
Abstract
The efficiency of in vitro culture systems for a premeiotic female germ cell is still low, mostly because of our incomplete understanding of the mechanisms controlling oogenesis and the obvious difficulties in reproducing the complex in vivo environment of such a process under in vitro conditions. Here we explored the possibility of recovering the developmental potential of mouse oocytes generated in vitro from premeiotic germ cells by transplantation under a kidney capsule of adult animals. To this aim, mouse embryonic ovaries of 12.5 days postcoitum cultured in vitro in a serum-free medium for 7 or 14 days, were transplanted beneath the kidney capsule of immunodeficient mice and analyzed after 21 (7+21 group) or 14 days (14+14 group). Cultured ovaries before transplantation showed delayed oocyte meiotic progression and follicle development. Interestingly, grafted ovaries of both groups, especially those of the 7+21 group, seemed able to restore the reproductive cycle of recipients. While the almost complete absence of primordial follicles was observed in grafted ovaries, oocytes from these ovaries showed transcript levels of genes associated to oocyte maturation similar to control. Moreover, the developmental stage of follicles and oocytes of the 7+21 group ovaries were comparable to that of 21 days post partum in vivo ovaries, whereas significant developmental delay were found in the 14+14 group ovaries. Nevertheless, oocytes retrieved from transplanted ovaries of both groups matured (around 80%) and were fertilized in vitro (around 20%–45%). Two-cell embryos from the fertilized oocytes developed to hatching blastocysts (about 50%) or gave rise to healthy live offspring (from 6% to 10%) when transplanted in a host mother. In conclusion, our results indicate that premeiotic female germ cells cultured in vitro up to primordial/primary follicle stages preserve their capability to complete oogenesis and can be fertilized and generate live pups after transplantation into a suitable in vivo environment.
Introduction
D
In the last few decades, in vitro culture systems for female germ cells from always more earlier developmental stages have been devised, including germinal vesicle (GV) oocytes from follicles at various developmental stages [16 –19], oocytes from fetal ovaries [20 –23], migratory and postmigratory PGCs [6,24,25], and putative female germ cells generated from embryonic stem cells [26 –28], induced pluripotent stem cells (iPSCs) [29], or even adult stem cells [30 –33]. These systems have provided valuable information about several aspects of germ cell formation and oogenesis in mammals and offer intriguing perspectives for future clinical applications [34]. Many obstacles must be still overcome to increase the efficiency of these in vitro culture methods and to reproduce in vitro the complex in vivo environment necessary for the production of functional and normal oocytes. In particular, the development of female PGCs and fetal or postnatal oocytes is quite difficult to be correctly and completely reproduced in vitro [17,23,35,36]. Similarly, while the formation of PGCs from pluripotent stem cells seems a relatively efficient process and the generation of haploid male germ cells in vitro or after in vivo transplantation have been reported [37 –40], the formation of mature oocytes derived from the stem cell has not been described so far [29,30,32,41 –43]. In this regard, it should be particularly important to establish culture conditions for female germ cells, which are able to reproduce at least some stages, if not all of such process, without compromising their capability to give rise to mature oocytes.
The low efficiency of current in vitro culture systems for germ cell formation, development, and maturation results from our incomplete understanding of oogenesis or folliculogenesis, and the obvious difficulties in reproducing the complex in vivo environment under in vitro conditions. Relative to in vitro induction and culture, the in vivo environment had proven its high efficiency and adaptability on supporting germ cell development. The ovarian orthotopic in vivo environment supports the renewal and differentiation of long-term cultured thecal stem cells [44] and female germ line stem cells [12,13]. The adult ectopic in vivo environment, such as the kidney capsule, supports the maturation of oocytes from purified fetal germ cells through aggregation and transplantation procedures [45], and living offspring had obtained from 12.5 dpc fetal mouse ovaries after ectopic transplantation [46]. Notably, recipient mice seem able to support the full-term development of female germ cells, despite sex differences in the hormonal environment of the recipients [46,47]. Finally, both embryonic and adult stem cell-derived germ cells obtained in vitro appear able to form developing follicles after aggregation with ovarian somatic cells and transplanted beneath the kidney capsule of recipients [28,48].
Based on our previous in vitro culture studies on mouse fetal ovaries [22,23,25], the aim of the present study was to explore the possibility of recovering the developmental potential of in vitro cultured premeiotic female germ cells by transplantation under the kidney capsule.
Materials and Methods
Animal care and mating
All mice used were housed and bred under controlled lighting conditions (12L: 12D). Timed mating was produced by housing females with males overnight, and then checking for vaginal plugs the next morning; 0.5 dpc=noon of the day when the vaginal plug was found. All procedures involving animals were approved by the Institutional Animal Care and Use Committees of the Qingdao Agricultural University (No. 20090617).
Fetal mouse ovaries isolation, culture, and transplantation
Fetal ovaries were obtained from 12.5 dpc pregnant CD-1 or chicken beta-actin promoter/enhanced green fluorescence proteins (CAG/EGFP) transgenic mice line (Stock TgN (GFPU) 5 Nagy) carrying a ubiquitously expressed EGFP) [49]. The sex of the isolated gonads was determined by their morphological appearance, and the attached mesonephroses cut away under a dissecting microscope. The ovaries were transferred in a 600 μL (310-thick) prebalanced culture medium onto 2% agarose blocks placed in a 24-well plate (Costar) [50,51]. The agarose block was made by adding 1 mL of liquid 2% agarose, and then removing it quickly, so the agarose formed a very thin layer block (less than 50 μm thick) and could prevent the cultured ovaries from adhering to the bottom of the well. The cultures were carried out at 37°C in a saturated humidity incubator (Thermo) infused with 5% CO2 in air. The medium was refreshed by exchanging 300 μL of used medium with 350 μL of prebalanced fresh medium every other day. The culture medium consisted of a 1:1 combination of DMEM/F12 (Hyclone) and α-MEM (Hyclone), supplemented with 1 mg/mL of Fetuin (Merck 341506), 3 mg/mL of bovine serum albumin (BSA, roche A8020), 1% Insulin-Transferrin-Selenium-A (ITS, Gibco, 51300), 0.23 mM sodium pyruvate (Hyclone), and 1% penicillin–streptomycin (Hyclone). The cultured ovaries were randomly divided into 2 groups as follows (Fig. 1):

Experimental design: 12.5 dpc (day postcoitum) mouse ovaries were isolated and in vitro cultured for 7 days or 14 days, and surgically transplanted beneath the kidney capsule of bilaterally ovariectomized immunodeficient mice. Ovaries were then collected after 21 days (7+21 group) or 14 days (14+14 group) for morphological analysis. Moreover, retrieved oocytes were subjected to in vitro maturation (IVM), in vitro fertilization (IVF), amd embryo transfer analysis.
Group 1: ovaries were in vitro cultured for 7 days before transplantation under the kidney capsule and recovered after 21 days (7+21 group).
Group 2: ovaries were in vitro cultured for 14 days before transplantation under the kidney capsule and recovered after 14 days (14+14 group).
For transplantation, 8–10-week-old SCID Beige female mice (Vital River) were intraperitoneally anesthetized with 240 mg/kg avertin (Sigma, T48402), and bilaterally ovariectomized. Donor ovaries were picked up with a mouth controlled glass Pasteur pipette and implanted beneath the right kidney capsule of recipient mice in less than 3 min [28,52 –54]. For each recipient, 8–10 cultured fetal ovaries were implanted as the number of germ cells decreased during in vitro culture. After grafting, the edges of the body wall and back skin of recipient mice were aligned and closed with suture. Grafts were collected from the recipient mice at the day described. Five IU pregnant mare serum gonadotropin (PMSG) was intraperitoneally injected to the recipient mice 48 h before collection [47].
Evaluation of meiotic prophase I stages
The meiotic prophase I stages were evaluated by cytospreads of oocytes isolated from ovaries obtained from 13.5, 15.5, 17.5, and 19.5 dpc CD-1 embryos or from in vitro cultured 12.5 dpc ovaries after 1–7 days of culture. Briefly, ovaries were disaggregated into single cells with 0.25% trypsin plus 0.02% EDTA (Hyclone). After neutralization with 10% fetal bovine serum (FBS; Gibco, 10099-141), the cells were treated in 1% trisodium citrate (Sigma) hypotonic solution for 20 min at room temperature, then fixed in 1% paraformaldehyde (PFA, Beyotime), spread onto poly-L-lysine (Sigma) precoated slides, and dried at 37°C. Slides were blocked in ADB (3% BSA, 1% normal goat serum, and 0.005% Triton-100 in tris buffered saline, TBS) for 30 min at room temperature and incubated overnight with 1: 400 diluted rabbit anti-SCP3 antibody (Novus Biologicals; NB300-232) at 37°C, then rinsing 3 times in TBS and subsequently incubated with 1: 400 diluted R-phycoerythrin-conjugated goat anti-rabbit secondary antibody (Sigma, P9537) for 1.5 h at 37°C in dark and finally counter stained with 1 μg/mL Hoechst33342 (Sigma, B2261) for 2 min. The meiotic prophase stages were determined under the Olympus BX51 fluorescence microscope with Cell Sens Ver.1.5 software by observing the characteristic patterns of SCP3 immunostaining of the oocyte chromosomes.
Histological analysis, immunohistochemistry, and follicle counts
At the end of the indicated time, in vitro cultured or grafted ovaries were fixed in the Bouin's solution overnight at 4°C and processed for standard histology. The samples were serially sectioned at 6 μm and subjected to hematoxylin–eosin staining or immunohistochemistry and examined for follicle count and development stages.
For immunohistochemistry, sections were heated at 60°C for 2 h. After de-paraffinization, sections were re-hydrated in a series of graded ethanol/water solutions, and boiled in 0.01 M sodium citrate (pH 6.0) at 96°C for 10 min. After incubation in 3% hydrogen peroxide (H2O2) for 10 min, the tissues were blocked in BDT (3% BSA, 10% normal goat serum in TBS) for 30 min and incubated with rabbit anti-STAT3 polyclonal antibody (Santa, SC-482) (a germ cell marker) [55] at a dilution of 1: 200 overnight at 4°C. Finally, after carefully rinsing in TBS, the sections were incubated with the horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Beyotime; A0208) at a dilution of 1: 50 for 45 min at 37°C, washed 3 times, and transferred in 3, 3′-diaminobenzidine (DAB, Beyotime, P0203). Samples were analyzed under the Olympus BX51 fluorescence microscope. The number of primordial, primary, secondary, antral follicles, and multioocyte follicles (MOF) was then scored every 4 slices.
Hormonal assay
Recipient mice, age-matched recipient mice control with or without bilaterally ovariectomization, and 21 dpp donor mice control, were anesthetized with avertin as described above. From each mouse, about 1 mL of blood samples were collected by heart puncture using a 1-mL injector. The concentration of E2 in each sample was measured by a chemiluminescence immunoassay analyzer.
In vitro maturation of GV stage oocytes
Unless otherwise indicated, all chemicals used in in vitro maturation (IVM) and in vitro fertilization (IVF) were purchased from Sigma Chemical Co. All medium drops used in IVM, IVF, and embryo cultures were covered with mineral oil and prebalanced in 37°C, 5% CO2, saturated humidity incubator for at least 6 h before use.
Antral follicle GV oocytes from transplanted or 21 dpp in vivo control (DMIV group) CD-1 ovaries were released by puncturing the ovaries by fine needles. Oocytes with or without cumulus cells were collected by a mouth controlled glass Pasteur pipette and washed twice in the M2 medium and 3 times in the maturation medium before transferring into maturation medium drops. The maturation medium consisted of TCM-199 (Gibco; 11150-059) supplemented with 10% (v/v) FBS (Gibco; 10099-141), 1 μg/mL β-estradiol 17-hemisuccinate (Sigma; E9252), 24.2 mg/L sodium pyruvate, 0.05 IU/mL follicle-stimulating hormone (FSH; Sigma, F2293), 0.05 IU/mL luteinizing hormone (LH, Sigma, L5259), and 10 ng/mL epidermal growth factor (EGF, Sigma, E4127) [19]. Oocytes were cultured in groups of 20–25 in 100 μL drops of medium for 14–16 h. At the end of the culture, the oocyte maturation stage of germinal vesicle (GV), germinal vesicle breakdown (GVBD), and meiotic metaphase II (MetII), was determined under a stereomicroscope.
The diameter of the cumulus-free GV oocytes was measured by a Nikon TE2000-U inverted microscope equipped with NIS-Elements BR 3.0 software. The diameters of spherical oocytes were defined as the length of a line that goes through the center of the oocyte sphere from the 2 sides of the zona pellucid edge. The diameters of ellipsoidal oocytes were defined as the average of length of minor and major axis diameter (Supplementary Fig. S1; Supplementary Data are available online at
IVF and embryo culture
After being cultured in the IVM medium for 14 h, GVBD or MetII oocytes of each experimental group and the DMIV control group were subjected to IVF. Oocytes subjected to IVF were derived from cumulus–oocyte complexes (COCs), since the zona pellucida hardening of cumulus-denuded oocytes (DOs) during IVM [57], prevented IVF and reduced the rate of blastocyst development [19,46,58].
Sperm were released from the caudae epididymis of 10-12-week-old CD-1 fertile male mice in 0.5 mL drop of the human tubal fluid (HTF) medium. After 20-min dispersion in the incubator at 37°C and 5% CO2 in air, highly motile sperm were transferred into 100 μL drops of HTF (fertilization drops) and diluted at the final concentration of approximately 106/mL. Sperm were allowed to capacitate for 40 min in the incubator under the same conditions as above. After being washed 3 times in HTF, the in vitro matured COCs were transferred in groups of 20–25 in 100 μL drops of the fertilization medium. Six hours after insemination, oocytes were washed in KSOM/AA media and observed under a stereomicroscope. Fertilization was determined by the presence of 2 pronuclei and 2 polar bodies.
Fertilized oocytes were cultured in groups of 30–35 in 100 μL drops of the KSOM/AA medium washed and transferred into mKSOM/AA [53,59,60] for further development up to the blastocyst stage. Parallel experiments carried out with superovulated CD-1 oocytes to validate our IVF and embryo development culture conditions resulted in about 85% fertilization and 90% development to the blastocyst stage.
Embryo transfer
Pseudopregnant mothers were produced by mating 8–10-week-old wild-type (WT) CD-1 virgin females with vasectomized CD-1 males on the day before embryo transfer. Females showing a vaginal plug the next morning were used for embryo transfer in the afternoon. For each group, about 18–20 2-cell stage CAG/EGFP embryos in the prebalanced M2 medium were surgically transferred into the right oviduct of a pseudopregnant recipient with a mouth controlled glass pipette in less than 3 min [19,53]. The recipients were housed singly in cages until parturition. The offspring were checked for EGFP expression using FluorChem® Q System, Supplementary Fig. S1F showed the image of kindy from either EGFP transgenetic or WT mouse captured by this system. As a negative control, none of the other 9 pseudopregnant mice without embryo transfer were observed pregnant.
Spindle assembling analysis
The in vitro matured MetII stage cumulus-DOs were fixed in 4% PFA for 30 min at room temperature and permeabilized for 20 min in 0.5% TritonX-100 in PBS (MPs). Oocytes were then blocked for 1 h in a blocking buffer (1% BSA, 0.1% Tween-20 in MPs), and then incubated in the FITC-conjugated mouse anti-á-tubulin antibody (Sigma; F2168) at a dilution of 1: 400 for 2 h at room temperature. After 3 washings in MPs containing 0.1% Tween-20 for 5 min each and counter stained with 1 μg/mL Hoechst33342 (Sigma; B2261) for 2 min, the oocytes were finally mounted on glass slides and examined using a confocal laser scanning microscope (Zeiss, LSM 850) [61].
Single-cell quantitative real-time polymerase chain reaction
Cumulus-free GV stage oocytes were collected and washed in the M2 medium until no somatic cells were visible as described above. A single oocyte with of approximately 90 μm diameter was picked up by a mouth controlled glass pipette and transferred into 7 μL RNase-free ddH2O supplemented with 2 IU/μL RNase inhibitor (TaKaRa, D2313A) and 5 mM DL-Dithiothreitol (Sigma; 43815) in a polymerase chain reaction (PCR) tube and immediately frozen at −80°C.
For reverse transcription, samples were boiled and vortexed for 1 min, respectively. After adding 1 μL recombinant DNase I (5 IU/μL, TaKaRa, D2270A) and 1 μL 10× DNase I buffer, the tubes were incubated at 25°C for 15 min and 65°C for 10 min to eliminate the genomic DNA. Then cDNA was constructed using the PrimeScript RT reagent kit (TaKaRa, DRR047A) according to the manufacturer's protocol with minor modification: cDNA was constructed at 25°C for 10 min and 37°C for 15 min.
Primers used for quantitative real-time PCR, specific for the oocyte genes, have been previously described [48,62 –64]. Their specificity was confirmed by BLAST search in GenBank. Primer sequences are shown in Table 1. Relative quantification analysis was performed in a LightCycler 480 II (Roche) Real-Time PCR apparatus using a SYBR® Premix Ex Taq™ II (TaKaRa, DDR081A) according to the manufacturer's instructions. Each reaction mixture consisted of 2 μL of cDNA and 18 μL of PCR mix containing 10 μL of SYBR Premix, 0.4 μL (20 μM stock) of each primer, and 7.2 μL of nuclease-free water per sample. The PCR protocol used was as follows: 10 min at 95°C, followed by 55 cycles at 95°C for 10 s and 60°C for 30 s and finally, a cooling step at 4°C. Each sample had 2 technical replicates and reactions were performed in triplicate for each gene. The expression of each gene was normalized to GAPDH expression using the following formula: 2−(target gene CT value− GAPDH CT value). As negative control, the scarcely transcriptional expression of Kitl in oocytes, which is specifically expressed in granulosa cells in follicles, confirmed the validity of our sample collection and analysis procedure.
Statistical analysis
The total number of samplings for each sample was represented in corresponding tables. Data are represented as mean±standard deviation (SD). Statistical significance was determined with Excel (Microsoft) using the unpaired Student's t-Test with 2-tailed distribution of 3-sample of unequal variance. P<0.05 denoted a statistically significant difference, while P<0.01 denoted a highly significant difference.
Results
Delay of oocyte meiotic progression and impaired folliculogenesis in embryonic ovaries cultured in vitro
We established a suspension in vitro culture method for the fetal ovaries, more suitable for transplantation than the adherent culture (Supplementary Fig. S2), allowing the in vitro development of mouse 12.5 dpc ovaries for 7 and 14 days, periods that we estimated comparable to about 0 and 7 dpp (19.5 dpc=0 dpp), respectively (Fig. 1).
Chromosome cytospreads carried out in parallel in oocytes isolated from in vitro cultured ovaries after 1, 3, 5, and 7 days of culture and from in vivo ovaries of comparable age (13.5–19.5 dpc) (Fig. 2), showed no significant difference in the oocytes capability to enter meiosis, but revealed a clear delay of meiotic progression in cultured oocytes (Fig. 2B–E).
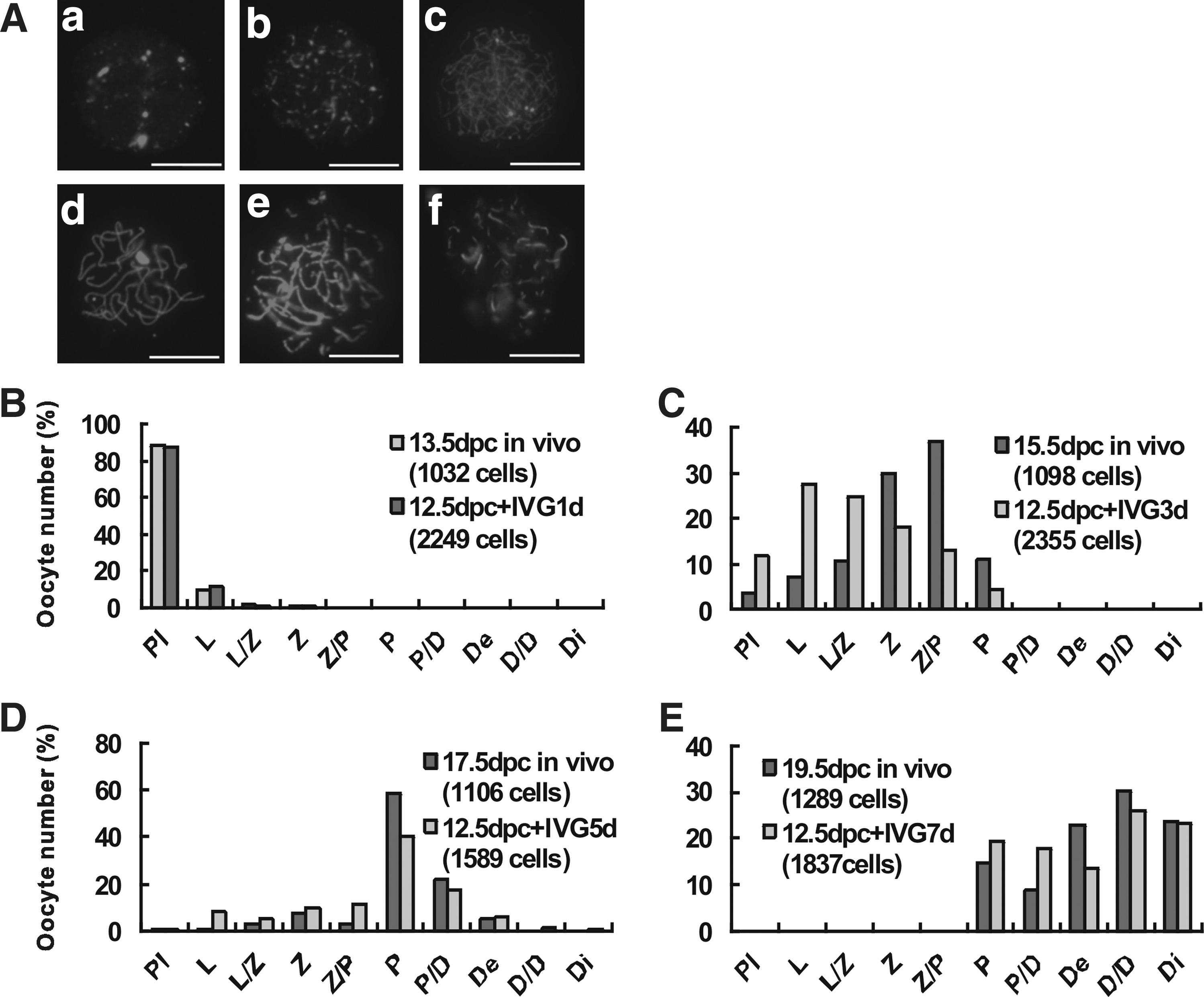
Delay of meiotic progression during in vitro culture of embryonic ovaries. Mouse embryonic ovaries of 12.5 dpc were cultured for 1, 3, 5, 7 days, and oocytes analyzed for the progression through the stages of meiosis prophase I by characteristic patterns of SCP3 immunostaining in cytospreads
In the in vitro cultured 12.5 dpc mouse embryonic ovaries, oocytes became visible under an inverted microscope from 7 days of culture onward (Fig. 3A). At this time, the staining of the oocytes with the anti-STAT3 antibody in tissue sections evidenced their prevalent ovary cortex localization in germ cell cysts or inside primordial follicles (Fig. 3A). At 14 days of culture, the oocyte diameter was clearly increased and preantral follicles at various stages of development were present in the ovarian medulla, including early stage of secondary follicles with 1–2 layers of granulosa cells (Fig. 3B). In ovaries of comparable in vivo stages, oocytes appeared localized both in the cortex and medulla within primordial follicles at 0 dpp and within primordial and secondary follicles with 2–4 layers of granulosa cells at 7 dpp (Fig. 3C). In contrast, when gonadal ridges from 11.5 dpc female embryos were isolated and cultured under the same conditions for 8 days in vitro, only a few oocytes were observed (Supplementary Fig. S1C red arrow).
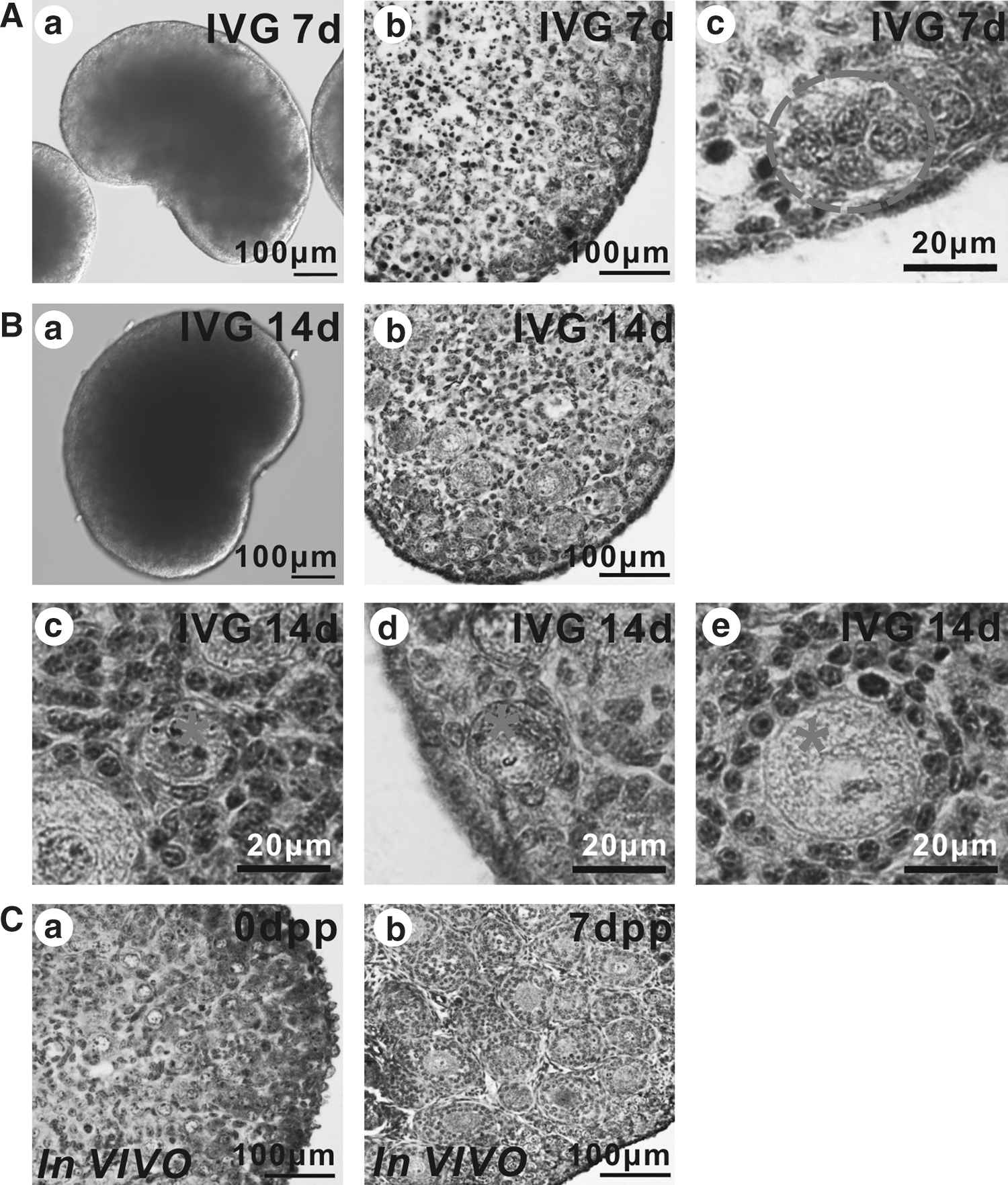
Defects in early folliculogenesis in cultured embryonic ovaries. 12.5 dpc ovaries were isolated and cultured for 7 days
Follicle development in cultured embryonic ovaries after kidney capsule transplantation
Despite the delay in oocyte meiotic progression and folliculogenesis observed in the cultured ovaries, we verified if these defects could be recovered after in vivo transplantation and normal oocytes could be produced. To this aim, 12.5 dpc ovaries cultured in vitro for 7 or 14 days were transplanted beneath the kidney capsule of immunodeficient ovariectomized adult females and after 21 days (7+21 group) or 14 days (14+14 group) recovered for analyses (Fig. 4A and Table 2). Forty eight hours before the retrievement of the transplanted ovaries, recipients and control females were injected with PMSG to induce estrus. Control mice included ovariectomized SCID recipient (OR), SCID recipient (RM), and donor 21 dpp CD1 mice (DMIV). The estrogen (E2) levels were examined in blood samples obtained from mice of all groups. As Fig. 4B shows, the E2 levels in the females of both experimental groups were higher compared with the OR group. The difference in the E2 levels between 14+14 and OR groups was, however, not statistically significant. In contrast, the E2 levels of the 7+21 group was significantly higher both than those of OR and 14+14 groups (P<0.01), but lower than those of the DMIV group.
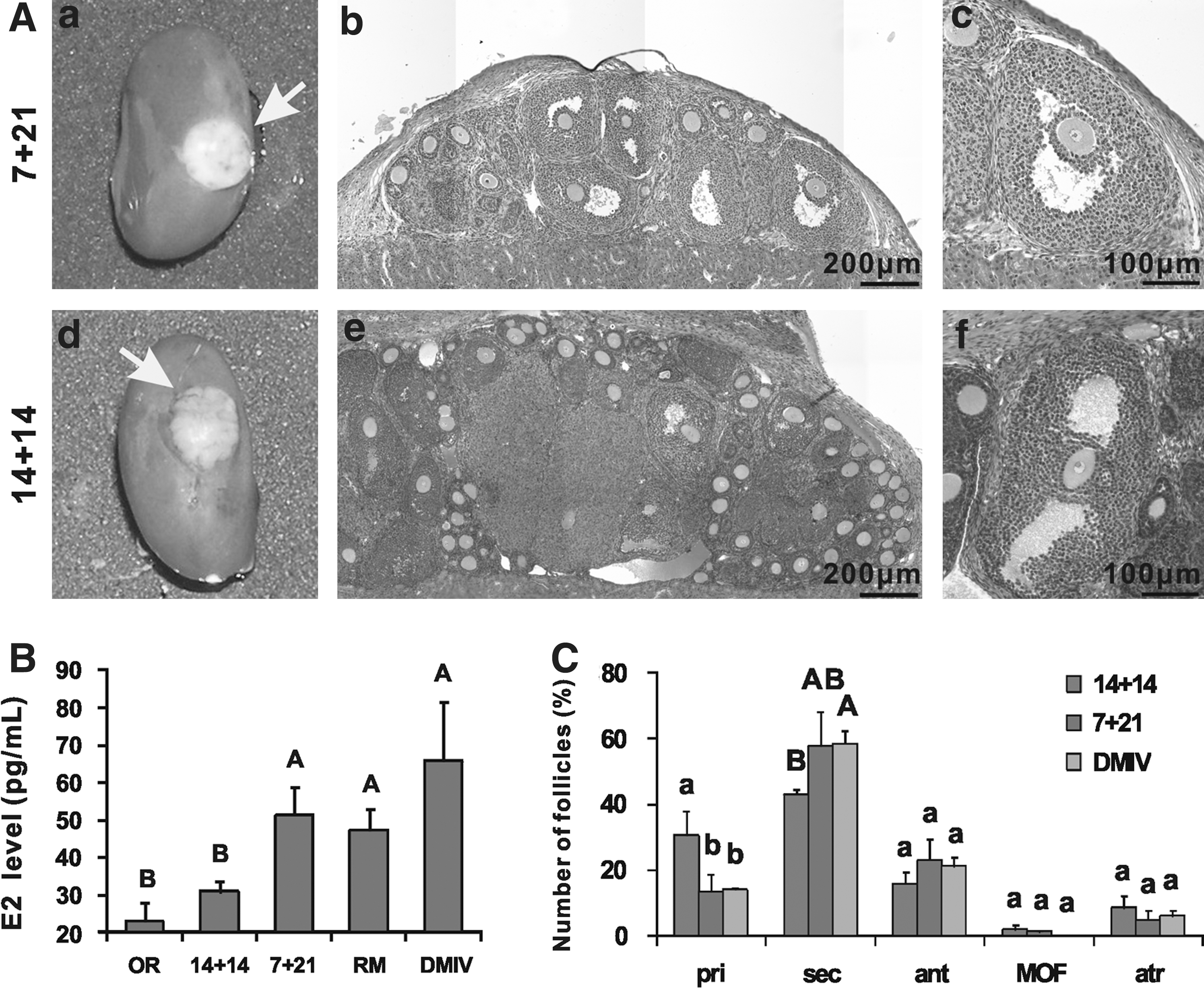
Folliculogenesis in grafted ovaries and estrous cycle in host ovariectomized females.
On the whole, these results indicated that the grafted in vitro cultured ovaries, namely, those of the 7+21 group, were able to restore estrus and possibly the reproductive cycle in the ovariectomized recipient females.
After surgical exposure of the kidney, vascularization was observed in grafted ovaries, indicating the reconstruction of blood supply in the grafts. Furthermore, grafted ovaries were found increasing in size with many visible follicles on the surface (Fig. 4A). In hematoxylin–eosin stained tissue sections, fully developed antral follicles were present in grafted ovaries of both experimental groups. More preantral follicles were observed in the grafted 14+14 group than in the 7+21 group (Fig. 4A). On the other hand, corpora hemorrhagica were observed in sections of the retrieved grafts, indicating the occurrence of previous ovulations (Supplementary Fig. S3A).
To quantify the differences in follicle development between the 2 experimental groups and in vivo control DMIV ovaries, follicle counts were performed. Whereas about 57% of follicles in DMIV ovaries were primordial follicles, this type of follicles were hardly found in either 7+21 or 14+14 transplanted ovaries (Supplementary Fig. S3B). On the other hand, excluding primordial follicles, the percentage of primary, secondary, and antral follicles in the ovaries of the 7+21 group (14.16%±5.35%, 60.51%±10.08%, and 24.46%±6.87%, respectively), was substantially the same compared with DMIV ovaries (14.99%±0.2%, 62.35%±2.99%, and 22.65%±3.19%, respectively) (Fig. 4C). In the ovaries of the 14+14 group, however, the percentage of primary follicles was significantly higher (P<0.05), while the percentage of secondary and antral follicles was lower (P<0.01 for secondary follicles) compared with the DMIV group. It is unlikely that such reduction is due to a higher atretic follicle rate in 14+14 ovaries, since the percentage of morphologically distinguishable atretic follicles was similar in all groups (7+21=4.83%±3.02%; 14+14=8.53%±3.81%; DMIV=6.11%±1.75%). Finally, more MOF were found, but differences were not statistically significant, in the 14+14 group (2.13%±1.35%) than the 7+21 group (0.87%±0.76%), while none was found in the DMIV group.
Taken together, these last results clearly show that the pool of primordial follicles was severely compromised in the grafted ovaries, but that some waves of folliculogenesis occurred in such ovaries after transplantation. On the other hand, prolonged in vitro culture (14+14 group) appeared to reduce the waves of folliculogenesis.
Single-cell analysis of oocyte-specific gene expression
We next compared the level of transcripts of 13 genes known to be expressed during oocytes maturation in GV oocytes of approximately 90 μm diameter (see Materials and Methods), retrieved from the antral follicles of the transplanted and control DMIV ovaries (Fig. 5). Supplementary Fig. S4 illustrates the relative expression of the analyzed genes.
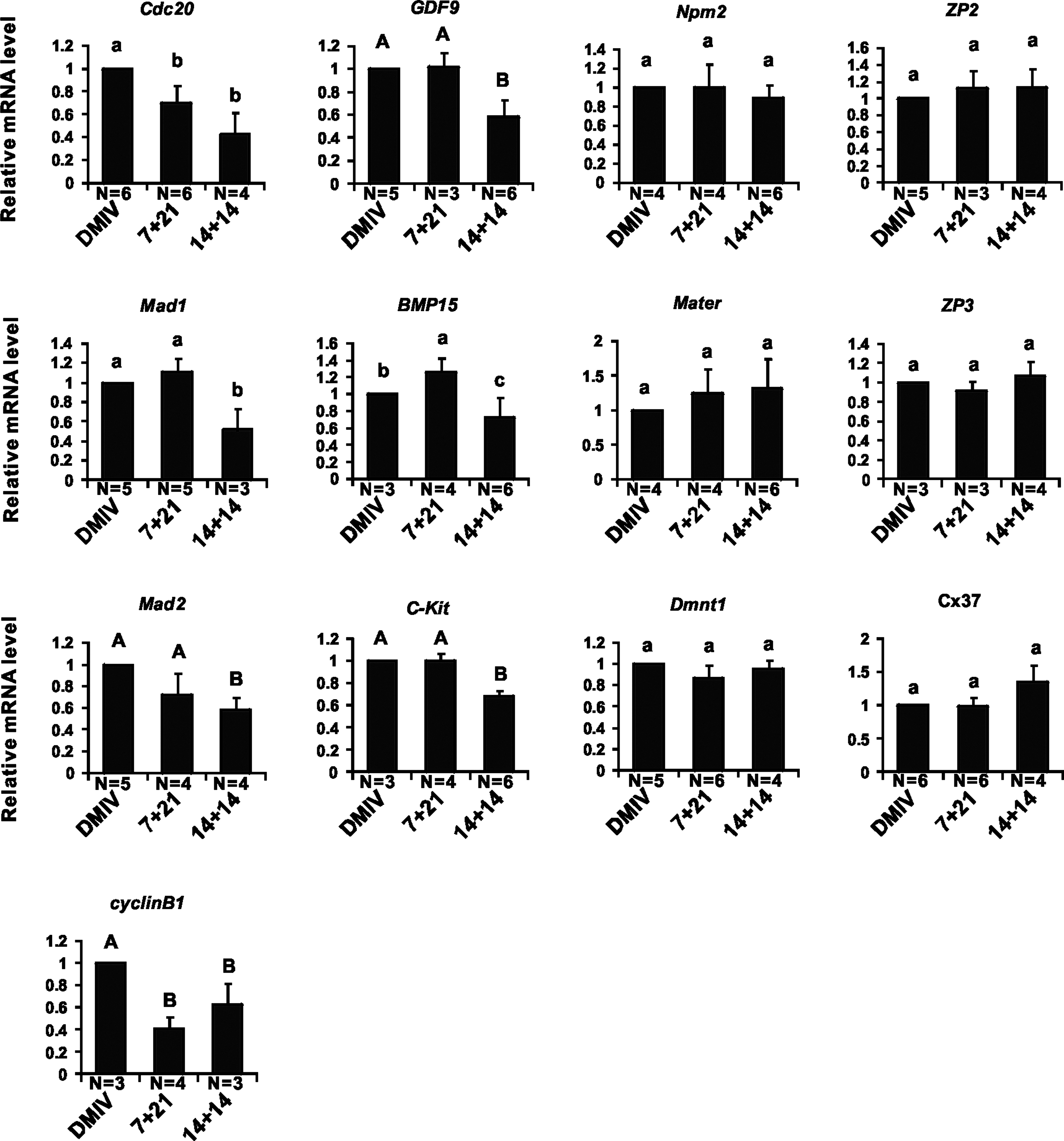
Single-cell quantitative real-time PCR analyses of mRNA levels of genes associated to oocyte maturation. Single germinal vesicle (GV) oocytes were collected from grafted or DMIV ovaries. Four groups of oocyte genes, including cell cycle related genes (Cdc20, Mad1, Mad2, Cyclin B1), oocytes maturation related genes (GDF9, BMP15, c-Kit), zona pellucida proteins (ZP1, ZP2, ZP3) and connexin gene (Cx37), and maternal genes (Npm2, Mater1, Dmnt1), were analyzed. Significant difference (P<0.05) among groups is indicated by little letters
GV oocytes of all groups scarcely expressed Zp1 (Supplementary Fig. S4), while no significant differences were observed in Zp2, Zp3, Cx37, Npm2, Mater, and Dnmt1 transcript levels among the all groups (Fig. 5). Transcripts for GDF9, c-Kit, and Mad2 were significantly lower in 14+14 oocytes than in 7+21 and DMIV oocytes (P<0.01) (Fig. 5). Moreover, DMIV oocytes showed levels of Cdc20 and CyclinB1 transcripts significantly higher of both 14+14 and 7+21 oocytes (P<0.05 and P<0.01, respectively) (Fig. 5). Finally, Bmp15 transcripts in 7+21 oocytes were significantly higher than both in 14+14 and DMIV oocytes (P<0.05), whereas 7+21 oocytes expressed significantly higher levels of Mad1 only in comparison to the 14+14 group (P<0.05) (Fig. 5).
In conclusion, only a few analyzed genes appeared misregulated in the oocytes of the 7+21 ovaries (namely, Cdc20, CyclinB downregulated, and Bmp15 upregulated) in comparison to control. Lower transcription levels for more genes were evident in 14+14 oocytes in comparison to control, suggesting a delay in the maturation progression of these oocytes.
Oocytes retrieved from grafted ovaries are able to complete meiosis and undergo early embryo development in vitro
To verify whether oocytes generated in the transplanted ovaries were competent to resume and complete meiosis and could be fertilized, they were isolated from antral follicles and placed in the maturation medium (Fig. 6). Oocytes were also isolated from DMIV ovaries and subjected to IVM for comparative purpose (Fig. 6A). Although the mean diameter of the oocytes of the 7+21 group (94.2±3.1 μm) was not significantly different from that of both DMIV (96.0±1.3 μm) and 14+14 (91.4±1.6 μm) oocytes, the diameter of 14+14 oocytes was significantly smaller compared with DMIV oocytes (P<0.05) (Fig. 6B). At the end of IVM, the percentage of GVBD of the oocytes of the 7+21 group (80.8%±1.71%) and of the 14+14 group (84.73%±2.67%) were similar, but the percentage of GVBD of the 14+14 group was significantly lower compared with the oocytes of the DMIV group (89.3%±1%) (P<0.05) (Fig. 6C). The percentage of oocytes of the 14+14 group (54.47%±3.43%), which reached the MetII stage was significantly lower than either that of the 7+21 group (74.40%±8.98%) (P<0.01) or the DMIV group (74.9%±2.69%) (P<0.01) (Fig. 6D).

IVM, fertilization, and early embryo development of oocytes retrieved from grafted ovaries. Oocytes retrieved from grafted ovaries are able to complete meiosis, can be fertilized, and give rise to preimplantation embryos in vitro
The analyses of spindle and chromosome morphology in MetII oocytes from transplanted ovaries did not reveal any apparent abnormality in comparison to control oocytes (Fig. 6E).
GVBD and MetII stage oocytes derived from COCs of each group were then subjected to IVF after IVM. Oocytes from the transplanted or control DMIV ovaries showed a fertilization rate between 25% and 45% 6 h after insemination. The cleavage rate of the fertilized oocytes of the 2 experimental groups and control was similar (90.16%±1.9% for 14+14 group, 91.42%±3.0% for 7+21 group and 93.39%±3.8% for DMIV group) (Fig. 6A and Table 3). Furthermore, the percentage of embryos reaching the blastocyst stage for the 14+14 group (47.51%±7.5%) was lower, although not in a statistically significant way, compared with the 7+21 group (49.39%±2.7%) or the DMIV group (50.85%±9.7%) (Fig. 6A; Table 3; Supplementary Table S1). As note, we observed a MOF-derived oocyte with a tightly adhered apoptotic oocyte, which after IVM was fertilized and underwent development to hatching blastocyst (Supplementary Fig. S5E–H).
Data presented as mean±S.D. and values within the same column with same superscript (a) are not significantly different (P>0.05).
Living offspring obtained from oocytes of transplanted embryonic mouse ovaries
To examine the developmental potential of oocytes retrieved from transplanted ovaries, 2-cell embryos obtained after IVF expressing EGFP (Fig. 7A–D), were transferred to the oviducts of pseudopregnant WT recipients. From 92 (7+21 group) and 39 (14+14 group) embryos transferred, 6 pups (6.52%, 2 males and 4 females) (Fig. 7E) and 4 pups (10.26%, 2 males and 2 females) (Fig. 7F), were delivered, respectively (Table 4). As in vivo control, 40 embryos were transferred, and twelve pups (30%, 7 males and 5 females) were obtained (Table 4). All pups were EGFP-positive (Fig. 7G, H) and able to grow up to adulthood with normal fertility when mated with mice of the opposite sex.
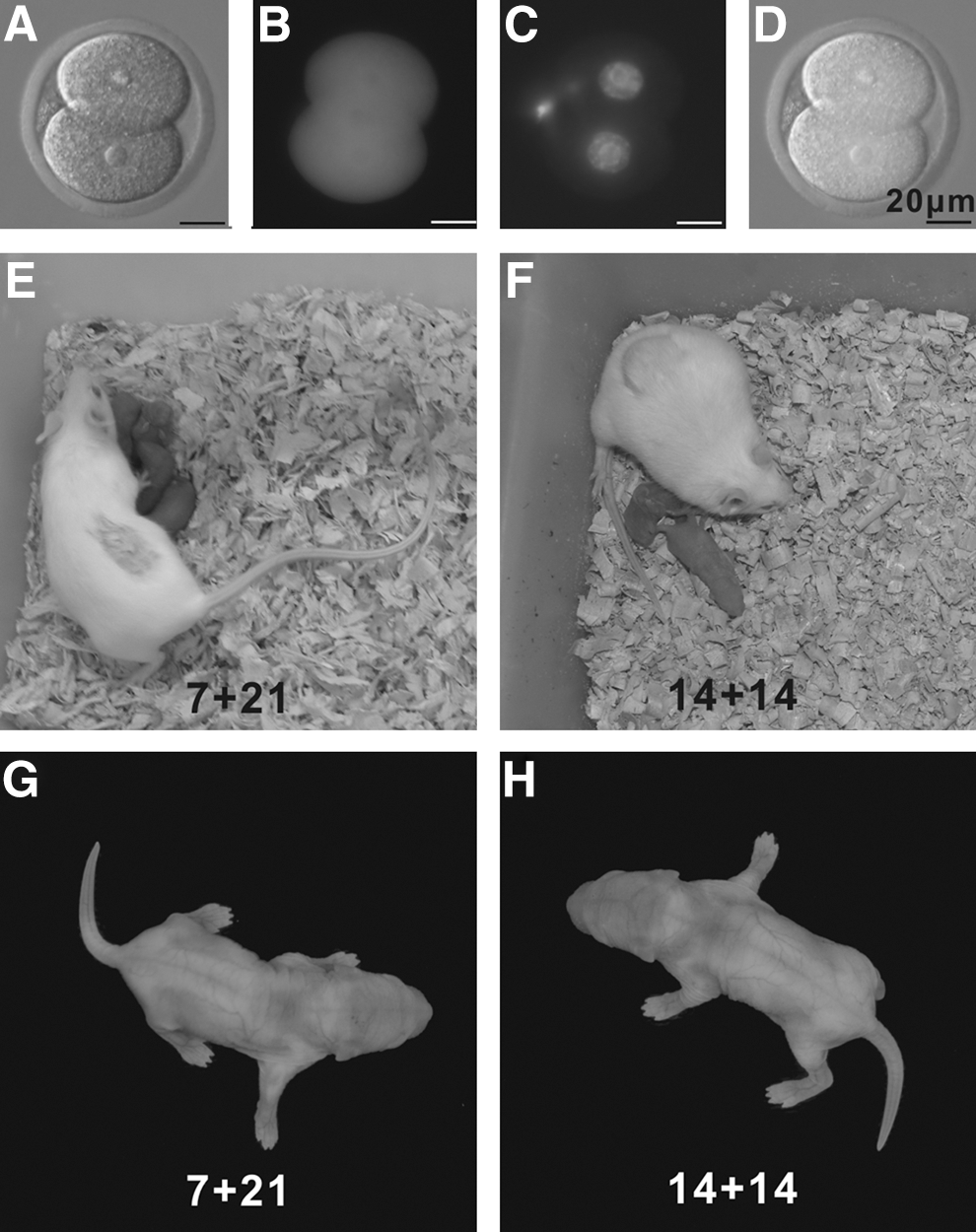
Healthy live offspring obtained from oocytes retrieved from grafted ovaries and fertilized in vitro. 2-cell embryos were checked under fluorescence microscope to confirm the expression of enhanced green fluorescent protein (EGFP) and the presence of normal nucleus
Discussion
In the present study, we adopted the strategy to culture embryonic ovaries under suspension serum-free culture conditions for a period of 7 and 14 days, estimated sufficient to generate primordial and primary/secondary follicles, followed by transplantation under the kidney capsule of recipient females. The aim of our work was to verify if the cultured germ cells passing in vitro through the crucial stages of the meiotic prophase I and early folliculogenesis maintain the capability to generate functional oocytes after a period of further maturation in the ectopic site in vivo. The possibility to generate in vitro oocytes, which can be fertilized and give rise to normal embryos and offspring, is one of the most fascinating perspectives of the modern reproduction and germ cell biology. While several early and new works clearly demonstrated that in vitro several processes of gametogenesis can be reproduced and oocyte-like cells can be generated from stem cells [26 –33], so far complete in vitro oogenesis has not been reported. In particular, previous studies proved that successful production of functional oocytes from premeiotic female germ cells could be obtained only after transplantation of embryonic ovaries or germ cells aggregated with ovarian tissues under the kidney capsule or the ovarian bursa of host animals [45 –47].
We found that under our in vitro culture conditions, before transplantation, germ cells entered normally into meiosis, but their meiotic progression up to the dictyate stage and early folliculogenesis were delayed. Moreover, we obtained physiological and morphological evidence that after transplantation, some waves of folliculogenesis ending with ovulation occurred in the transplanted ovaries. In another work in which isolated female germ cells were aggregated with ovarian cells before transplantation, follicles developed synchronously and exhibited only a single oogenesis wave [46]. This could also occur when stem cell-derived PGCs or germ cells are aggregated with ovarian cells [28,45,48,65]. This study, along with past work [12,13,44], emphasized the importance of the integrity of the ovarian structure in controlling the number of growing follicles in each oogenesis wave.
We observed, however, that a rapid depletion of the follicle reserve occurred in the transplanted ovaries of both experimental groups. In fact, primordial follicles were hardly found in such ovaries after 3 or 2 weeks from transplantation, respectively. Although the reason of this depletion was not investigated in detail, it might be due to reduced proliferation of the PGCs/oogonia, increased oocyte apoptosis, or impaired formation of primordial follicles within the ovaries in vitro. These defects might be germ cell intrinsic or be a consequence of inefficient proliferation/differentiation of pregranulosa cells. Finally, although, we do not have evidence about, accelerated activation of the primordial follicles may be also possible.
It is worthy to note that when in our experiments, we used gonadal ridges from 11.5 dpc female embryos, only a few oocytes were scored both in the in vitro cultured and transplanted ovaries (data not shown). Since at 11.5 dpc, most of PGCs have already migrated into the gonadal ridges, it is likely that the culture conditions are unable to sustain their normal proliferation and/or survival. Moreover, between 11.5 and 13.5 dpc, crucial events for the beginning of meiosis occur in PGCs [66] that might be not properly reproduced in vitro. In line with these observations, postmigratory and meiotic prophase I stages are difficult to be correctly reproduced in putative PGCs obtained from various kinds of stem cells [29,65,67,68] In addition to these possible defects, here we observed a higher number of oocytes remaining in cysts in cultured ovaries in comparison to in vivo controls, which indicated that the progress of follicle assembly was also delayed in vitro. Since about 2 thirds of oocytes undergo apoptosis during primordial follicle assembly if they are not enclosed by granulosa cells [8,69] and inefficient proliferation of follicular cells has been proved to be one of the main causes of in vitro follicle development deficiency [23,70], it seems likely that such processes are also responsible for the reduction of the follicle reserve observed in the present study.
It is also important to note that, nourishing by the serum-free medium, which contained no additional cytokine, female germ cells could undergo the developmental process from the postmigrate stage to the neonatal stage in vitro, and preserve their capability to complete oogenesis, but showed delayed development as the period of in vitro culture extended. These data indicate that postmigrate female germ cells within the gonadal environment have the competence to undergo meiotic entry and arrest, as well as primordial follicles assembly and activation, but further development, such as secondary follicle formation, could not be fully supported by our culture conditions.
Despite the observed oogenesis/folliculogenesis defects, whatever the reasons for the rapid depletion of the follicle reserve and the restriction of our in vitro culture conditions, it was important to verify the quality of the oocytes remained in the transplanted ovaries and their capability to give rise to viable pups after fertilization. Overall, the expression level of several oocyte-specific genes in oocytes retrieved from transplanted ovaries revealed quite a normal pattern, despite some exception mainly for oocytes, which have been cultured for the longer 14-day period. The oocytes from both experimental groups showed, however, similar capability to undergo meiotic maturation and to be fertilized in vitro. Most importantly, most of the fertilized oocytes develop to blastocyst that after transplantation in foster mothers gave rise to viable pups.
In conclusion, the present study reveals a number of critical factors affecting in vitro oogenesis by comparing the differences between in vivo and in vitro ovaries, such as postmigrate proliferation/survival, integrity of the ovarian structure, loss of the primordial follicle pool, and delayed follicle development during in vitro culture. By exploring the possibility of recovering the developmental potential of in vitro cultured germ cells through ectopic transplantation, we provide a clear demonstration that, despite the observed defects, oocytes within primordial/primary follicles generated in vitro from postmigrate PGCs preserve their capability to complete oogenesis and can be fertilized and generate live pups, which open the way to 2-step strategies in which embryonic female germ cells are first collected or generated in vitro, and then transplanted to obtain fertilizable oocytes.
Footnotes
Acknowledgments
This work is supported by the National Basic Research Program of China (973 Program, 2012CB944401, and 2007CB947401), the National Nature Science Foundation (31001010, 31171376, and 31101716), the Foundation of Distinguished Young Scholars (JQ201109), the Doctoral Foundation (BS2010NY010), and the Taishan Scholar Foundation of Shandong Province. We gratefully acknowledge Ping Zhou, Karen Wigglesworth, Wen-Bo Liu, Nan He, Shu-Tao Qi, Zhao-Jiao Ge and Xing-Jiang Yu for the assistance of helpful discussions about experimental techniques used in this study. Thanks are due to Shi-Wen Li for the assistance in preparing the photomicrographs of spindle assembling analysis.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.