
Abstract
The protein tyrosine phosphatase, SHP2, is widely expressed; however, previous studies demonstrated that hematopoietic cell development more stringently requires Shp2 expression compared to other tissues. Furthermore, somatic gain-of-function SHP2 mutants are commonly found in human myeloid leukemias. Given that pharmacologic inhibitors to SHP2 phosphatase activity are currently in development as putative antileukemic agents, we conducted a series of experiments examining the necessity of SHP2 phosphatase activity for human hematopoiesis. Anti-sense oligonucleotides to human SHP2 coding sequences reduced human cord blood- and human cell line, TF1-derived colony formation. Expression of truncated SHP2 bearing its Src homology 2 (SH2) domains, but lacking the phosphatase domain similarly reduced human cord blood- and TF1-derived colony formation. Mechanistically, expression of truncated SHP2 reduced the interaction between endogenous, full-length SHP2 with the adapter protein, Grb2. To verify the role of SHP2 phosphatase function in human hematopoietic cell development, human cord blood CD34+ cells were transduced with a leukemia-associated phosphatase gain-of-function SHP2 mutant or with a phosphatase dead SHP2 mutant, which indicated that increased phosphatase function enhanced, while decreased SHP2 phosphatase function reduced, human cord blood-derived colonies. Collectively, these findings indicate that SHP2 phosphatase function regulates human hematopoietic cell development and imply that the phosphatase component of SHP2 may serve as a pharmacologic target in human leukemias bearing increased SHP2 phosphatase activity.
Introduction
T
Particularly highlighted in the context of human disease, both the SHP2 phosphatase and adapter function appear to be necessary for its positive signaling effect, best demonstrated by the apparent paradox that PTPN11 mutations causing increased phosphatase activity are found in the human congenital disorder, Noonan syndrome [15], while PTPN11 mutations destroying SHP2 phosphatase activity are found in the human congenital disorder, LEOPARD syndrome [16,17], which phenotypically mimics Noonan syndrome [18]. Somatic gain-of-function mutations are commonly found in the childhood myeloproliferative neoplasm, juvenile myelomonocytic leukemia (JMML) [8,19], providing functional evidence that dysregulated SHP2 phosphatase function regulates hematopoietic cell differentiation and proliferation. However, while several knockout and knockdown studies have demonstrated that Shp2 is particularly important in hematopoietic development and hematopoietic stem cell function [20 –26], none of these has directly examined the role of SHP2 phosphatase function in hematopoiesis. This is particularly of relevance, as pharmacologic inhibitors of SHP2 phosphatase activity are currently in development and have demonstrated activity in oncogene-induced myeloproliferative neoplasms [27 –29]. Thus, the following studies utilized various SHP2 mutants either lacking the SHP2 phosphatase domain or bearing a loss-of-function point mutation or a gain-of-function point mutation reducing or increasing SHP2 phosphatase function, respectively, to define specifically the role of SHP2 phosphatase function in hematopoiesis.
Materials and Methods
Retroviral plasmids and vector production
A truncated version of the SHP2 cDNA, including the N-SH2 and C-SH2 domains, but lacking the phosphatase domain (referred to as SHP2SH2) was cloned into the pLXSN retroviral plasmid [30] to generate pL-SHP2SH2-SN (Fig. 1C). The ecotropic GP+E86 [31] and amphotropic PA317 [32] retrovirus packaging cell lines were used to generate retroviral supernatants using the pLXSN and pL-SHP2SH2-SN plasmids. Transduction efficiency of transduced human cord blood-derived colonies was assessed using genomic DNA obtained from individual colonies and amplified using PCR for the presence of an amplified fragment from pLXSN. Alternatively, the full length SHP2 cDNA (wild-type or point mutants SHP2 D61Y and SHP2 C463A) tagged at the carboxy terminal end with an enhanced yellow fluorescent protein (EYFP) cloned into the MSCV retroviral plasmid (Clontech). All constructs were confirmed by sequencing. Amphotropic retroviral supernatants were generated by cotransfecting MSCV-based retroviral plasmids and the pan-tropic pVSV-G envelope plasmid into GP2-293 packaging cells (Clontech). Following transduction, cells were subjected to fluorescence-activated cell sorting (FACS) to enrich for EYFP-expressing cells. Studies in our laboratory indicate that EYFP-tagged SHP2 functions similarly to untagged SHP2 in methylcellulose-based and biochemical assays, as previously published by us [33].
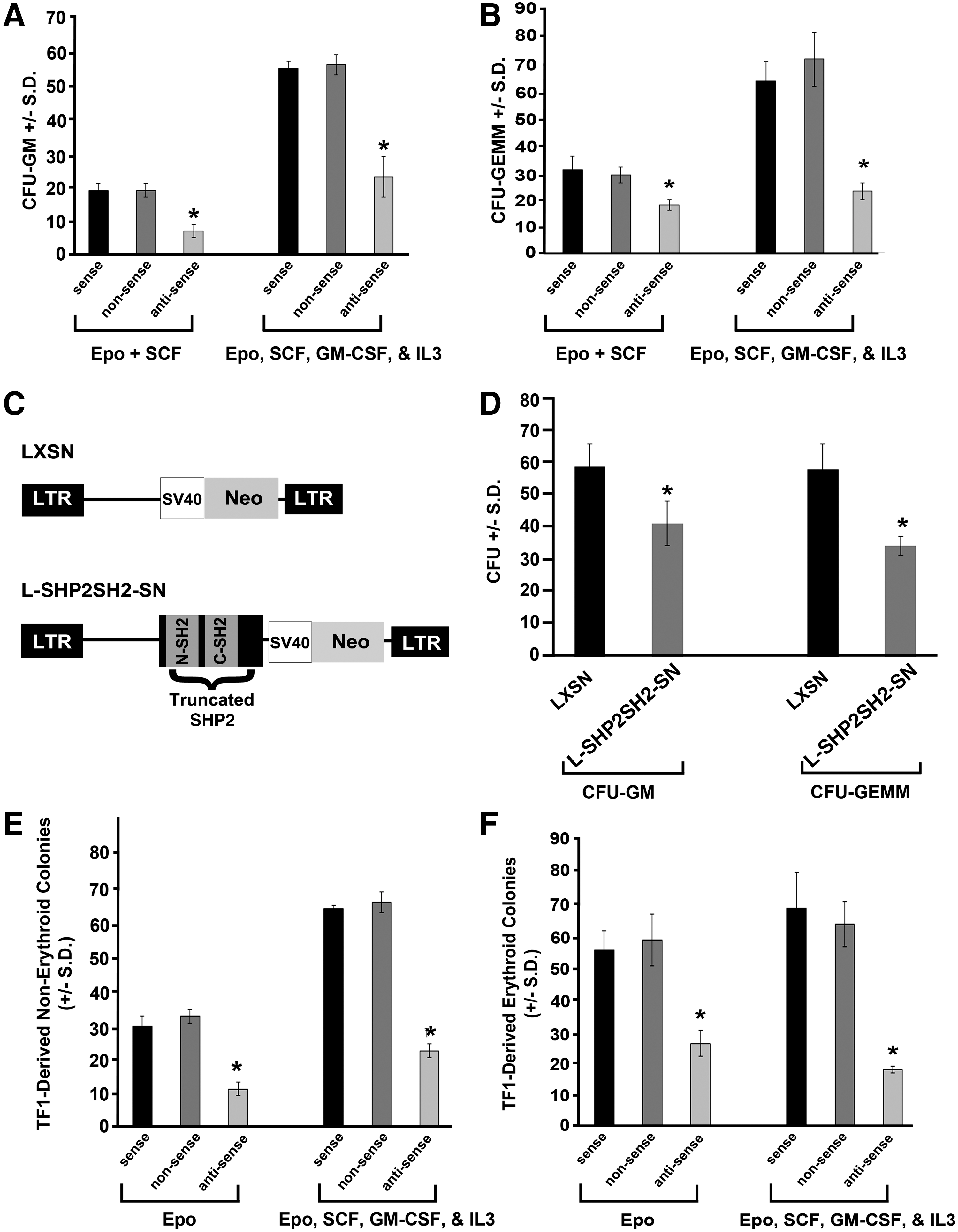
Pretreatment with antisense oligonucleotides (ODN) to SHP2 or expression of truncated SHP2 reduces colony formation. CD34+ cord blood cells were pretreated with antisense, sense, and nonsense ODN as previously described [37] in 1% methylcellulose culture medium with 30% fetal bovine serum (FBS) plus EPO (2 U/mL) and SCF (50 ng/mL) minus and plus GM-CSF (20 ng/mL) and IL-3 (20 ng/mL) at 400 cells/mL. Colonies were scored after 12 days of incubation for
Human umbilical cord blood collection, CD34+ cell separation, and retroviral transduction
Cord blood samples were obtained with institutional review board approval from normal human umbilical cord blood scheduled for discard after delivery of the infant. Cord blood was diluted in phosphate-buffered saline (1:1) and low-density mononuclear cells were isolated on Ficoll-Paque TM Plus (GE Healthcare). CD34+ cells were purified either by staining with fluorescein isothiocyanate-labeled anti-human CD34 followed by FACS for the top 20% highest CD34-expressing cells (≥98% CD34+), as previously described [34], or by utilization of the MACS separation kit (Miltenyi Biotec) following the manufacturer's instructions (≥94% CD34+). After isolation, human cord blood CD34+cells were prestimulated in human growth factors followed by retroviral transduction, as previously described [35]. The transduced cells were assayed for colony formation in the presence of different growth factor combinations at the following concentrations: 2 U/mL rhuEpo, 20 ng/mL rhuGM-CSF, 20 ng/mL rhuIL-3, and 50 ng/mL rhuSCF. Fourteen days after plating, the number of colonies was counted. In some experiments, single transduced cells were sorted into one well of a 96-well microtiter plate using an auto-clone device (Coulter) containing 1% methylcellulose, 30% fetal bovine serum (FBS), and indicated growth factors.
Culture and transduction of TF1 cells
The human growth factor-dependent cell line, TF1, was originally obtained from Dr. T. Kitamura [36] and is available from ATCC (CRL-2003). TF1 cells were grown in the RPMI 1640 medium containing 10% fetal calf serum in the presence of 1.5 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF). TF1 cells were transduced with LXSN (negative control) or with the L-SHP2SH2-SN retroviral vector in the presence of polybrene 8 μg/mL and GM-CSF 1.5 ng/mL followed by selection in G418 1 mg/mL until resistant colonies were obtained. G418-selected TF1 cells were evaluated in colony assays and in biochemical studies, as indicated.
Oligonucleotides design and cell treatment
Three 20-mer oligonucleotides (ODN) were designed against the coding nucleotide sequence for human PTPN11 for introduction into human cord blood CD34+ cells or into the human growth factor-dependent TF1 cell line as follows: (i) anti-sense ODN, 5′-CTC CGC GAT GTC ATG TTC CT-3′, which spans +14 to −6 relative to the start codon; (ii) sense ODN, 5′-GAG GAA CAT GAC ATC GCG GA-3′, which spans −7 to +13 relative to the start codon; and (iii) nonsense ODN, 5′-TGG GTG TGT CCA AGA GAA CT-3′. For pretreatment with ODN, human cord blood CD34+ cells were cultured in Iscove's modified Dulbecco medium with 2% FBS and TF1 cells were cultured in RPMI 1640 with 2% FBS in the presence of 500 μg/mL of each ODN for 2 h. Serum was then added to each culture to a final concentration of 10%. Ten hours later, the same concentration of ODN was added and the cells were plated for colony assays, as previously described [37].
Immunoprecipitation and immunoblots
Total cellular protein extracts were prepared from G418-selected TF1 cells and assayed using the Bradford Reagent and a standard curve, as previously described [38]. To examine expression of full length and truncated SHP2, the anti-SHP2 monoclonal antibody (BD Transduction Laboratories) was used for immunoblotting. To examine the SHP2-Grb2 interaction, SHP2 was immunoprecipitated with anti-SHP2 serum as previously described [4,6] and blots were probed with anti-Grb2 (BD Transduction Laboratories) and reprobed with anti-SHP2. Immunoreactive proteins were visualized by an ECL chemiluminescent reaction kit (Amersham).
Statistical analysis
Groups were compared using the unpaired, 2-tailed, student's t test or the χ 2 test.
Results
Human progenitor cell colony formation is inhibited by reduced SHP2 expression
While there has been a large body of work done to examine the function of Shp2 in hematopoiesis using mouse models and murine embryonic stem cell-derived hematopoiesis [20 –26], few studies have been conducted examining the role of SHP2 in human hematopoiesis. To address this, we reduced expression of SHP2 in human cord blood CD34+ cells using antisense ODN to SHP2 compared to sense and nonsense ODN. Human cord blood CD34+ cells preincubated with sense, nonsense, and anti-sense ODN to SHP2 were plated in methylcellulose-based colony forming assays in the presence of multiple cytokines to pick up the immature subsets of progenitors, and the colony forming unit granulocyte macrophage (CFU-GM) and the CFU-granulocyte erythroid macrophage megakaryocyte (CFU-GEMM) were scored. While the sense ODN did not influence colony formation compared to the nonsense ODN, the anti-sense ODN significantly reduced both CFU-GM- and CFU-GEMM-colony formation of cord blood cells compared to the sense ODN in response to cytokine cocktails (Fig. 1A, B). These findings indicate the necessity of SHP2 for optimal cytokine-stimulated human progenitor growth and proliferation.
Expression of truncated SHP2 lacking phosphatase domain impairs human progenitor colony growth
To evaluate how SHP2 functions to promote human progenitor cell growth, we retrovirally transduced a truncated SHP2, bearing the N-SH2 and C-SH2 domains, but lacking the phosphatase domain (SHP2SH2, Fig. 1C), into human CD34+ cord blood cells followed by colony forming assays in the presence of erythropoietin, interleukin (IL)-3, and stem cell factor (SCF). Compared to the empty vector (LXSN), transduction of the truncated SHP2 (L-SHP2SH2-SN) significantly reduced both CFU-GM- and CFU-GEMM-colony formation (Fig. 1D). To assess transduction efficiency, genomic DNA from individual colonies was subjected to PCR for an amplified fragment from pLXSN, and the PCR-positive colonies for the L-SHP2SH2-SN-transduced cells was 100%, indicating efficient transduction efficiency (data not shown). The influence of truncated SHP2 (SHP2SH2) was assessed in greater detail by evaluating its effect on single isolated CD34+ cord blood cell-derived colony formation. The percent of colonies formed per single cell plated was significantly lower in all colony types assessed, including CFU-high proliferative potential (CFU-HPP), CFU-GEMM, BFU-E, and CFU-GM (Table 1). Expression of truncated SHP2 had the greatest suppressive effect on formation of CFU-HPP, a very early cell within the stem/progenitor cell compartment, which is consistent with previously published studies demonstrating a stringent need of SHP2 for the support of hematopoietic stem cell and embryonic stem cell development and self-renewal [22,23,25,26,39]. Additionally, the observation of globally reduced single cell-derived colony formation upon expression of truncated SHP2 suggests that full SHP2 function is needed in a cell autonomous fashion for hematopoietic progenitor growth.
P<0.001 by χ 2 test compared to mock control and b P<0.01 by χ 2 test compared to mock control.
CFU-HPP, indicates high proliferative potential-colony forming cells; CFU-GEMM, colony forming unit–granulocyte erythroid macrophage megakaryocyte; BFU-E, burst forming unit erythroid; CFU-GM, colony forming unit–granulocyte macrophage.
TF1-derived colony growth is inhibited by reduced SHP2 expression and by expression of truncated SHP2
To further elucidate the role of SHP2 in human hematopoiesis, we utilized the human growth factor-dependent TF1 cell line, thus allowing us the potential for determining better mechanistic insight. Similar to effects noted using human CD34+ cord blood cells, preincubation with anti-sense ODN to SHP2 significantly reduced both TF1-derived erythroid and nonerythroid colonies in response to erythropoietin alone or in response to the cytokine combination erythropoietin, SCF, GM-CSF, and IL3 (Fig. 1E, F). To obtain mechanistic insight of how SHP2 promotes human progenitor cell growth, we next selected LXSN- and L-SHP2SH2-SN-transduced TF1 cells in G418, and demonstrated expression of truncated SHP2 (SHP2SH2) by immunoblot (Fig. 2A). Expression of SHP2SH2 inhibited TF1-derived erythroid and nonerythroid colonies in response to erythropoietin alone or in response to a cytokine cocktail (Fig. 2B, C), indicating that TF1 cells respond functionally in a similar manner as human CD34+ cord blood cells upon reduction of SHP2 expression or upon expression of truncated SHP2, indicating that these cells can serve as a useful surrogate model for primary human cells for biochemical analysis.
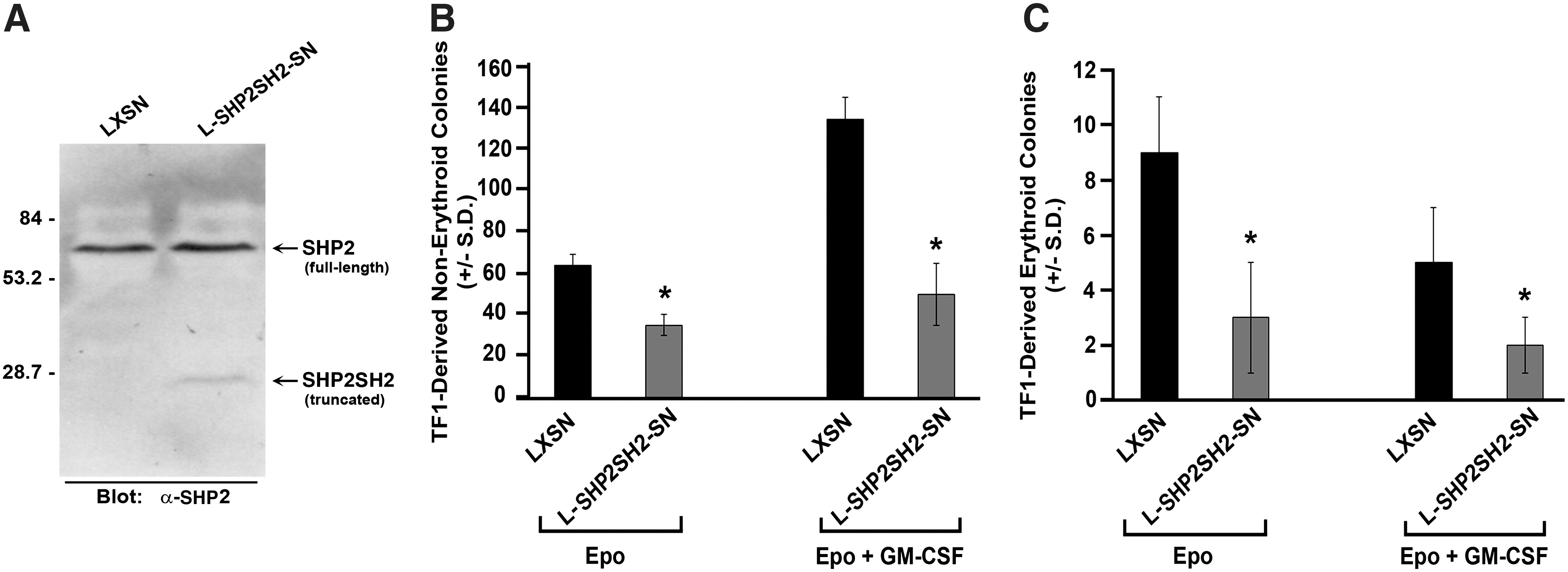
Influence of transducing human growth factor-dependent cell line TF1 with truncated SHP2, Shp2SH2. TF1 cells were retrovirally transduced with either empty vector (LXSN) or truncated SHP2 (L-Shp2SH2-SN) followed by selection in G418 1 mg/mL.
Expression of SHP2SH2 in TF1 cells inhibits the protein interaction between SHP2 and GRB2
In response to cytokine receptor stimulation, SHP2 has been shown to positively activate the Ras- MAPK pathway by biochemical interaction with the adapter protein, GRB2, and the guanine exchange factor, SOS1, and to positively activate the PI3K-AKT pathway by interaction with GRB2, GAB1/2, and the regulatory subunit of PI3K, p85 [9]. As GRB2 is implicated in SHP2-mediated activation of both the Ras-MAPK and PI3K-AKT pathways, we chose to examine the SHP2 interaction with GRB2 in TF1 cells transduced with LXSN and L-SHP2SH2-SN and selected in G418. We anticipated that expression of SHP2SH2 may interfere with the cytokine receptor-induced interaction between endogenous, full-length SHP2 and GRB2. Upon immunoprecipitation of SHP2 and immunoblotting with anti-GRB2, a clear reduction in GRB2 is found in the full-length SHP2-containing protein complex from cells stimulated with GM-CSF (Fig. 3). These findings suggest that ectopic expression of SHP2SH2, containing the N-SH2 and C-SH2 domains, reduces effective levels of interacting proteins that normally bring SHP2 and GRB2 into the same cytokine-induced protein complex to promote activation of the Ras-MAPK and PI3K-AKT pathways. This biochemical finding implies that the presence of full-length SHP2, including the phosphatase domain, is necessary within cytokine-induced signaling protein complexes for proficient cytokine-induced human hematopoietic progenitor cell growth.

Expression of truncated SHP2 reduces interaction between adapter protein, GRB2, and full-length SHP2. Immunoprecipitation of total cellular proteins from G418-selected TF1 cells with α-SHP2 followed by immunoblotting with α-GRB2 or α-SHP2 in the presence and absence of GM-CSF stimulation.
SHP2 phosphatase activity is needed for human progenitor colony growth
To specifically evaluate the role of SHP2 phosphatase function in human progenitor cell growth, we retrovirally transduced wild-type (WT) SHP2, a somatic JMML-associated gain-of-function SHP2 mutant with a known elevated phosphatase function, SHP2D61Y [19,40], or a phosphatase dead mutant, SHP2C463A [41], into human CD34+ cord blood cells. To enrich transduced cells, cells were sorted for expression of EYFP, tagged in frame to the 3′ end of each SHP2 cDNA. Transduced, sorted cells were plated into colony forming assays with GM-CSF alone to pick up the mature subset of CFU-GM and evaluate for GM-CSF hypersensitivity of the SHP2D61Y mutant [33,42,43], or in a cytokine cocktail, including GM-CSF, erythropoietin, IL-3, and SCF to evaluate effects on the more immature subsets of progenitors. As expected, SHP2D61Y-transduced cells generated substantially higher numbers of colonies in GM-CSF alone compared to WT SHP2-transduced cells, consistent with JMML-associated somatic gain-of-function SHP2 mutants inducing GM-CSF hypersensitivity (Fig. 4A) [33,42,43]. At similar concentrations of GM-CSF, SHP2C463A-transduced cells generated significantly fewer colonies than WT SHP2-expressing cells and did not produce increased colony formation in response to increasing concentrations of GM-CSF (Fig. 4A). These findings indicate that SHP2 phosphatase function specifically is needed for GM-CSF-stimulated human progenitor colony growth. In response to the cytokine cocktail, both CFU-GM- and CFU-GEMM-colony formation were increased in the SHP2D61Y-transduced and decreased in the SHP2C463A-transduced cells, respectively (Fig. 4B). Notably, however, the phosphatase dead mutant, SHP2C463A, more substantially inhibited growth of immature CFU-GEMM colonies compared to CFU-GM colonies, again highlighting the importance of SHP2 phosphatase function in the early hematopoietic stem/progenitor cell compartment.

Expression of phosphatase gain-of-function SHP2D61Y increases, while expression of phosphatase dead SHP2C463A decreases human cord blood CD34+ cell-derived colony formation by myeloid progenitor cells.
Discussion
While the protein tyrosine phosphatase, SHP2, is ubiquitously expressed, previous studies demonstrated that hematopoietic cell development is relatively more dependent on SHP2 than other tissues [21]. Further, somatic gain-of-function SHP2 mutations are commonly found in the childhood disease, JMML [19], and SHP2 has been found to be overexpressed in adult leukemias [44]. Very recent studies demonstrate that reduced SHP2 expression decreases human cord blood progenitor-derived colony growth and development [24], consistent with our findings and several studies performed in murine models. The current studies specifically address how different functional components of the SHP2 molecule, specifically the phosphatase function, influence human cord blood progenitor-derived colony growth, thus providing implications for SHP2 phosphatase inhibition as a novel therapeutic approach in hematologic malignancies.
Expression of truncated SHP2 (SHP2SH2, Fig. 1C), bearing the N-SH2 and C-SH2 domains, but lacking the phosphatase domain, in human cord blood CD34+ cells resulted in reduced colony formation at all levels of the hematopoietic hierarchy (Fig. 1D and Table 1), indicating that the adapter function of SHP2 in the absence of SHP2 phosphatase function works as a dominant negative in cytokine-stimulated colony formation, and suggesting that SHP2 phosphatase function is imperative for the activating function of SHP2 in human hematopoiesis. To evaluate the functional and biochemical effects of SHP2SH2 further, we utilized the human growth factor-dependent TF1 cell line, which behaved similarly to the human cord blood CD34+ cells upon reduced SHP2 expression (Fig. 1E, F) or expression of truncated SHP2 (SHP2SH2, Fig. 2). Given the dominant negative effect of truncated SHP2 in the absence of its phosphatase function, we hypothesized that SHP2SH2 sequesters necessary signaling molecules from full-length SHP2, culminating in reduced cytokine-stimulated signaling and subsequent reduced cytokine-stimulated colony growth (Figs. 1D and 2, and Table 1). To evaluate this hypothesis biochemically, we examined GM-CSF-stimulated interaction between full-length SHP2 and the adapter protein, GRB2, which is common to SHP2-containing protein complexes in the GM-CSF-stimulated Ras-MAPK and PI3K-AKT signaling pathways. As predicted, a reduced level of interaction between full length SHP2 and GRB2 was observed in GM-CSF-stimulated SHP2SH2-expressing cells (Fig. 3). These findings mechanistically demonstrate that the SHP2 phosphatase function is needed within GM-CSF-stimulated protein complexes for the suitable support of human progenitor growth.
Given that the studies utilizing SHP2SH2 strongly pointed toward the necessity of SHP2 phosphatase function for cytokine-stimulated human progenitor-derived colony growth, we next directly examined the role of SHP2 phosphatase function by utilizing both phosphatase gain-of-function and phosphatase dead SHP2 mutants. SHP2D61Y is a well-described gain-of-function mutant commonly found in children with JMML with increased phosphatase activity compared to WT SHP2 [19,40,45]. SHP2C463A is a loss-of-function, catalytically inactive mutant with replacement of the conserved catalytic cysteine with alanine [41]. The phosphatase gain-of-function SHP2 mutant promoted, while the phosphatase dead SHP2 mutant reduced, human cord blood colony formation, providing strong evidence that the phosphatase function of SHP2 is fundamental for cytokine-stimulated human progenitor development and growth. These findings imply that pharmacologic inhibition of SHP2 phosphatase function may be applicable in hematologic malignancies bearing SHP2 hyperactivation, either due to gain-of-function PTPN11 mutations or due to SHP2 overexpression.
Due to the highly conserved active site shared by all mammalian phosphatases, identification of specific SHP2 phosphatase inhibitors has been difficult; however, within recent years, pharmacologic inhibitors of SHP2 phosphatase function have been identified [29,46 –48], and have demonstrated activity in mutant KIT-, gain-of-function PTPN11-, and FLT3-ITD-induced myeloproliferative neoplasms [27 –29], all of which bear hyperactivation of SHP2. Based on these promising advancements, we anticipate that inhibition of SHP2 phosphatase activity using small pharmacologic molecules may yield a valuable adjuvant approach to therapies for acute myeloid leukemias and myeloproliferative neoplasms bearing SHP2 hyperactivation. Notably, SHP2 phosphatase inhibition, which we address in the context of hematologic malignancies, may additionally be applicable in solid tumors, as gain-of-function SHP2 mutations are observed in solid tumors [49] and SHP2 plays a critical role in Gab2-induced breast carcinogenesis [50,51]. Collectively, the findings presented herein refine our understanding of how SHP2 contributes to hematopoiesis and support the premise that inhibition of SHP2 phosphatase activity may provide a novel approach to hematologic malignancies and solid tumors bearing hyperactive SHP2 phosphatase function.
Footnotes
Acknowledgments
The authors gratefully acknowledge the In Vivo Therapeutics Core of the Indiana University Simon Cancer Center as well as the nursing staff and Dr. Arthur Baluyut at the St. Vincent Hospital (Indianapolis, IN) for providing a portion of the umbilical cord blood samples for this study. The authors thank the administrative assistance of Mrs. April Maines and Ms. Linda S. Henson. These studies were supported by the Public Health Service Awards NIH RO1 HL56416, NIH RO1 HL67384, and PO1 DK90948 (to H.E.B.), NIH RO1 CA173444 and the Riley Children's Foundation (to R.J.C.), and the Indiana University Showalter Trust Fund (to X.J.L.).
Author Disclosure Statement
The authors have nothing to disclose.