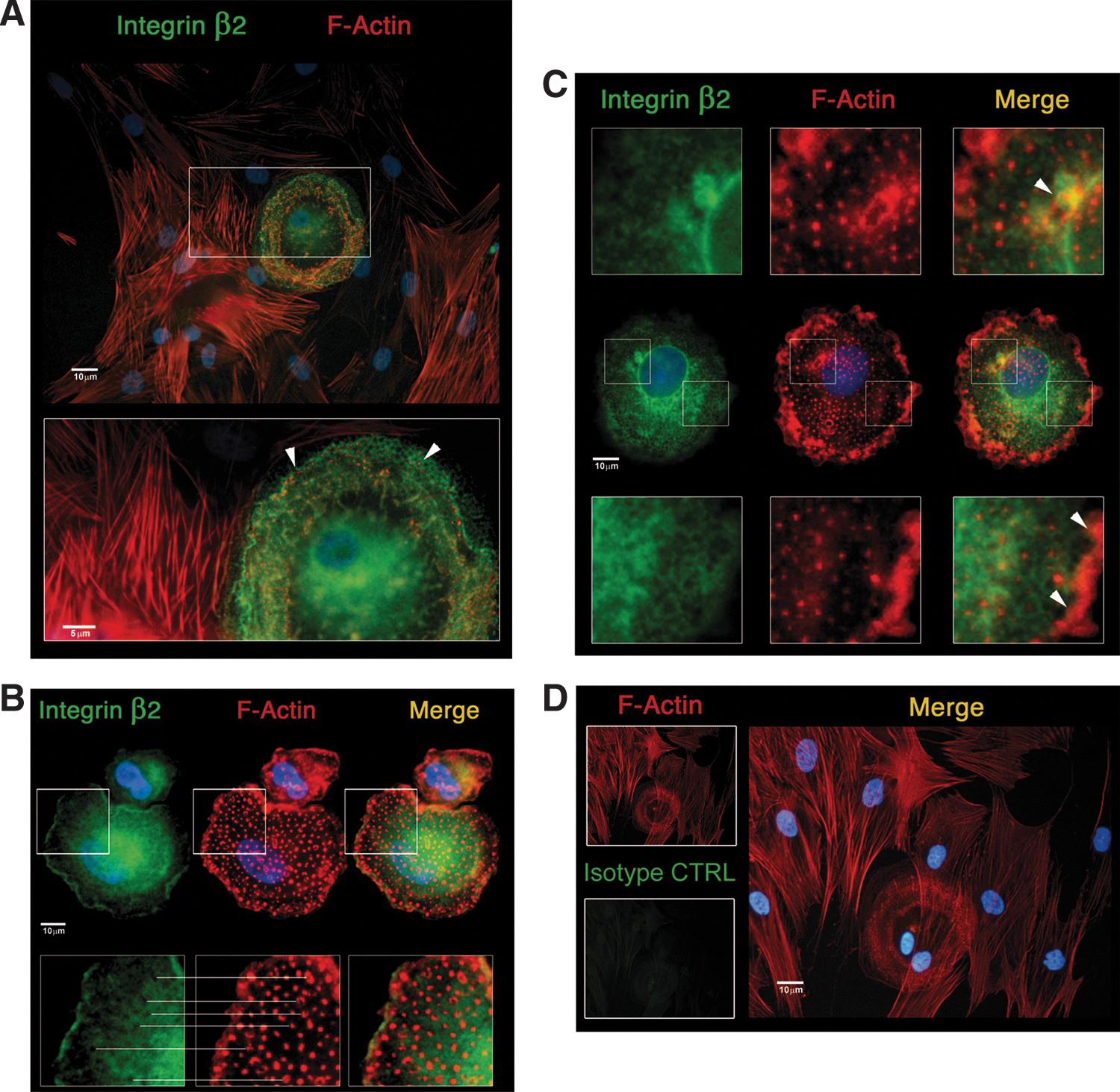
Tricolor immunofluorescent detection of Integrin-β2.
Functional studies on endothelium adhesion revealed that MPCs were able to interact with activated HUVECs as shown by significantly high-fluorescence signals (Fig. 6A, P<0.001). No difference with controls was found in P2-MSCs, indicating that most cells were washed out because of their inability to adhere, independently by the endothelial activation treatment (Supplementary Fig. S5). MPCs showed adhesion properties regardless of HUVEC treatments (Fig. 6B), demonstrating that MPCs were able to interact also with nonactivated endothelium.
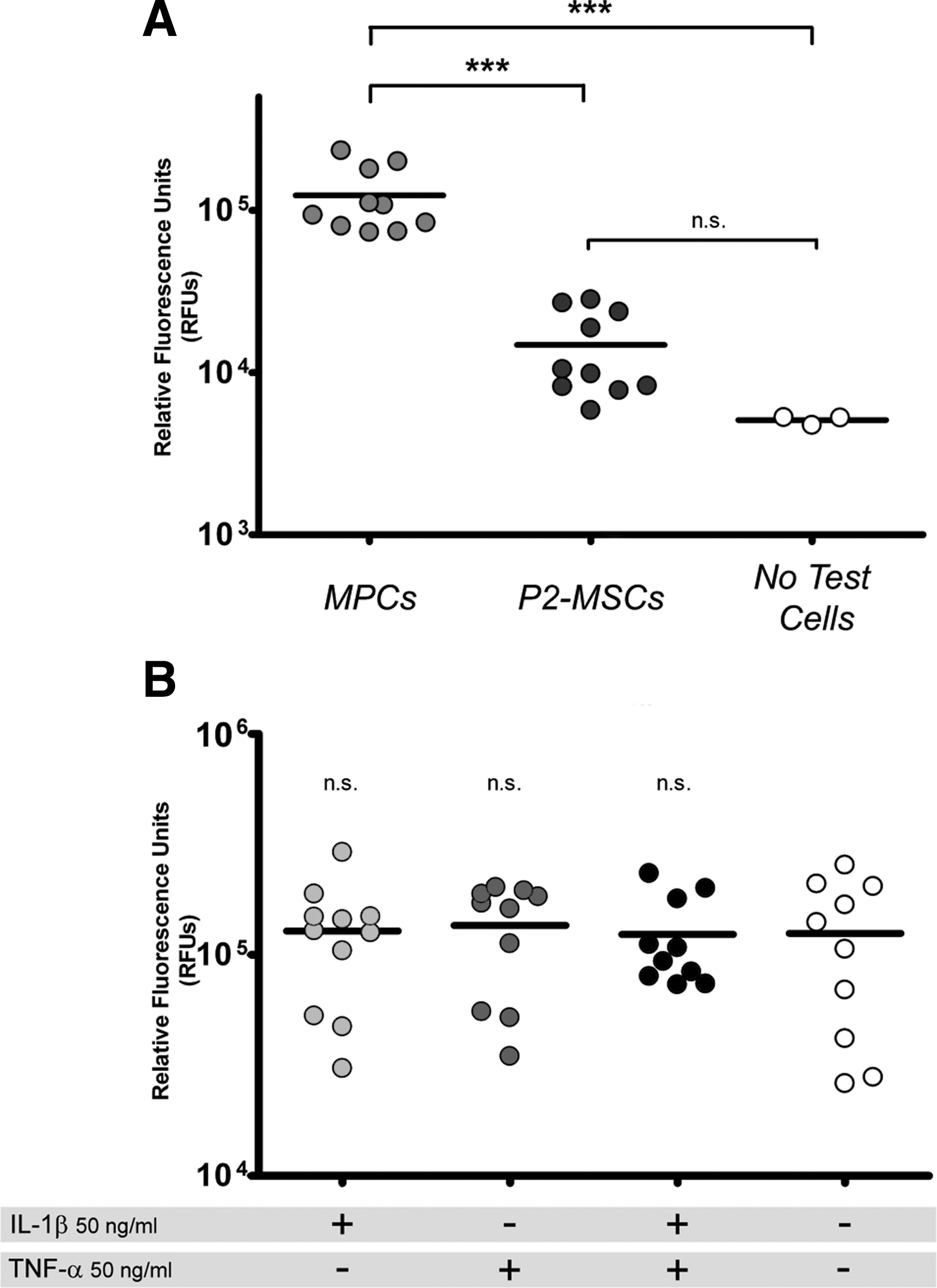
Static endothelial cell adhesion assay.
Quantitative RT-PCR of CXCR4 revealed consistent expression of both CXCR4 v1 and CXCR4 v2 variants in MPCs only (Supplementary Fig. S6A). These data were confirmed by cytofluorimetric analysis where all MPC events (SSClow/highCD90negCD105+) showed a positive stain for CXCR4, while MSCs (SSClowCD90+CD105+) were completely negative (Supplementary Fig. S6B).
Discussion
Our group previously identified MPCs as MSC precursors in human bone marrow cultures, showing the possibility to obtain purified cell preparations suitable for specific analysis [18]. Being aware that several cell populations suggested to be the precursor of MSC have been reported, we described the putative differences among these cell populations in a previous article [19]. We believe that different cell culture conditions may influence the phenotypic and even functional differences among the reported mesenchymal precursors; however, the peculiar characteristics of this ex vivo cultured cell population allow us to identify MPCs as a specific cell population. MPCs express a number of genes usually associated to pluripotency, and they are able to differentiate, in vitro, toward mesenchymal or endothelial lineages while retaining a robust proliferative capability. In the present article, we further characterized MPCs and showed that these cells, which are highly adherent cell population, feature a distinctive pattern of surface ITGs as compared to MSCs. In particular, CD11a, CD11b, CD11c, and CD18 were exclusively expressed in MPCs.
Immunofluorescence analysis showed the presence of a number of podosome-like adhesion structures in MPCs. Podosomes are highly dynamic actin-rich membrane structures presenting a dense F-actin core surrounded by a ring of AMs [24]. They are involved in the attachment to the ECM, in various cell types, and can assemble to form clusters (rosettes). These surface structures, when associated with focal ECM protein degradation [25] and membrane protrusions, are also referred as invadosomes, which are reported to be present on the surface of many different cell types, including osteoclasts, macrophages, epithelial, endothelial and vascular smooth muscle cells, and subsets of leukocytes. Here we provided data demonstrating that MPC podosome-like structures are sustained by Integrin-β2 in heterodimers with Integrin-αX, αL, or αM, to form complement receptor 4 (CR4), lymphocyte function-associated antigen 1, and macrophage antigen 1 (CR3/Mac-1), respectively.
In leukocytes, focal basement membrane protein degradation is driven by the formation of protrusive invadosomes, where secreted MMP-9 is reported to specifically complex with β2-Integrin and in particular in the αMβ2 heterodimer [26,27]. Interestingly, MPCs express consistent levels of MMP-9. On the contrary, MSCs showed a tangential cell–matrix interaction mediated by FAs, which are known to provide the principal sites of attachment to the underlying or surrounding substrate. This result was consistent with MSC-specific expression of a number of matrix proteins as well as different MMP mRNAs.
In our highly selective experimental setting, we found CXCR4 to be specifically expressed by the totality of MPCs. Wynn et al. [28] reported a very low percentage (from 1.0 to 3.9%) of CXCR4-positive stromal cells in FBS-cultured bone marrow, thus suggesting MPC contamination at the earliest passages of their MSC cultures.
In our hands, MSCs exhibited activation of FA complexes, higher cell motility, and reduced or absent adhesiveness onto endothelial cells, suggesting for them a matrix remodeling vocation. The present results clearly indicated that MPCs carry surface structures highly similar to podosomes, specifically involving Integrin-β2, αX, αM, and αL, and they possess the ability to adhere with activated as well as nonactivated endothelium. Conversely, MSCs do not significantly adhere to endothelium. In a recent article, Teo et al. elegantly demonstrated that a subpopulation of human MSCs (hMSCs) cultured in 15% FBS (from passage 3 to passage 7) are able to interact with TNFα-treated endothelial cells [29]. Nonetheless, the authors reported that only small portions of the seeded hMSCs were not washed out during static EC adhesion assays, while single-cell analysis of the adhesion mechanisms revealed heterogeneity of the cell-to-cell interaction that do not involve podosomes, which are not detected on hMSCs. Most of the cells showed integration associated to EC monolayer retraction that the authors ascribed to the poor/unhealthy adult EC monolayer used. On the contrary, some authors reported that murine MSCs show very low interaction with activated endothelium in static conditions. Nonetheless, these groups demonstrated that EC adhesion could be activated by shear stress [30,31].
We first demonstrated that the MPC population is able to rapidly give rise to a mesenchymal offspring when cultured in FBS or human cord blood serum, and is able to differentiate toward the endothelial lineage, and thus the cells were named MPCs. We also reported that different percentages of PhABS and FBS in the culture medium and the variability of the used batches of sera could lead to different percentages of MPCs coisolated in the BM-MSC preparations [18]. Further, according to Fazzi et al. [17], we characterized two different populations of MSCs: early MSCs and late MSCs, after the MPC induction. On the basis of the expression of CD105, CD90, and SSEA-4, we were able to describe a first step of induction with the contemporary presence of MPCs (CD105+CD90negSSEA-4+), early MSCs (CD105+CD90+SSEA-4+), and late MSCs (CD105+CD90+SSEA-4neg). These data support the hypothesis that most of the controversies in the field of the MSC biology could be ascribed to the intrapopulation heterogeneity of the cell preparation and to the not stringent criteria of characterization [32,33]. Further, interpopulation variability introduced by different origins and cell isolation protocols is actually far to be correctly evaluated, especially in terms of percentage of MPCs, early MSCs, or late MSCs. The lack of SSEA-4 detection, in the widely applied phenotype characterization, and moreover the lack of the investigation of the CD90-negative population could lead to incorrect or absent evaluation of the MPC counterpart, which shows a wider differentiation capability, as well as specific adhesion properties, erroneously attributed to the generically defined MSCs.
According to the hierarchical model, we proposed that the MPCs as the in vivo putative progenitor of the mesenchymal and endothelial lineage, present in the BM-MNC population from 1% to 3%, when seeded in an FBS-containing medium, these cells could rapidly undergo to mesenchymal, as well as endothelial, differentiation giving rise to the so-called MSC culture after few days, and at the end, generating the misinterpretation of the genuine identity of the cultured and highly heterogeneous MSC population. Further, MPCs show resistance to trypsin digestion during detachment, and consequently, these cells could be lost in the course of passaging, lowering the proliferation/differentiation potential of the subcultures.
The influence of culture conditions on the BM-derived progenitor cell isolation is well documented [32] and could also represent an obstacle to the correct data interpretation about the identity of another interesting precursor cell population, endothelial progenitor cells (EPCs). Controversies about origins of EPCs and their in vivo identity are still far to be elucidated mainly due to the in vitro methods applying to isolate EPCs and to assay angiogenesis [34]. EPCs showed endothelial differentiation potential sharing with some early hematopoietic markers as CD34 and CD133, sustaining the concept of the existence of a multipotent precursor termed hemoangioblast. Similarly, MPCs carry some endothelial characteristics as PECAM-1 expression and capillary formation potential in Matrigel®, alongside the capability to form mesenchymal tissues [18,19] that could sustain the emerging concept of the existence of a multipotent precursor, distinct from the hemangioblastic lineage, termed mesangioblast [35].
Concluding, in this article, we demonstrated that culturing BMMNCs in media with different serum supplementations (PhABS or FBS) could lead to a different percentage of the MPC/MSC counterparts that show completely different actin distribution, FAK-ERK1/2 activation, and cytoskeleton structure/remodeling alongside a peculiar ITG, AM, and MMP expression pattern. We believe that future studies on adhesion properties, endothelium interaction, and ECM remodeling functions of cultured BM-derived multipotent cells should take in consideration the heterogeneity of the cell preparations, evaluating the percentage of CD105+CD90neg MPCs.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.