
Abstract
Embryonic pluripotent stem cells (ePSCs) can generate all somatic cell types, as well as functional gametes. In mouse and rat, derivation of ePSCs from the early epiblast is promoted by the double inhibition (“2i”) of mitogen-activated protein kinase kinase (MAP2K), antagonizing fibroblast growth factor signaling (FGF), and glycogen synthase kinase 3 (GSK3), stimulating the WNT pathway. However, it has remained unclear whether this culture regime is applicable to nonrodent livestock species. Here we report the generation of bovine ePSCs under minimal conditions. Inner cell masses (ICMs) were immunosurgically isolated from in vitro fertilized bovine blastocysts and cultured feeder-free in 2i medium. Dual kinase inhibition primed bovine ICMs for stem cell derivation and sustained expression of epiblast-specific pluripotency markers SOX2 and NANOG, while repressing the hypoblast marker GATA4. Following mechanical passage, 2i supported limited proliferation for several weeks. Continuously cultured ePSC lines expressed discriminatory markers of naïve pluripotency and primordial germ cells, but not of primed epiblast stem cells. In female ePSCs, most OCT4-positive cells lacked epigenetically silenced X-chromosomes, displaying a diagnostic feature of naïve pluripotency. Bovine ePSCs maintained a normal karyotype and differentiated into derivatives of all three germ layers in suspension culture. This culture system provides a screening platform for factors that maintain long-term proliferation of pluripotent embryonic cattle cells without genetic intervention.
Introduction
Under chemically defined conditions, ESCs will self-renew or differentiate, respectively, in response to the cytokine leukemia inhibitory factor (LIF) or fibroblast growth factor (FGF)/mitogen-activated protein kinase (MAPK or ERK) signaling [11]. In contrast, epiSCs do not respond productively to LIF and can be stably propagated in the presence of FGF and ACTIVIN [4]. Upon FGF/ACTIVIN withdrawal and ectopic expression of either Klf2, Klf4 [12], Nanog [13], Nr5a [14], or a JAK/STAT3 activating receptor [15], epiSCs can revert to naïve ground state pluripotency. Analogous to these overexpression approaches, naïve PSCs can also be generated by delivering ectopic pluripotency-inducing genes into adult somatic cells [induced PSCs (iPSCs)] [16].
Until recently, naïve chimera-competent ESCs could only be derived from a few inbred mouse strains and not from other species. This obstacle has been reproducibly overcome by using culture media supplemented with small molecules that alleviate differentiation cues and stabilize signaling pathways maintaining pluripotency [11]. Using their original protocol, Ying et al. [11] converted mouse epiSCs into ESCs [12] and partially into fully reprogrammed iPSCs [17] by double inhibition (“2i”) of MAP2K (MAPKK or MEK), neutralizing FGF signaling, and glycogen synthase kinase-3 beta (GSK3B), stimulating the WNT pathway. To maintain a high clonogenicity and growth rate, as well as directly improving the reprogramming process, this 2i medium also contained the self-renewal cytokine LIF [15]. Notably, the 2i/LIF medium led to the first derivation of bona fide ESCs from previously refractory mouse strains [18] and rat embryos [19,20]. While human embryo-derived PSCs cannot be derived or continuously maintained in 2i/LIF alone [21], it helped to establish a mouse ESC-like molecular profile in the presence of forskolin [22]. As a result, human cells with limited proliferation potential, but an epigenetic and transcriptional profile similar to naïve ESC lines could be established [22]. For ethical reasons, full pluripotency cannot be assessed in human-derived PSCs. Thus, their exact status, based solely on teratoma formation, presently remains elusive. To distinguish such cells from naïve rodent ESCs, we refer to them more generically as ePSCs.
Over the past 3 decades, all attempts to derive cattle ESCs have failed [23,24]. Cells have been cultured according to established mouse [25,26] or human protocols [27 –29], including feeders, serum, and supplementation with LIF or FGF2. The resulting ESC-like ePSCs vary in morphology, antigen profile, and in vitro differentiation ability [25,29,30]. By contrast, we used chemically defined feeder-free culture without exogenous growth factor supplementation. Under these minimal conditions, we explore the existence of a pluripotent ground state, stabilized through shielding from FGF/MAPK signaling and WNT activation, in bovine inner cell mass (ICM)-derived cultures.
Materials and Methods
Chemicals were purchased from Sigma-Aldrich (Auckland, New Zealand), unless specified otherwise. Embryo and cell culture was carried out in a humidified atmosphere of 5% CO2, 7% O2, and 88% N2 at 38.5°C.
In vitro production of embryos
In vitro-matured oocytes were derived from slaughterhouse ovaries of mixed breed dairy cows as described [31]. Spermatozoa were prepared from commercially obtained frozen–thawed semen from a sire with proven in vitro fertility. X-sorted sperm from one bull (103579X; CRV Ambreed, Hamilton, New Zealand) was used for the generation of female bovine embryos. The contents of two 0.25-mL straws were layered upon a Percoll gradient (45%:90%) and motile spermatozoa were collected after centrifugation at 700g for 20 min at room temperature. The sperm concentration was adjusted to 1×106 sperm per mL (4×105 for sexed sperm) and insemination performed 22–24 h poststart of maturation in 50 μL in vitro fertilization (IVF)-synthetic oviduct fluid (SOF) as described [31]. After 18–24 h, presumptive zygotes were washed in 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES)-buffered SOF (HSOF) and cultured for 7 days (D0: fertilization) in sequential the early SOF and late SOF (LSOF) medium [32]. For some experiments, the culture medium was supplemented with the MAP2K inhibitor PD0325901 (10 mM stock, a 0.4 μM final concentration; Stemgent, Cambridge, MA) and the GSK3B inhibitor CHIR99021 (10 mM stock, a 3 μM final concentration; Stemgent) (“2i”). All inhibitor stocks were dissolved in dimethyl sulfoxide (DMSO) and solvent controls contained 0.03% DMSO in the culture medium. Embryo cultures were overlaid with mineral oil and kept in a humidified modular incubation chamber (ICN Biomedicals Inc., Aurora, OH). Embryos were morphologically graded late on day (D) 7 or early on D8 [33].
Isolation and culture of ICMs
Expanded (X) blastocysts (B) of morphological grade 1 to 2 (XB1-2) were selected for immunosurgery and treated with 0.5% pronase to remove the zona pellucida. They were washed three times in HEPES-buffered transfer LSOF with 0.1 mg/mL cold soluble poly(vinyl alcohol) (PVA) (Mr : 10-30000) (THSOF-PVA), and then 8–10 embryos were incubated in rabbit anti-bovine serum (1:4) for 40 min at 38.5°C. After washing twice with THSOF-PVA, they were placed into the guinea pig complement (1:4) for 20–30 min. Isolated ICMs were individually seeded on glass coverslips coated for 1 h at 5–7 μg/cm2 with natural mouse laminin (Life Technologies, Auckland, New Zealand) or overnight with gelatin [1 mg/mL in phosphate buffered saline (PBS)] followed by laminin. Cells were cultured in 4-well plates, with each well containing 500 μL of an ePSC medium comprising Dulbecco's modified Eagle medium (DMEM)/F12 (e Life Technologies) + N2 (Life Technologies), mixed 1:1 with Neurobasal medium (Life Technologies) + B27 (Life Technologies), 1 mM L-glutamine [N2B27], and supplemented with 3 μM CHIR99021 and 0.4 μM PD0325901 [11]. For some experiments, the medium was supplemented with 10 μg/mL of human recombinant LIF (ORF Genetics, Kopavogur, Iceland). After 6–8 days of culture, colonies and surrounding outgrowths were dissected by either using two 30-gauge needles or a splitting blade (ESE 020; Bioniche Animal Health, Athens, OH) mounted to a micromanipulator (MO-188; Nikon Narishige, Tokyo, Japan). Fragments were seeded onto 4-well plates (1–2 fragments/well). This process was repeated every 4–5 days for routine passaging.
Cell proliferation assays
Nuclei numbers were counted after Hoechst 33342 DNA stain (5 μg/mL), either on complete confocal z-stacks (Olympus FluoView FV1000 with FV-10 ASW 1.4) or on deconvolution images (Leica DMI 6000 B with LAS AF6000). Both methods used ImageJ, either for manual (via a point-selection tool) or semiautomatic counting (via Image > Adjust > Threshold > Auto-threshold; Analyze > Analyze particles [Size (pixel^2): 0-Infinity; Circularity: 0.00–1.00; Show: outlines]). Since counts from both methods differed by <1%, which was nonsignificant, results were pooled. DNA synthesis was assessed using a click-iT® 5-ethynyl-2′-deoxyuridine (EdU) cell proliferation assay (Life Technologies). Cells were grown in the presence of 10 μM EdU for 30 min or 24 h. After labeling, cells were washed with PBS, fixed with 4% paraformaldehyde (PFA)/4% (w/v) sucrose in PBS followed by permeabilization with 0.5% Triton X-100 in PBS. The reaction cocktail was prepared fresh each time and detection carried out according to the manufacturer's instructions. Cells stained without EdU labeling served as a negative control.
Detection of apoptosis
Apoptosis was examined with the click-iT TUNEL Alexa Fluor® Imaging Assay (Life Technologies). Cells were fixed, permeabilized, and stained according to the manufacturer's instructions. Cells stained without EdU-incorporation provided a negative control. HeLa cells, treated with 0.5 μM staurosporine for 4 h, provided a positive control. Cleaved poly-ADP ribose polymerase (PARP) was detected in apoptotic cells with a rabbit anti-cleaved PARP antibody (ab4830; 1:100; Abcam, Cambridge, MA) and visualized using an Alexa Fluor 488 goat anti-rabbit IgG antibody.
RNA extraction and reverse transcriptase polymerase chain reaction
Samples were lysed in TRIZOL™ (Life Technologies) and cDNA synthesized as described [34]. Reverse transcriptase was omitted in one sample, each time a batch was processed for cDNA synthesis (-RT). A Mastercycler Gradient (Eppendorf, Hamburg, Germany) was used for endpoint polymerase chain reaction (PCR) amplification using primers shown in Table 1. Primers were designed using LightCycler® Probe Design 2.0 or NCBI/Primer-BLAST. PCR was performed using the following conditions: one cycle denaturation at 95°C for 5 min, followed by 35 cycles of 15 s at 95°C, 30 s at 52°C–63°C (see Table 1 for primer-specific annealing temperatures), 60 s at 72°C; 7 min extension at 72°C, and cooling to 4°C. For quantitative PCR, a LightCycler (Roche, Auckland, New Zealand) was used. All reactions were performed with the LightCycler FastStart DNA MasterPLUS SYBR Green I Kit. The ready-to-use Hot Start LightCycler reaction mix consisted of 0.4 μL of each primer (10 μM), 2.0 μL LightCycler SYBR Green I master mix, 5.2 μL diethylpyrocarbonate water, 1.0 μL DMSO if required, and 1–2.0 μL cDNA template. The following 4-segment program was used: (1) denaturation (10 min at 95°C); (2) amplification and quantification (20 s at 95°C, 20 s at 56°C–60°C, followed by 20 s at 72°C with a single fluorescent measurement repeated 45 times); (3) melting curve (95°C, and then cooling to 65°C for 20 s, heating at 0.2°C/s to 95°C, while continuously measuring fluorescence); and (4) cooling to 4°C. The product identity was confirmed by gel electrophoresis and melting curve analysis. For relative quantification, external standard curves were generated from serial 5-log dilutions of bovine blastocyst cDNA for each gene in duplicate. One high efficiency curve (3.6≥slope≥3.1, R 2>0.99) was saved for each target gene and imported for relative quantification as described [34].
Includes 4% dimethyl sulfoxide for RT-PCR.
Immunofluorescence
The following antigens were analyzed: SOX2 (AF2018; R&D Systems, Minneapolis, MN), OCT4 (sc-9081), GATA6 (sc-9055), SSEA-1 (sc-21702), SSEA-3 (sc-21703), SSEA-4 (sc-21704; all Santa Cruz Biotechnology Inc., Santa Cruz, CA), NANOG (eBioscience 14-5768, San Diego, CA), CDX2 (MU392A-UC; BioGenex, Fremont, CA), KLF4 (Ab72543), TRA-1-60 (Ab16288), TRA-1-81 (Ab16289; all Abcam), and H3K27me3 [35]. Cells were fixed in 4% (w/v) PFA/4% (w/v) sucrose in PBS for 15 min at 4°C, washed three times in PBS, quenched in 50 mM ammonium chloride in PBS for 10 min, permeabilized in 0.1% (v/v) Triton X-100 in PBS for 10 min at room temperature, and blocked in 5% donkey or goat serum, 5% bovine serum albumin in PBS for at least 30 min. Primary antibodies were incubated overnight at 4°C, washed in PBS, and incubated with Alexa Fluor 488 or 568 donkey anti-mouse, anti-rat, anti-rabbit, or anti-goat secondary IgG antibodies (all Life Technologies) for 30 min at 37°C. All antibodies were diluted in a blocking buffer. DNA was counterstained with 5 μg/mL Hoechst 33342. Preparations were washed thrice in PBS and once in water before mounting (DAKO, Med-Bio Ltd., Auckland, New Zealand). Images were taken on an epifluorescence microscope (AX-70; Olympus, Auckland, New Zealand) equipped with a Spot RT-KE slider charge-coupled device camera (Diagnostics Instruments Inc., Sterling Heights, MI) or on a confocal microscope (Olympus FluoView FV1000).
Alkaline phosphatase activity
For alkaline phosphatase (AP) staining, cells were washed with PBST (0.05% Tween 20 in PBS), and fixed with 4% PFA for 2 min at room temperature. After washing 3 times in PBST, cells were stained in 1 mL NTMT buffer [10 mM Tris (pH 9.5), 100 mM sodium chloride, 50 mM magnesium chloride, 1% Tween 20] with 3.38 μL of 100 mg/mL nitro blue tetrazolium reagent (Roche) and 3.5 μL of 50 mg/mL 5-bromo-4-chloro-3-indolyl-phosphate reagent (Roche) for 5–10 min in the dark. Once blue color developed, cells were washed with PBS and stored at 4°C in the dark.
Karyotyping
Karyotyping was performed at Passage 3 as described [36]. Briefly, ePSC colonies were treated with the KaryoMAXColcemid® solution (0.1 μg/mL; Life Technologies) for 1 h. The pellet was resuspended in a 0.56% potassium chloride solution, incubated at 37°C for 15 min, and fixed in −20°C methanol:acetic acid (3:1) at 4°C for 30 min. Washing with fresh fixative was repeated twice before resuspending the pellet in 500 μL of ice-cold fixative, spreading onto chilled microscope slides, and staining with 10% KaryoMAX Giemsa in Gurr buffer (pH 6.8; BDH, Auckland, New Zealand) for 10–15 min followed by washing under a gentle stream of tap water. Metaphase spreads were photographed using a 100×oil immersion objective and a Spot RT-KE slider camera.
Embryoid body formation
Bovine ePSC colonies were mechanically removed from the substrate and disaggregated into small clumps using two 30-gauge needles. The dissociated single cells or small clumps were transferred to bacterial Petri dishes (BD Falcon, Franklin Lakes, NJ) containing the N2B27 medium. The cells were fed every 3 days by tilting the plate, allowing cells to settle, and carefully removing the medium. After 14 days, embryoid bodies (EBs) were collected and processed for RNA extraction.
Statistical analysis
All values are presented as mean±SD, unless indicated otherwise. Statistical significance was accepted at P<0.05 and determined using the two-tailed t-test with equal variance.
Results
Primary bovine ePSCs sustain pluripotency markers in 2i
We first examined the effect of the 2i medium on culturing and explanting ICMs from IVF-derived embryos. Freshly isolated ICMs were transferred into the DMEM/F12/N2B27-based culture medium onto laminin-coated substrates. ICM attachment was determined 6 days after seeding and did not depend on 2i supplementation (62/90=69% with 2i vs. 48/82=59% without 2i, respectively, n=6, P=0.21). We previously identified gelatin as a preferred plating substrate for bovine ICM cells over collagen and poly-L-lysine (data not shown). Since mouse ESCs benefit from laminin for maintaining pluripotency and proliferation [37], we tested a combination of gelatin with laminin. Plating efficiency in 2i, scored as the number of attached out of the seeded ICMs after 6 days in vitro, was increased over laminin alone (111/136=82% vs. 208/329=63%, n=21 and n=24 repeat experiments, respectively, P=0.0001; Supplementary Fig. S1A; Supplementary Data are available online at
To assess functionality of the LIF pathway in cattle, we supplemented the 2i medium with human recombinant LIF. In direct comparisons with nonsupplemented controls, we found no effect of LIF on ICM attachment (28/36=78% vs. 24/37=65%, n=5), primary colony formation (18/28=64% vs. 15/24=63%, n=5; Supplementary Fig. S1E), outgrowth diameter (1011±193 vs. 1113±169 μm, n=3), and primary colony size (81±24 vs. 77±4 μm, n=4; Supplementary Fig. S1F). Morphologically, LIF-supplemented cultures were indistinguishable from controls. We conclude that LIF did not promote colony expansion in bovine ePSC cultures.
Primary colonies were fixed at D6 and D8 postplating and analyzed by immunostaining. While the 3i medium has an antiapoptotic effect during ESC derivation in mouse [38], bovine colonies showed no qualitative differences in cell death before passaging (Supplementary Fig. S2). Epiblast-specific pluripotency markers NANOG and SOX2 were confined to the central colony and absent from the surrounding outgrowth. NANOG tended to be mutually exclusive with the putative hypoblast marker GATA6, which was present in all nuclei of the outgrowth (Supplementary Fig. S3A). SOX2 was also restricted to the central colony where it did not overlap with the putative trophoblast marker CDX2 (Supplementary Fig. S3B). Occasionally, CDX2 expression also extended into the outgrowth (data not shown). Discriminatory pluripotency surface markers SSEA-1 and TRA-1-60/-81 were restricted to the colony and absent from the outgrowth, whereas others, such as SSEA-3/4, as well as pluripotency-related transcription factors OCT4 and KLF4, were present in both (Fig. 1A). Most pluripotency-related markers, including AP activity, OCT4, KLF4, SSEA-1/3/4, and TRA-1-60/-81, as well as the putative hypoblast and trophoblast markers GATA6 and CDX2, respectively, were not qualitatively affected by the presence of 2i (Fig. 1A). However, SOX2, which was still present in colonies on D6, had largely disappeared by D8 in the absence of 2i (D6: 9/12=75% vs. 8/11=73% and D8: 15/25=60% vs. 2/16=13% in 2i vs. no 2i, respectively, P<0.01; Fig. 1B). Likewise, NANOG-positive colonies, which were still present on D4, appeared significantly less frequently by D6 in the absence of 2i (D4: 3/4=75% vs. 3/3=100% and D6: 21/28=60% vs. 6/18=33% in 2i vs. no 2i, respectively, P<0.05; Fig. 1B). 2i culture also significantly increased NANOG and concomitantly decreased GATA4 expression (P<0.005; Fig. 1C), confirming our previous observations in bovine blastocysts [39].
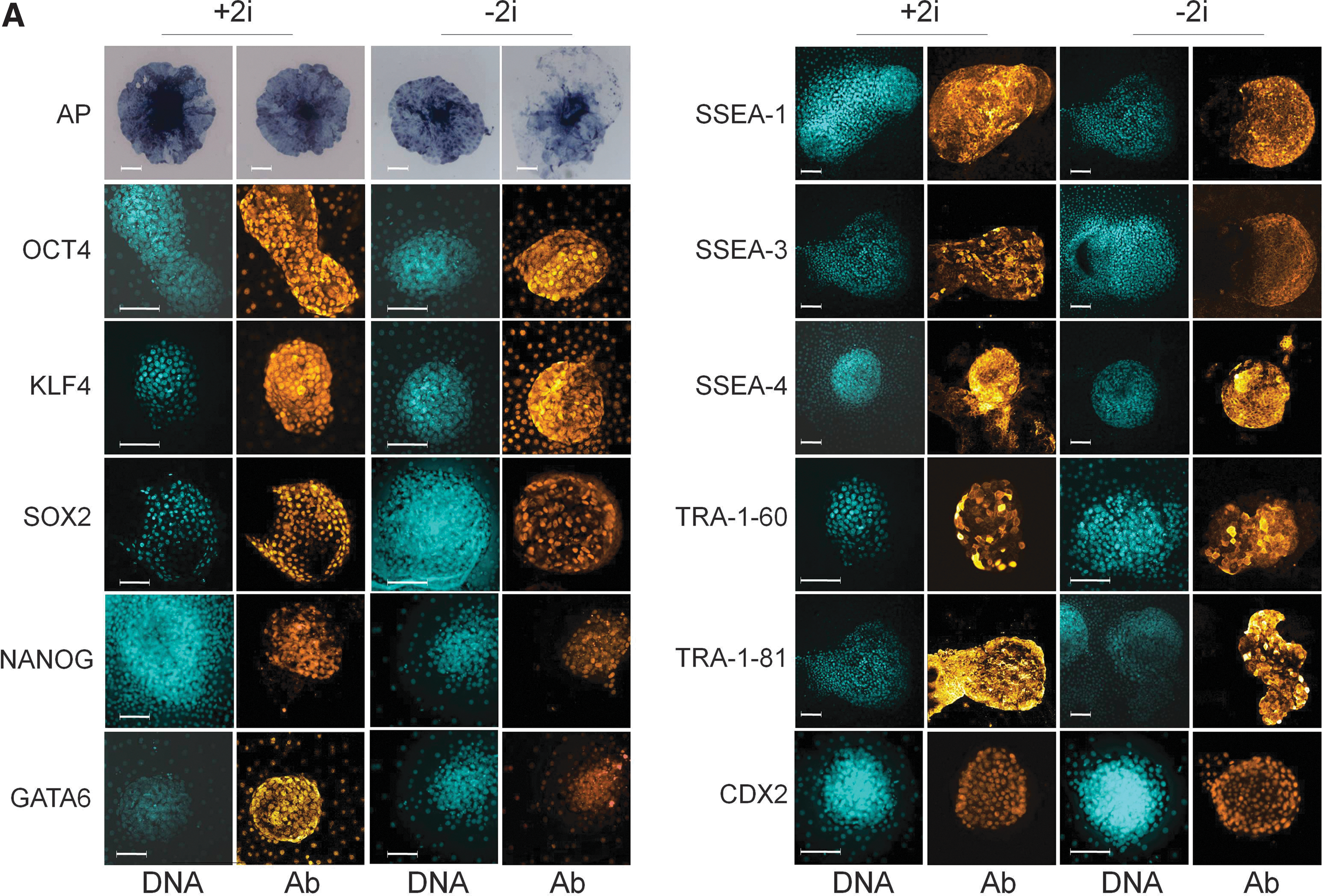
Molecular characterization of primary bovine inner cell mass (ICM)-derived outgrowths in 2i medium.
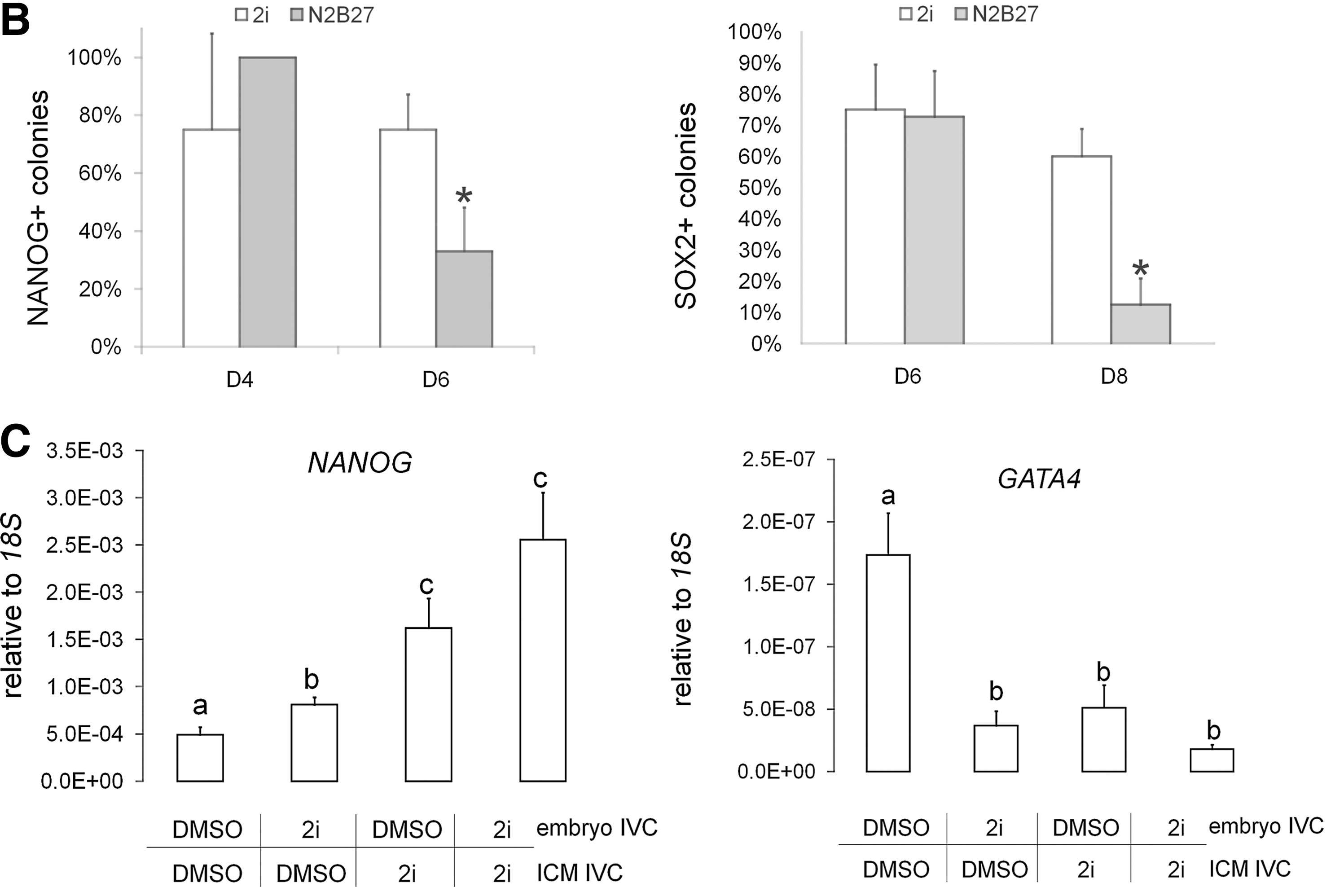
To test whether 2i biased blastocysts for subsequent ePSC derivation, we cultured embryos in either 2i or an equal DMSO concentration from the zygote stage. On D8, ICM cultures were derived and cultured for another 6 days in either 2i or DMSO before gene expression analysis (n=4 replicate experiments; Fig. 1C). NANOG transcription in primary ICM cultures (N=16) was increased by the presence of 2i in the embryo culture medium (P<0.05). Continuous exposure to 2i for a total of 14 days resulted in the highest NANOG levels; however, this effect was no longer significant compared to including 2i only in the ICM culture medium (N=12 colonies, P=0.15). GATA4 expression (N=16 colonies) was suppressed when 2i was included in the embryo culture medium (P<0.005) and this effect was not reversible, even when 2i was removed during subsequent ICM culture. Continuous exposure to 2i for a total of 14 days resulted in the lowest GATA4 levels, but this effect was again no longer significant compared to just including 2i in the ICM culture medium (N=12 colonies, P=0.18). Taken together, this suggests that 2i-treated blastocysts irreversibly reprogrammed some hypoblast into epiblast, priming them for subsequent ePSC derivation.
Finite ePSC lines proliferate in 2i
We next attempted to obtain ICM-derived cell lines through subculture of primary colonies. Individual colonies were cultured for one week, mechanically dissociated into small fragments, and replated into fresh wells. Since 2i sustained expression of pluripotency markers SOX2 and NANOG, while suppressing the hypoblast marker GATA4, we carried out all passaging experiments in this medium. In 14 independent IVF experiments, we established 64 different primary cell lines from as many blastocysts. All lines were mechanically passaged 8 times, corresponding to about 35 days in continuous culture, before cultures were discontinued. Colonies did not survive dissociation into single cells by either mechanical trituration or enzymatic treatment with accutase, pronase, trypsin, or dispase. Reattachment after dissection was around 50% and nonattached cell clumps were not further analyzed. On average, about 50% of the reattached colonies were passaged again, resulting in an overall passaging efficiency of about 25%. Passaged cultures grew in multilayered clumps of cells, overall resembling the flat morphology of human embryo-derived stem cells (Fig 2A, a). Confocal z-scans of Hoechst 33342 stained colonies showed up to 3 layers of nuclei stacked on top of each other in the core of each colony (Fig. 2A, b). Within each colony, cells were often so tightly packed that individual nuclear and cytoplasmic compartments or cell borders were not easily discernible (Fig 2A, c and d). As a proxy for cell proliferation, we quantified DNA synthesis following different EdU-incorporation protocols (Fig. 2B). EdU pulse-labeling for 30 min revealed that in steady-state, 20%–30% of cells were in S-phase in primary and passaged colonies (67/283=24% and 43/138=31%, respectively; Fig. 2C). After cumulative labeling for 24 h, almost all primary and passaged cells were cycling (322/322=100% and 249/261=95%, respectively; Fig. 2C). In 2i, explanted ICMs expanded from 51±4 to 1102±55 cells per colony and surrounding outgrowth within 6 days of culture, equivalent to ∼4–5 population doubling (PD) or a PD time of ∼1–1.5 days (Fig. 2D). The cell number per colony and outgrowth remained fairly constant upon regular passaging every 4–5 days.
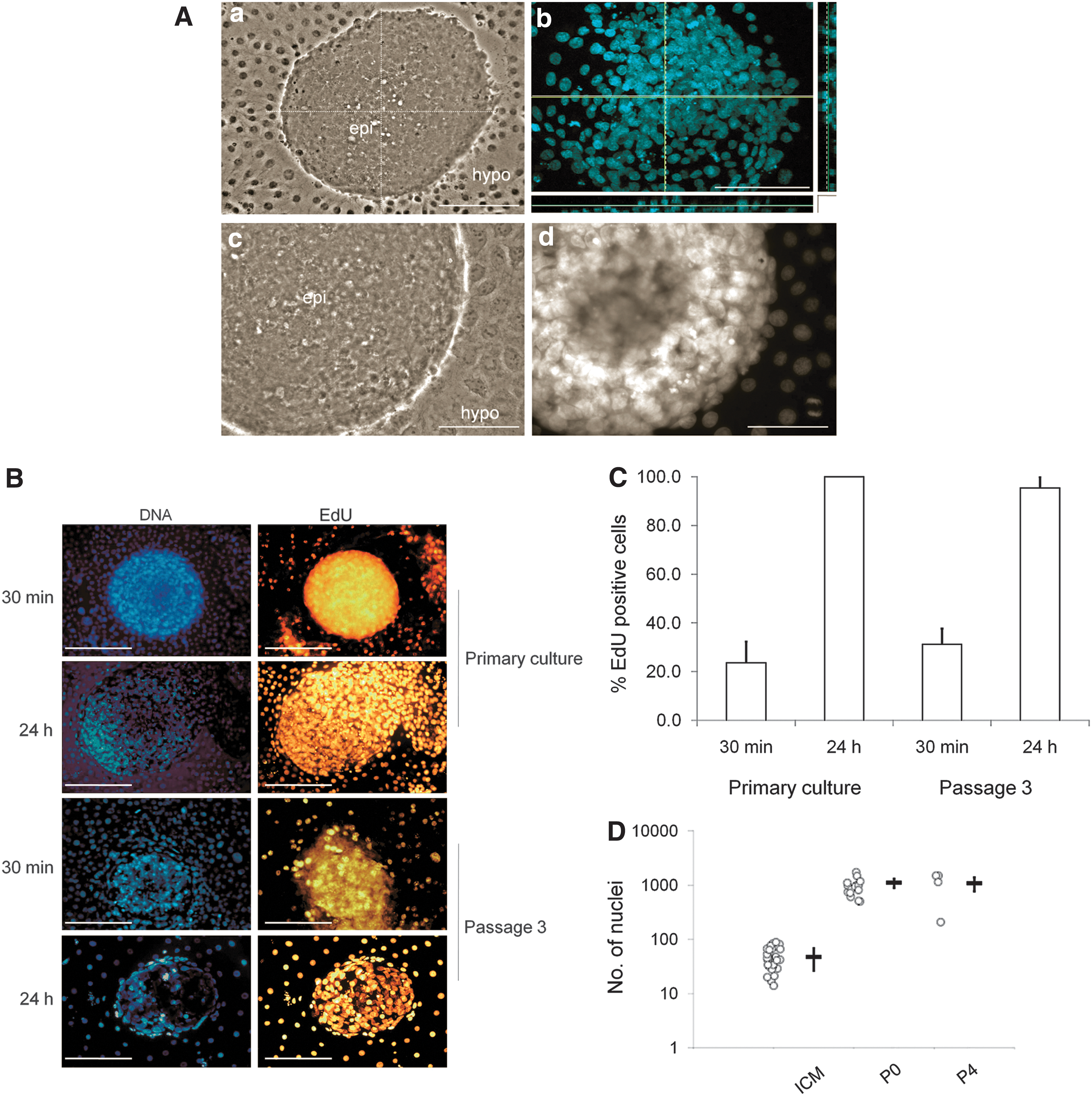
Primary ICM-derived colonies proliferate and survive in 2i medium.
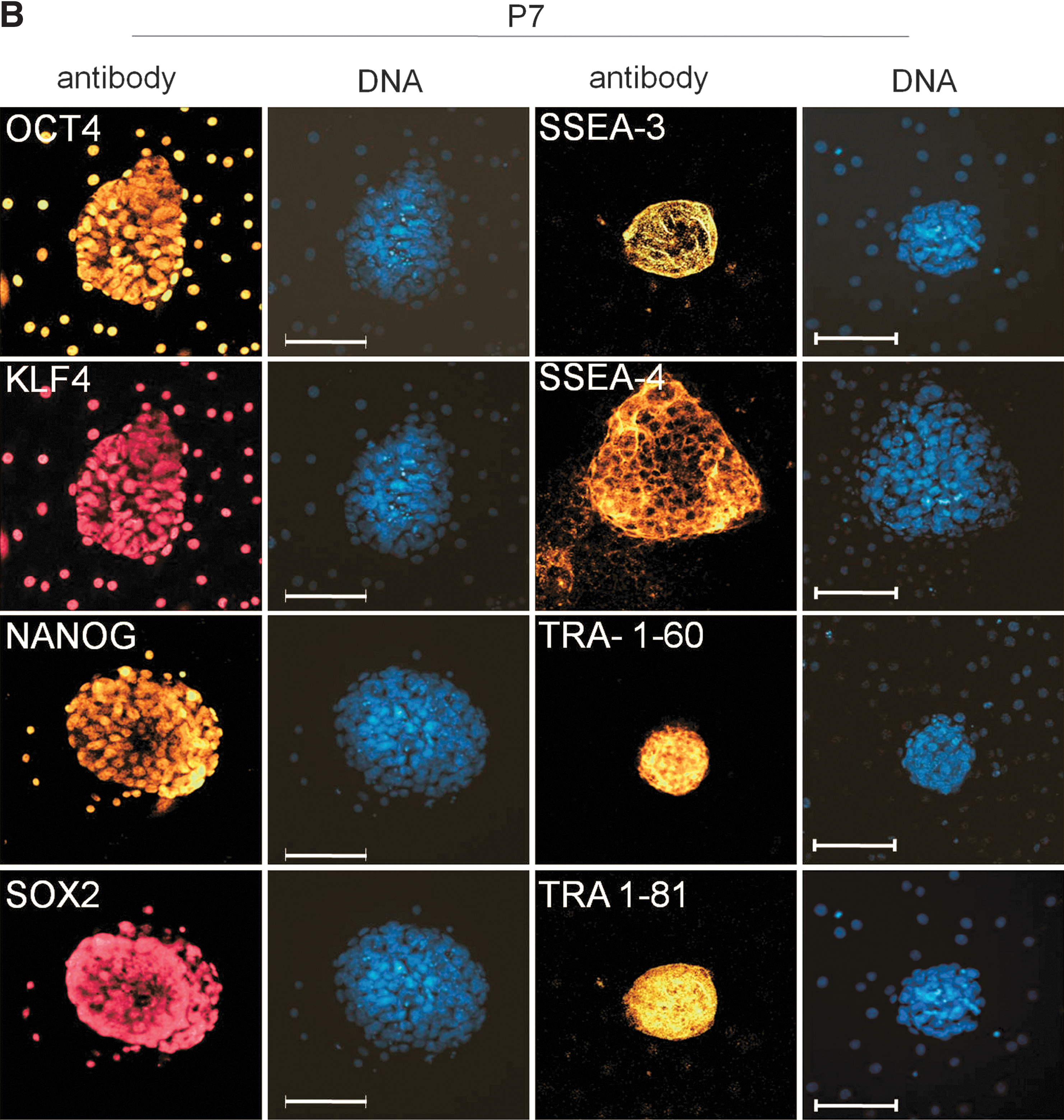
Molecular characterization of ePSC lines in 2i
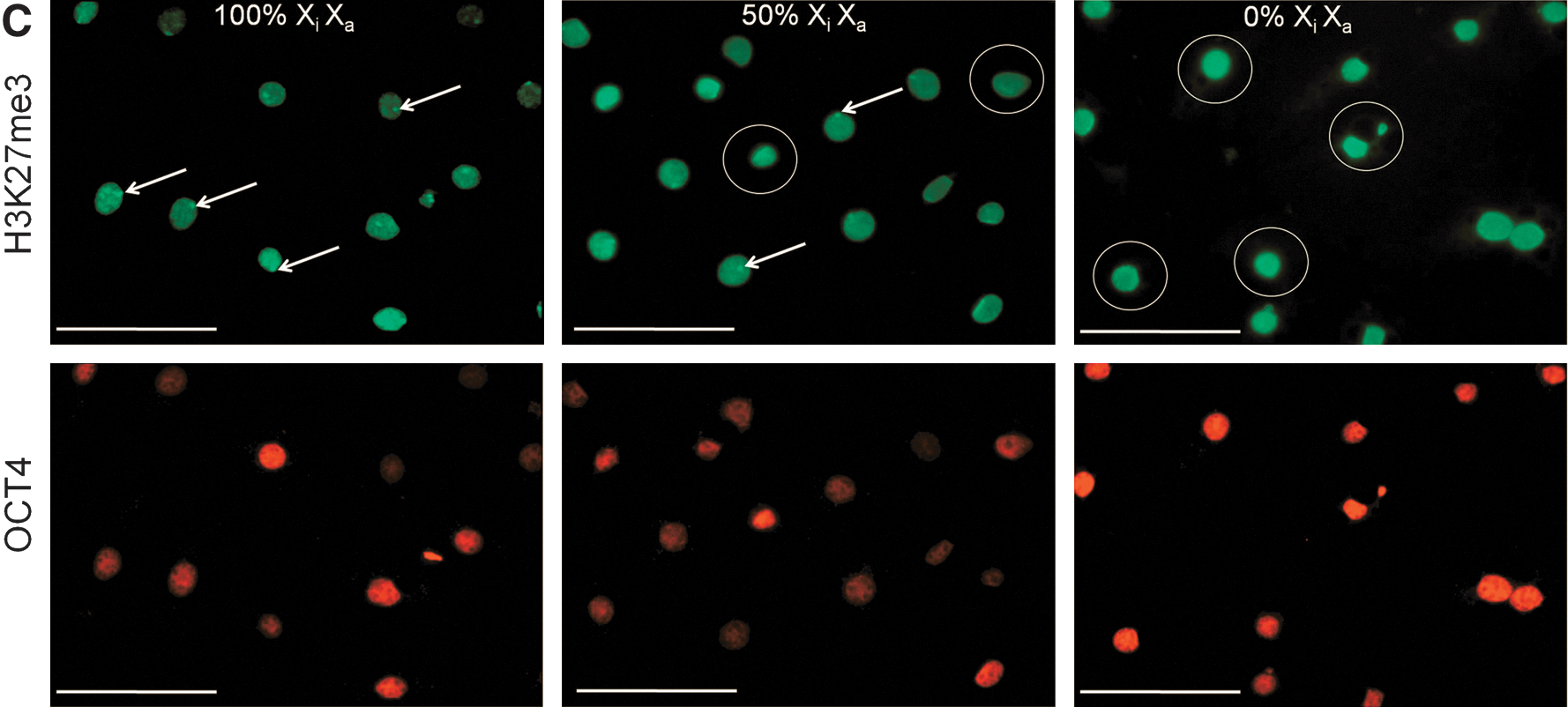
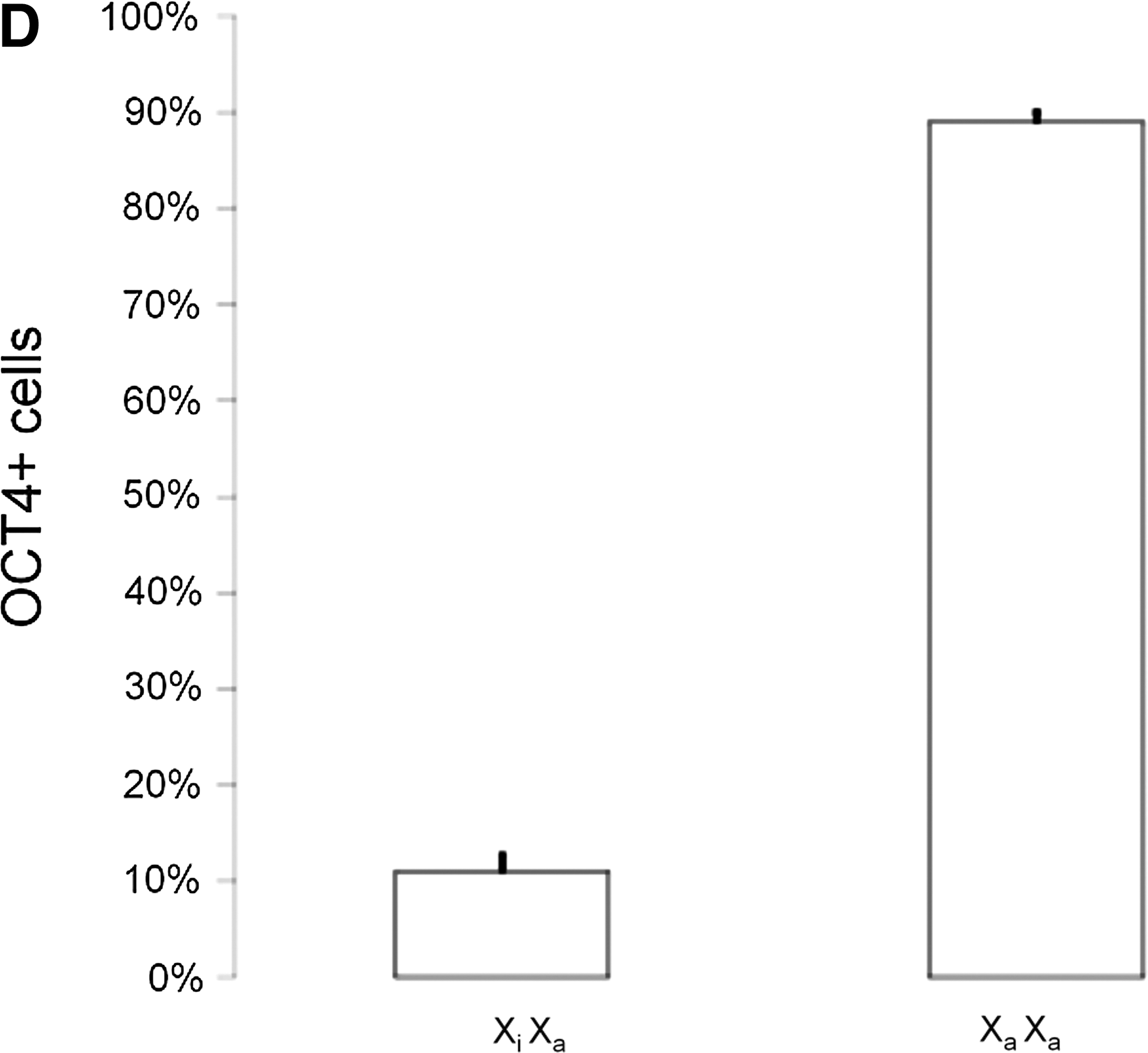
Following subculture, cells were analyzed for key pluripotency and lineage commitment markers at Passage (P) 4 and P7, corresponding to ∼D20 and ∼D30 in culture, respectively (Fig. 3A). At P4 (data not shown) and P7, ePSC lines still expressed a repertoire of core pluripotency-related factors (CDH1, OCT4, SALL4, SOX2, and TCF3), including markers enriched in naïve pluripotent cells (DPPA3, KLF4, LIN28, NANOG, SOCS3, and STAT3) and primordial germ cells (IFITM3). Genes that are downregulated in primed pluripotent cells were either undetectable (FGF5 and T-BRACHYURY) or downregulated (LEFTY) after passaging. These mRNA results were confirmed on the protein level where OCT4, KLF4, SOX2, and NANOG, as well as SSEA-3/4 and TRA-1-60/-81, but not SSEA-1 (data not shown), remained widely expressed (Fig. 3B). A diagnostic feature of mouse ESCs is the simultaneous presence of two active X-chromosomes and OCT4. This distinguishes them from OCT4-positive epiSCs carrying one epigenetically silenced X-chromosome (Xi) and somatic cell types that do not express OCT4 [40]. In female ePSCs, no nuclear foci of intense H3K27me3, indicating XCI, were found within the central colony (0/771, n=6 colonies). Within the outgrowth, nuclear H3K27me3 bodies were rare in OCT4-positive cells and loss of Xi was more common (184/1699=11% vs. 1515/1699=89%, n=12; Fig. 3C, D). Thus, most ePSCs exhibited major epigenetic erasure, a unique feature of the early rodent epiblast and female ESCs.
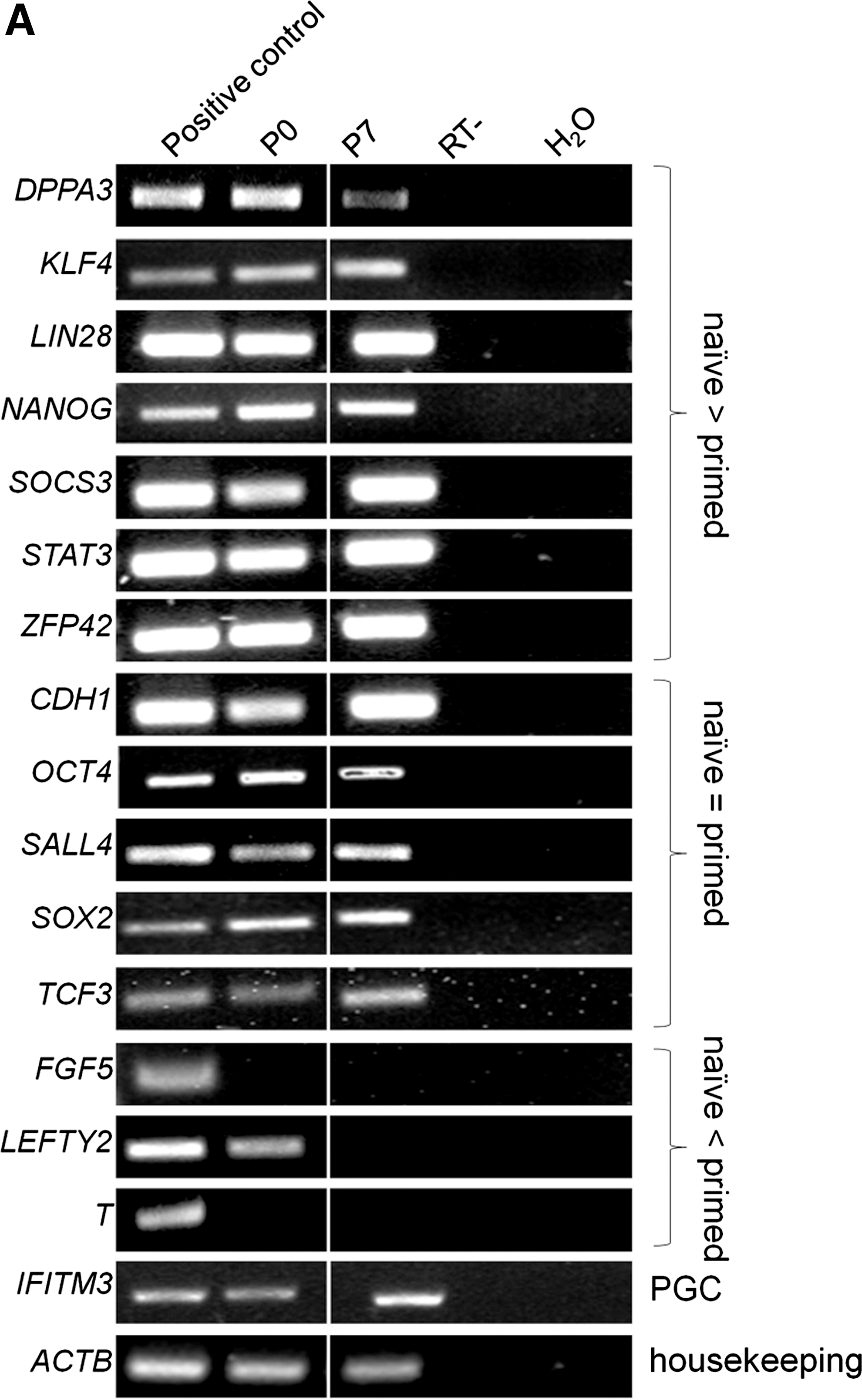
Molecular characterization of passaged bovine embryonic pluripotent stem cell colonies.
ePSC lines differentiate in vitro
Karyotyped bovine ePSCs after 3 passages showed a normal number of 60 chromosomes in most cells (17/18=94%; Fig. 4A). To determine their differentiation potential in vitro, bovine ePSCs were cultured in suspension without 2i. Within 1–3 weeks, solid and cystic EBs formed (Fig. 4B). Negative controls, cultured in 2i, did not form cystic EBs (data not shown). Gene expression analysis showed that these EBs expressed neuroectoderm (TUBB3, GFAP, and NES), endoderm (AFP and HNF4A), and mesoderm (MEF2C and MYF6) markers (Fig. 4C).
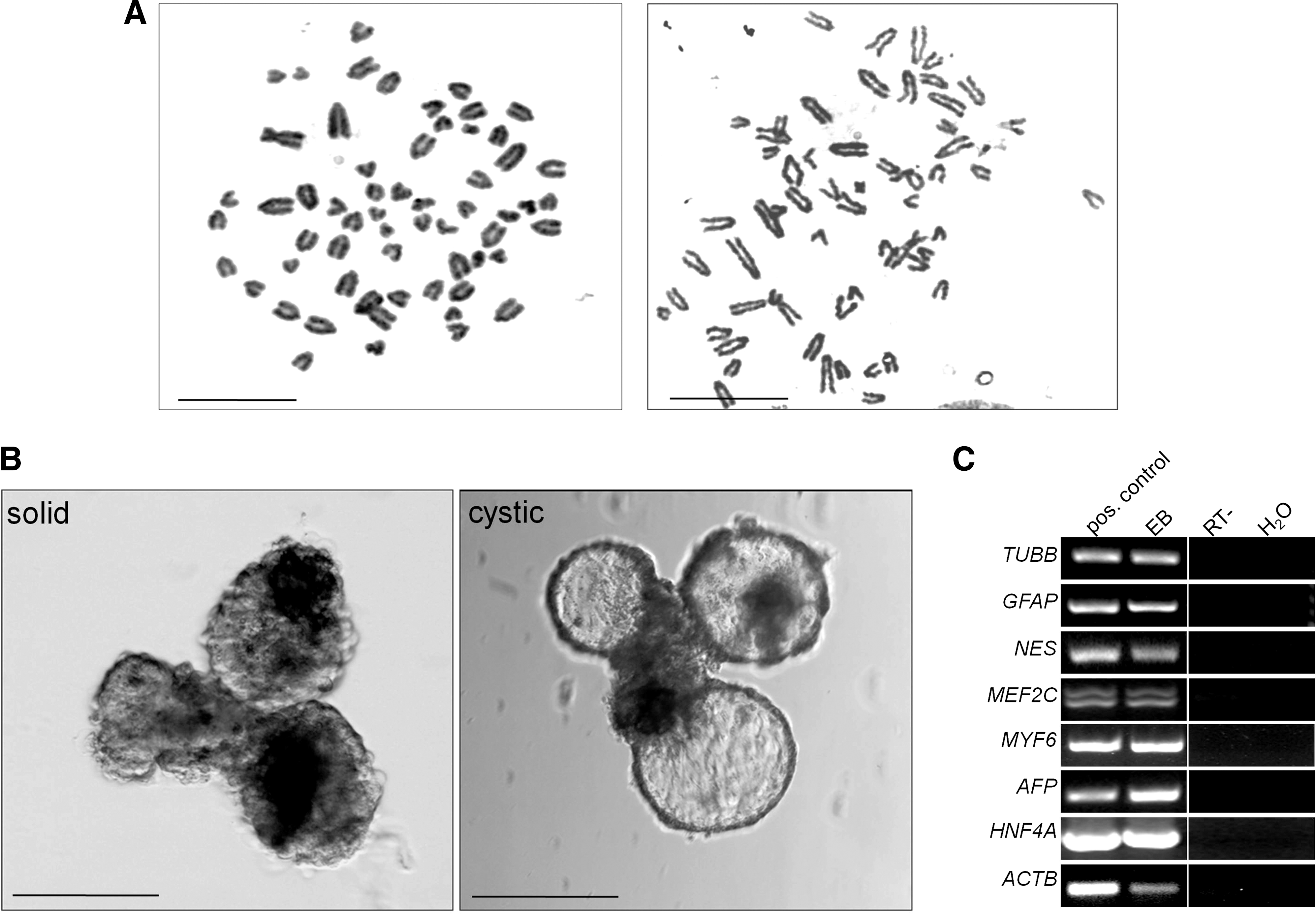
Differentiation of bovine embryonic pluripotent stem cell (ePSC) colonies in vitro.
Discussion
We demonstrate that 2i medium enables the derivation of ICM-derived cell lines under chemically defined conditions. We refer to these cells as ePSCs, rather than ESCs, as their potential for germline chimerism remains unproven. Cells exhibited molecular and functional features of pluripotency, including expression of diagnostic markers of mouse naïve epiblast-derived cultures, suppression of hypoblast and primed pluripotency markers, robust proliferation, and in vitro differentiation into 3 germ layer derivatives.
We first considered physical factors of ePSC culture. Physiological O2 levels in the oviduct are lower than atmospheric concentrations [41]. Low-O2 culture increases blastocyst survival and proliferation [42], protects against DNA damage [43], suppresses differentiation [44], prevents precocious XCI, and enhances pluripotency [45]. Considering these beneficial effects, all cultures were conducted under hypoxic conditions. Epiblast cells naturally adhere to a basal lamina [46] whose central component is laminin. By combining laminin with gelatin, the hydrolyzed form of collagen, ICM adhesion was increased to >80% without compromising proliferation. Being epithelial, epiblast cells are connected by robust tight junctions and desmosomes [47]. Consequently, colonies did not survive dissociation into single cells, using either enzymatic or chemical (e.g., ethylenediaminetetraacetic acid, Ca2+/Mg2+-free PBS) means, and were mechanically subdivided for expansion [23].
In previous studies, bovine ePSCs were derived from different founder tissues, including zygotes [25], blastocysts [30,48 –50], and prestreak epiblasts [46]. Here we used expanded perihatching in vitro production (IVP) blastocysts as the starting material. At this stage, the ICM has initiated segregation into hypoblast and epiblast, marked by nonoverlapping expression domains of GATA4/6 and NANOG, respectively [51]. We chose this stage in analogy to the mouse, where ESCs directly derive from microdissected single epiblast cells at high efficiency, suggesting that trophoblast and hypoblast removal facilitates ESC generation [52]. Isolation of pure epiblast tissue can be problematic since immunosurgery does not destroy all trophoblast cells [53] and leaves the hypoblast intact [54]. Accordingly, presence of the trophoblast marker CDX2 in the central colony was likely due to persistent expression in the ICM, where it would have been expressed before isolation [39] and protected from immunosurgery. Trophoblast cells are morphologically distinct from hypoblast and epiblast cells through the accumulation of cytoplasmic lipid droplets [23]. Following passage, we rarely observed this morphology or CDX2-positive nuclei in the outgrowth, indicating that trophoblast proliferation was not stimulated by 2i, similar to the situation in mouse [55]. Instead, outgrowth cells almost exclusively displayed the web-like cytoskeletal network characteristic of hypoblast cells [23] and were immunoreactive for GATA6. This abundance of hypoblast cells was unexpected since in mouse MAP2K inhibition reprograms almost all hypoblast into epiblast by blocking Gata4 activation and lifting its inhibitory effect on Nanog expression [55]. Bovine hypoblast formation and proliferation also depends on FGF signaling. For example, exogenous FGF stimulates hypoblast proliferation in outgrowth cultures [56] and converts most epiblast into hypoblast by increasing GATA6 and repressing NANOG expression [51]. Abolition of fibroblast growth factor receptor (FGFR) activation has the reverse effect of respecifying GATA4- into NANOG-expressing cells [56], even though this effect was not apparent on the protein level [51]. 2i does not block other pathways downstream of FGFR, such as PI3K activation, and only leads to a modest level of lineage conversion in bovine blastocysts [39,51]. Taken together, this suggests that some downstream effectors of FGFR, such as the PI3K cascade, may be beneficial for pluripotency or that GATA6 expression may also be activated via FGF/MAPK-independent signals. While 2i-mediated repression of GATA4 transcription was not sufficient to impair bovine hypoblast expansion in culture, it did maintain NANOG and SOX2 expression in the central colony. This confirms our previous findings in 2i-treated blastocysts [39]. Since both transcripts are usually rapidly downregulated in bovine ICM-derived cultures [49], the 2i regime clearly promoted progression into pluripotency. We speculate that this increased transcriptional activity may have been due to MAP2K inhibition stimulating nuclear accumulation of TBX3, a T box transcription factor activating Nanog and, to a lesser extent, Sox2 in mouse [57]. This may contribute to activation of the second Nanog allele and increased expression, a phenomenon that has been observed in the naïve epiblast and 2i-cultured ESCs [58]. How might increased Nanog expression be beneficial for pluripotency? It has been proposed that the unique function of NANOG is to switch on the naïve pluripotent program in mammalian cells [59]. In mouse, NANOG is required for specification, but not maintenance, of the naïve epiblast and its expression coincides with the establishment of an uncommitted pluripotent ground state [13]. In the presence of other key factors, specifically OCT4, SOX2, and KLF4, increased Nanog levels stabilize the pluripotent state against differentiation cues and finalize reprogramming into iPSCs [59]. It is not yet fully elucidated how NANOG coordinates the pluripotency network. Depending on the sequence conservation between mouse versus bovine, NANOG and its closely colocalizing interaction partner SOX2 [60], would establish pluripotency through direct repression of target genes, such as GATA4/6 [61] and the noncoding RNA Xist, which is critical for XCI. In mouse, direct suppression of Xist results in reactivation of the silenced X, which is important for later random XCI in early female epiblasts [62,63]. Transient reactivation of the inactive X in the naïve epiblast strictly correlates with Nanog expression in vivo [13] and provides a diagnostic feature of ground state pluripotency [64]. In bovine embryos, the occurrence, timing, and molecular mechanism of transient XCI reversal has not been described in detail. We found that XCI was rare within in the central epiblast colony or surrounding hypoblast outgrowth, indicative of major epigenetic erasure. It remains to be determined whether this condition is stably propagated upon continuous passaging and critically depends on the presence of the 2i medium. During the transition into primed pluripotency, Nanog and Zfp42 are downregulated [65], resulting in one X chromosome undergoing random XCI [66]. This was not apparent in cultured bovine ePSCs where genes associated with naïve pluripotency continued to be expressed. In addition to NANOG and ZFP42 [67], this included STAT3 [15] and SOCS3 [12,22], as well as their downstream target KLF4 [68], which, in turn, lies upstream of Nanog and Sox2 [69]. Consistent with this, markers of primed pluripotency, such as FGF5, LEFTY, and T-BRACHYURY, were absent during prolonged culture.
In mouse, STAT3-mediated SOCS3 activation after LIF treatment is elevated in naïve ESCs [12,15] and, in co-operation with KLF4, plays a crucial role in establishing ground state pluripotency [12,15,70]. The LIF pathway facilitates ex vivo capture of the mouse epiblast [52] and is absolutely required during mouse diapause when proliferation and developmental progression of the free-floating blastocyst is arrested [13,71]. A similar state does not exist in cattle where preimplantation embryos continue to proliferate and differentiate. Despite the presence of STAT3 and SOCS3, we did not observe a productive response to the heterologous LIF in bovine 2i cultures. This supports the notion that LIF signaling is not operational during bovine blastocyst culture [72,73], ePSC derivation [25,72,74], or early pregnancy [75]. The lack of the LIF activity to promote self-renewal or inhibit differentiation of epiblast cells likely compromises robust maintenance of the core pluripotency network in cattle.
Other potential downstream mediators of LIF-Stat signaling that could contribute to its reprogramming mechanism include genes involved in phosphorylation, and thereby inactivation, of GSK3 [76]. Partial GSK3 inhibition stabilizes c-Myc [77], a potent oncogene, and stimulates several ill-defined anabolic processes [78], augmenting cell proliferation and viability when MAPK is suppressed. We previously observed that GSK3 inhibition increased pluripotency markers in bovine iPS-like cells [79] and blastocysts [39]. Thus, WNT signals may play a more important role in specifying bovine epiblast and/or suppressing hypoblast formation.
While 2i effectively sustained pluripotency marker expression and prevented spontaneous differentiation in cultured cells, it did not favor extensive amplification of epiblast cells in the central colony over hypoblast cells in the outgrowth. This mirrors preimplantation stages in utero where a modest increase in epiblast cell numbers contrasts with a dramatic expansion of the hypoblast. In vitro, we also observed uncoupling of pluripotency and proliferation during the generation of bovine iPSCs under 2i conditions [79]. Likewise, human ePSCs cannot be derived or continuously maintained in 2i/LIF alone, even though it helps establishing a molecular profile resembling naïve mouse ESC lines [21,22]. It appears that unlike mouse ESCs, which intrinsically self-renew, nonrodent ePSCs require, as yet unknown, extrinsic signals for their proliferation. This should not pose a practical problem for breeding applications, such as multiplication of genomically selected embryos through conversion into ePSC lines [80]. Short-term culture systems for finite cell lines would be sufficient for this major application awaiting ePSCs in agriculture, which is why we did not extend the characterization of bovine ePSCs past about one month of continuous culture. Although complex genomic modifications can be routinely done in somatic cells [80], long-term proliferating ePSCs might still be useful for transgenic applications, as they are potentially more amenable to homologous recombination [81].
Bovine ePSCs readily formed EBs expressing markers of all 3 germ layers. Even though endoderm markers, such as AFP and HNF4A, may derive from the presence of contaminating hypoblast cells, this is less likely for early neural (NES, TUBB3, and GFAP) and late muscle differentiation markers (MEF2C and MYF6). However, ePSCs were unable to generate teratomas, even when whole expanded blastocysts or freshly isolated ICMs were injected into immunodeficient mice (B. Huang, unpublished results). While this may be due to the relatively small number of cells injected, it is consistent with earlier reports [25,82]. The limited differentiation potential of ePSCs is of concern, as it potentially limits their use for applications that require authentic pluripotency, for example, generation of germline chimeras or fertile ePSC-derived animals through IVP-based assisted reproductive technologies.
In summary, 2i/LIF alone was insufficient to achieve an ESC-like pluripotent ground state in cattle. The existence of a naïve pluripotent state as a conserved feature of mammalian embryogenesis therefore remains hypothetical. However, chemically defined 2i conditions provide a future platform to systematically screen species–specific signaling molecules that promote long-term proliferation and naïve pluripotency without genetic intervention in cattle.
Footnotes
Acknowledgments
The authors would like to thank P. Turner for EB markers and Dr. C. Smith for primers and epiblast tissue. We also thank Dr. D. Wells for helpful comments on the article. This work was funded by Ministry of Science and Innovation C10X1002 and AgResearch. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
B.H., P.K.K., and B.O. are employees of AgResearch. V.V. was affiliated with AgResearch at the time of the study. This does not alter the author's adherence to all the journal's policies on sharing data and materials. There are no patents, products in development, or marketed products to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.