
Abstract
Embryonic stem (ES) cells are a powerful model for the development of cells responsible for the cellular immune response. Therefore, we analyzed the defense and phagocytic capacity of embryoid bodies (EBs) derived from ES cells using in the vitro inflammatory conditions caused by Escherichia coli. Further, we used this phagocytic activity to purify activated immune cells. Our data show that spontaneously differentiated 18-day-old EBs of the cell line CGR8 contained immune cells, which were positive for CD45, CD68, CD11b, F4/80, and CD19. Exposure of these EBs to E. coli with defined infection doses of bacterial colony-forming units (CFUs) led to a significant time-dependent reduction of CFUs, indicating the immune responses exerted by EBs. This was paralleled by an upregulation of inflammatory cytokines, that is, IL-1β and TNF-α. Western blot analysis of infected EBs indicated an upregulation of CD14 and cytochrome b-245 heavy chain (NOX2). Silencing of NOX2 significantly reduced the antibacterial capacity of EBs, which was partially explained by reduction of F4/80-positive cells. To identify, isolate, and further cultivate phagocytic active cells from differentiated EBs, a cocultivation assay of differentiated ES cells with green fluorescent protein (GFP)-labeled E. coli was established. Colocalization of GFP-labeled E. coli with cells positive for CD45, CD68, and F4/80 revealed time-dependent phagocytotic uptake, which was underlined by colocalization with the LysoTracker-Red® dye as well as preincubation with cytochalasin D. In conclusion, a primitive immune response with efficient phagocytosis was responsible for the antibacterial capacity of differentiated EBs.
Introduction
E
The innate immunity is the first functional immune response of a growing organism if exposed to pathogens or pathogen-associated molecular patterns (PAMPs) [7]. This innate immunity develops during differentiation processes, since it was previously shown that undifferentiated ES cells display a significant lack of defending pathways, for example, the transcription profile of pattern recognition receptors (PRRS), including Toll-like receptors (TLRs) [7]. The best-investigated PAMP is lipopolysaccharide (LPS), which is derived from gram-negative bacteria, but also, further PAMPs such as flagellin, bacterial DNA, or virus dsRNA are known [8]. LPS, for example, activates the LPS receptor complex that consists of CD14 antigen, TLR4, and lymphocyte antigen 96 (LY96) [9]. TLR4 recruits adapter proteins, including MyD88, MAL, TRIF, and TRAM, to initiate signaling cascades leading to activation of NF-κB and the induction of further inflammatory genes [10]. Until now, there are 10 different TLRs identified in human cells [10]. Previously, it was shown that the CD14 antigen was identified in differentiated ES cell-derived embryoid bodies (EBs) at a certain stage of differentiation [11,12].
Host defense is a key function of cytochrome b-245 heavy chain (NOX2), a component of the NADPH oxidase of phagocytic cells, which is evidenced by severe infections observed in patients with chronic granulomatous disease (CGD) [13]. However, while a direct reactive oxygen species (ROS)-dependent killing was initially thought to completely explain the host defense function of NOX enzymes, it seems currently more obvious that the interplay of several mechanisms is necessary to achieve a successful oxygen-dependent killing of bacteria [13].
Recently, a connection between recognition of bacterial antigens and the induction of a bacterial killing process via activation of NOX2 was described to be mediated by the isoforms of the self-ligand and cell surface receptor (SLAM) [14]. These proteins were responsible for inducing the accumulation of phosphatidylinositol-3-phosphate, a regulator of both NOX2 function and phagosomal or endosomal fusion [14]. Nevertheless, also phagocytes possess a number of oxygen-independent killing mechanisms, for example, the release of microbicidal proteins from granules [15]. Indeed, studies with neutrophils from patients with CGD demonstrate that NOX2 is not required for the killing of many types of bacteria [16]. However, in the recent years, there are more hints that indicate an additional role of NOX2 within correct differentiation of CD11b+ cells in a respective mouse model [17].
In general, the increasing number of multiresistant bacteria strains and the rising number of infections during hospitalization require new strategies and test methods to study the inflammatory processes in vitro to face this severe problem in the future [18]. Therefore, we analyzed the so-far unknown antibacterial capacity of ES cell-derived EBs. The potential use of competent immune cells for therapy is often limited by a loss of function during antibody-based cell isolation techniques such as magnetic cell separation (MACS). In the present study, a novel cell-sorting strategy is introduced that is based on flow cytometry (FCM) of phagocytic cells containing green fluorescent protein (GFP)-positive bacteria in the lysosomal compartment. Moreover, our data illustrate differentiated EBs derived from pluripotent ES cells as a powerful tool to investigate the inflammatory processes.
Materials and Methods
ES cell cultures and EB formation
The pluripotent murine ES cell line CGR8 was cultured in a Glasgow minimal essential medium (Sigma-Aldrich) supplemented with 16% heat-inactivated (56°C, 30 min) fetal bovine serum (FBS; Sigma), 2 mM
Culture and growth of bacteria
Respective Escherichia coli strains (strain K-12 MG1655) were cultured either in a liquid culture or on agar plates made from conventional LB agar (Life Technologies). GFP or red fluorescence protein (RFP) cassettes with an ampicillin resistance were introduced by electroporation. Positive colonies were subsequently selected by eye and microscopic inspection using ultraviolet (UV) light. The E. coli strain with an extended spectrum of β-lactamase resistance (ESBL) was routinely cultured growing on MacConkey agar and further characterized by the inhibition zone method. In detail, the used E. coli strain was resistant against ampicillin, ampicillin/sulbactam, piperacillin/tazobactam, cefuroxim, cefpodixim, cefotaxim, ceftazidim, gentamicin, ciprofloxacin, moxifloxacin, and tetracycline. In general, for the most experiments, an initial multiplicity of infection of two was used (this was mentioned as initial low infection dose). For a high initial infection dose, the around 10-fold number of colony-forming unit (CFU) was added to the same cell number per mL of differentiated CGR8 EBs.
Immunocytochemistry for confocal laser-scanning microscopy and FCM
Respective cell samples were fixed for 30 min up to 1 h at 4°C in 4% paraformaldehyde in phosphate-buffered saline (PBS; pH 7.4). For further confocal laser-scanning microscopy analysis, the samples were then washed three times with PBS containing 0.01% Triton X-100 (PBST; Sigma-Aldrich). For FCM analysis, PBS + 2% sterile-filtered heat-inactivated FBS was used. The tissues for immunocytochemistry (ICC) against unspecific binding were blocked for 60 min with 10% fat-free milk powder (Carl Roth) dissolved in 0.01% PBST. As primary antibodies, rat anti-mouse CD45 (ICC: 1:100/FCM: 1:250; Millipore), rat anti-mouse CD68 primary bound to Alexa 647 (ICC: 1:100/FCC: 1:300; AbD Serotec), rat anti-mouse LY-6b.2 (neutrophil antigen/ICC: 1:100/FCM: 1:50; AbD Serotec), rat anti-mouse F4/80 antigen (ICC: 1:100/FCM: 1:500; AbD Serotec), rat anti-mouse CD11b (ICC: 1:100/FCM: 1:200; Becton Dickinson Pharmingen), rabbit anti-mouse CD19 (ICC: 1:50/FCM: 1:200; Cell Signaling Technology), rat anti-mouse CD14 (ICC: 1:100/FCM: 1:200; Becton Dickinson), and rabbit anti-mouse NOX2/gp91phox (ICC: 1:100; Abcam) were used. Tissues were subsequently incubated overnight at 4°C with the primary antibodies and thereafter washed with 0.01% PBST. Single cells for FCM were incubated for 20 min with the respective primary antibodies and thereafter washed with PBS supplemented with 2% FBS. Samples were reincubated with either a Cy3- or Cy5-conjugated secondary antibody (Millipore) at a dilution of 1:200 up to 1:500 using an exposure time of 60 min for tissues and 20 min for single cells. FCM analysis was performed with negative, secondary antibody, or isotype control. For example, Alexa-Fluor-647-conjugated rat IgG in the respective concentration was used as an isotype control. Fluorescence of respective EB tissues was recorded using a confocal laser-scanning microscope (LSM 510; Zeiss) connected to an inverted microscope (Axiovert 135; Zeiss). FACSCalibur (Becton Dickinson) was used for FCM analysis. A total number of 10,000 cells were analyzed.
Magnetic cell separation (MACS)
EBs were dissociated at day 17 of differentiation in PBS containing collagenase type II (2 mg/mL; PAA) at 37°C for ∼30 min. The separation procedure was carried out using anti-mouse CD45 and CD11b MicroBeads (dilution 1:10; Miltenyi). The selection procedure was carried out according to the manufacturer's guidelines. Cells from the negatively selected fraction were plated and incubated at 37°C and 5% CO2 for 24 h before starting the respective coincubation experiments.
Cell sorting of GFP+ cells derived from differentiated EBs
CGR8 EBs were generated and cultivated as described above. For cell-sorting experiments based on a coincubation with living GFP+ E. coli K12 bacteria, EBs were taken from the spinner flask, played at day 4 of differentiation on a 60-×15-mm2 tissue culture dish (around 30 EBs per tissue culture dish; BD Falcon) cultivated until day 18 and subsequently incubated with around 1e+4.5 CFUs per mL of GFP+ living E. coli. For EB differentiation, the medium exchange was carried out daily with a conventional antibiotic-free medium as described above. To obtain single cells from the differentiated and plated EBs for FCM analysis or cell sorting, the respective EBs were digested with collagenase type II (2 mg/mL; PAA) dissolved in PBS for around 20 min at 37°C. For cell sorting of cells containing GFP+ E. coli, a Cytomation MoFLo (Beckman Coulter) cell sorter was used. Purity of the sorted cell fraction was assessed by a conventional FCM analysis. GFP+-selected cells were plated on gelatin-coated glass plates and incubated at 37°C and 5% CO2 for 24 h up to one week for further analysis. After the sorting process and for further culture, 100 U/mL penicillin and 100 μg/mL streptomycin (Biochrom) were added to the isolated cells.
Western blot analysis
EBs were washed in PBS and dissolved in an ice-cold standard lysis buffer containing protease inhibitors and 1% phosphatase inhibitor cocktail (Sigma-Aldrich). The protein concentration was assessed by the method of Bradford. The proteins were separated in acrylamide Tris–HCl gels and transferred to a nitrocellulose membrane (Amersham). Unspecific bindings were blocked with 10% dried fat-free milk in PBS containing 0.1% Tween 20. Incubations with primary antibodies were carried out at 4°C overnight using rat anti-mouse CD14 (1:500; Becton Dickinson) and rabbit anti-mouse NOX2/gp91phox (1:500; Abcam). After washing, the membranes were incubated with an appropriate HRP-conjugated secondary antibody (1:2,000; Santa Cruz) for 1 h at 4°C. Protein expression was detected by ECL reagent (Amersham), and protein bands were visualized using Digital Imaging System LAS 3000 (Fujifilm). Rabbit anti-mouse GAPDH (1:1,000; Abcam) was used as the endogenous control.
Silencing of NOX2 using shRNA technique
For stable shRNA silencing, pLKO.1-puro- (Sigma-Aldrich) derivative plasmids carrying an individual shRNA targeting murine NOX2 were separately introduced into CGR8 cells. Cells transduced with the pLKO.1 vector (containing non-hairpin insert) were used as a negative control. Lentiviral particles were generated using human embryonic kidney 293FT-based amphotropic Phoenix packaging cells (Phoenix-Ampho; Invitrogen) cultured in Dulbecco's Modified Eagle Medium, 10% FBS, and 1% sodium pyruvate plus penicillin/streptomycin. For a 10-cm dish, lentiviral vector plasmids (10 μg) were cotransfected with plasmids encoding the HIV-Rev (5 μg), HIV-MDL (10 μg), and the ecotropic envelope (2 μg) in the presence of polyethylenimine (70 μg; Sigma-Aldrich). The supernatants were harvested after 24 and 48 h and filtered through 0.22-mm filters. Then, the supernatants were added to CGR8 cells, which were placed in a well of a six-well plate, centrifuged at 400 g in a cell culture centrifuge for 1 h, and replaced with a fresh medium on the following day. A total of three infection rounds were carried out within 48 h. On the following day, the cells were passaged and selected with 2 μg/mL puromycin (Sigma-Aldrich) for 10 days. The transduction efficiency was assessed via a GFP control vector (Sigma-Aldrich).
Statistical analyses
Data were given as mean values±standard deviation or±standard error of mean, with n denoting the number of experiments. All experiments were repeated at least three times. GraphPad InStat-3 software (GraphPad Software, Inc.) was applied for one-way analysis of variance or t-test unpaired data as appropriate. A value of p<0.05 was considered significant (*). Data are partially expressed as the percentage of expression relative to the control values, which were set to 100%.
Results
ES cell-derived EBs differentiate into cells of the innate immune response
Within 18 days of cell culture, EBs derived from the murine ES cell line CGR8 differentiate spontaneously without adding special growth factors next to other cell types into different inflammatory cells of the innate immune response. These cells were characterized by different surface markers, that is, the pan leukocyte marker CD45, the neutrophil antigen Ly-6b.2, CD68, which is a marker for monocytes, macrophages, and precursor cells, as well as F4/80, which is present on mature macrophages and potentially coexpressed on dendritic cells (Fig. 1A–C). Interestingly, also CD11b and the B-lymphocyte antigen CD19 were identified in differentiated CGR8 EBs (Fig. 1A–C). Depending on the grade of differentiation, inflammatory cells formed a defined percentage of the total cell population within differentiated EBs. Using FCM analysis, CD45+ cells represented ∼36%, neutrophil antigen+ cells ∼24%, CD68+ cells ∼34%, F4/80+ cells ∼24%, CD11b+ cells ∼18%, and CD19+ cells ∼12% of the total cell population (Fig. 1B, C).
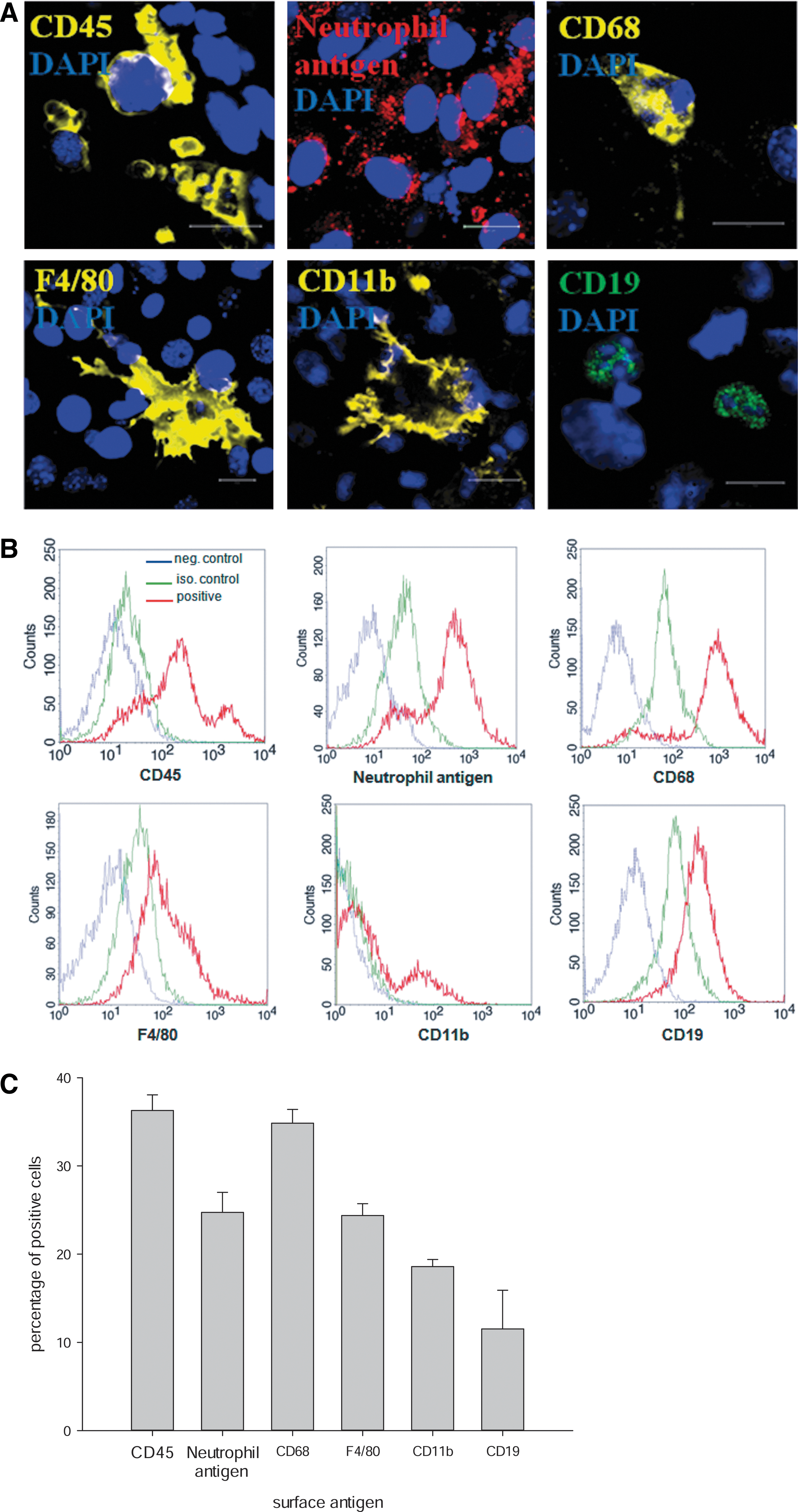
Differentiated 18-day-old embryoid bodies (EBs) derived from the embryonic stem cell line CGR8 spontaneously differentiate into different types of immune cells. These cells can be identified by different antigens such as CD45 (yellow), neutrophil antigen (red), CD68 (yellow), F4/80 (yellow), CD11b (yellow), or CD19 (green) using immunocytochemistry
Coincubation of E. coli, with differentiated CGR8 EBs, leads to a significant reduction of bacterial growth
To analyze the potential functionality of the observed immune cells differentiated from 18-day-old plated CGR8 EBs, coincubation with living E. coli in a defined initial infection dose of ∼1e+4.5 CFUs/mL was performed for 10 h (Fig. 2). EBs of day 18 were chosen, because it was previously described in the literature that leukocytes and macrophages were definitely detectable after day 12 of differentiation within the murine ES cell lines [11,19]. Interestingly, a significant reduction of E. coli CFUs was found in the presence of differentiated 18-day-old CGR8 EBs in comparison to E. coli culture without coincubation (Fig. 2 and Supplementary Fig. S1; Supplementary Data are available online at

Antibacterial capacity of 18-day-old differentiated EBs derived from the murine embryonic stem cell line CGR8 was analyzed by counting the number of colony-forming units (CFUs) in the respective group after 10 h of coincubation with living E. coli
Differentiated EBs derived from murine ES cells efficiently phagocyte E. coli
To further explain the observed reduction of E. coli outgrowth in the presence of differentiated 18-day-old CGR8 EBs, GFP+ E. coli (Supplementary Fig. S3) were coincubated with the corresponding differentiated ES cells. In coherence with the previously described reduction of CFUs in the presence of differentiated CGR8 EBs, a significant increase of the GFP+ CGR8 cells within 8 h during coincubation with GFP+ E. coli was observed, indicating phagocytic uptake of bacteria by differentiated ES cells (Fig. 3A, B). The percentage of GFP+ cells increased from ∼2% after 2 h by coincubation with a defined number of CFUs up to ∼25% after 8 h, which was ascertained by FCM (Fig. 3A, B). In the next step, it was shown that the uptake of GFP+ E. coli was clearly dependent on the differentiation state of the CGR8 cells. Our data indicated that there was no significant GFP+ E. coli uptake (<0.05%) if undifferentiated CGR8 ES cells, which were kept in the presence of LIF (day 0 of differentiation), were coincubated for 8 h with the respective bacteria (Fig. 3C). To further exclude a simple invasion of E. coli into the cells of differentiated EBs from the cell line CGR8, we used inactivated (by defined UV-light exposure) RFP+ E. coli and coincubated with differentiated 18-day-old CGR8 EBs. The results also indicated a significant uptake of these inactivated E. coli (Fig. 3D and Supplementary Fig. S4). To further exclude the possibility of a cell line-specific variation, some key findings were additionally verified using differentiated 18-day-old EBs derived from the murine cell line CCE (Supplementary Fig. S5). Our results suggest that these differentiated ES cells also showed a defined uptake of living GFP+ E. coli. Nevertheless, the percentage of GFP uptake was less in comparison to cells from the cell line CGR8 (Supplementary Fig. S5).
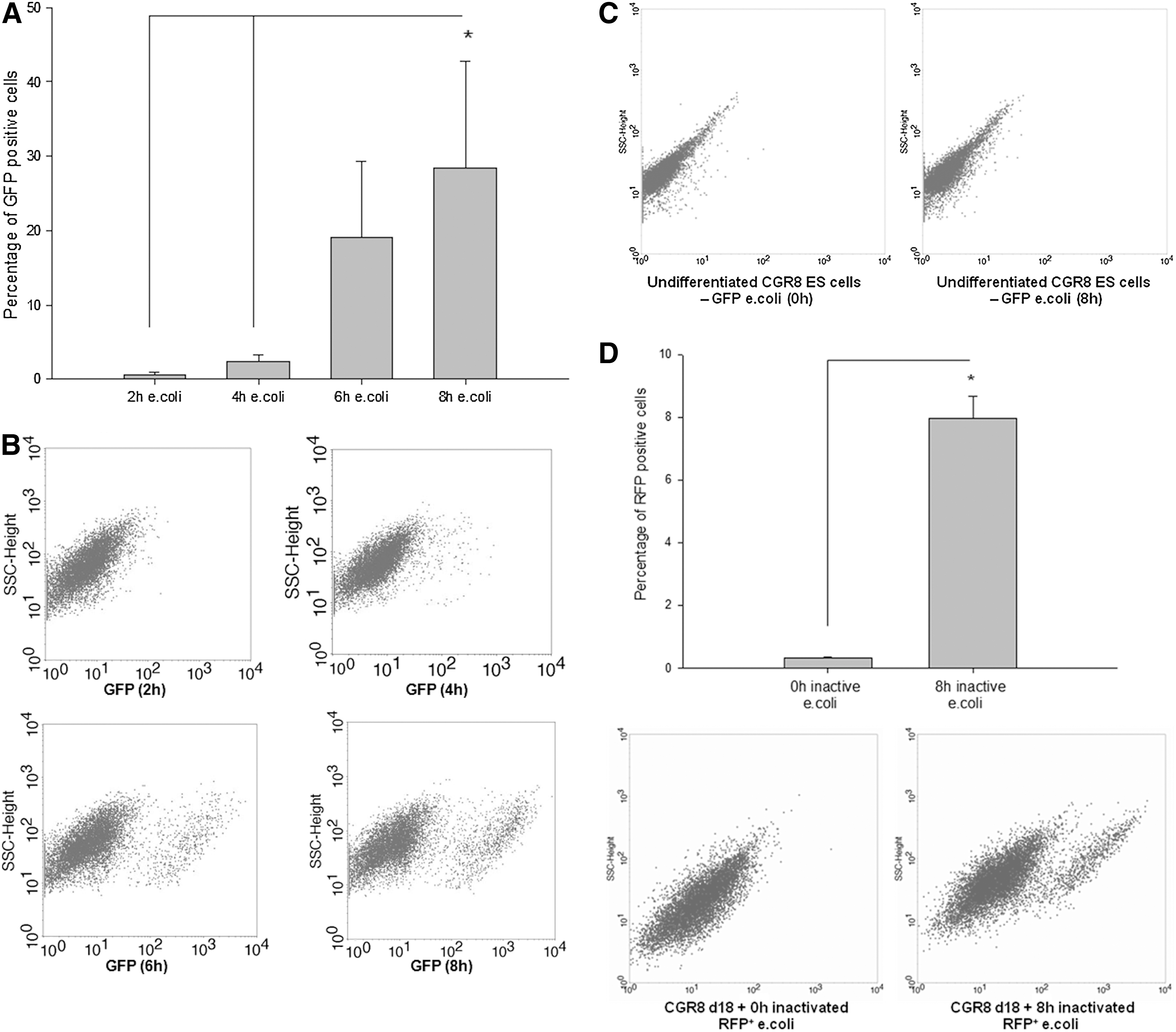
Phagocytosis assay of 18-day-old differentiated EBs derived from the murine embryonic stem (ES) cell line CGR8 analyzing the percentage of green fluorescent protein (GFP+) cells within the population of EBs at 2, 4, 6, and 8 h after coincubation of the respective cells with GFP+ E. coli
GFP+ E. coli are mainly endocytosed by leukocytes and macrophages derived from differentiated CGR8 EBs
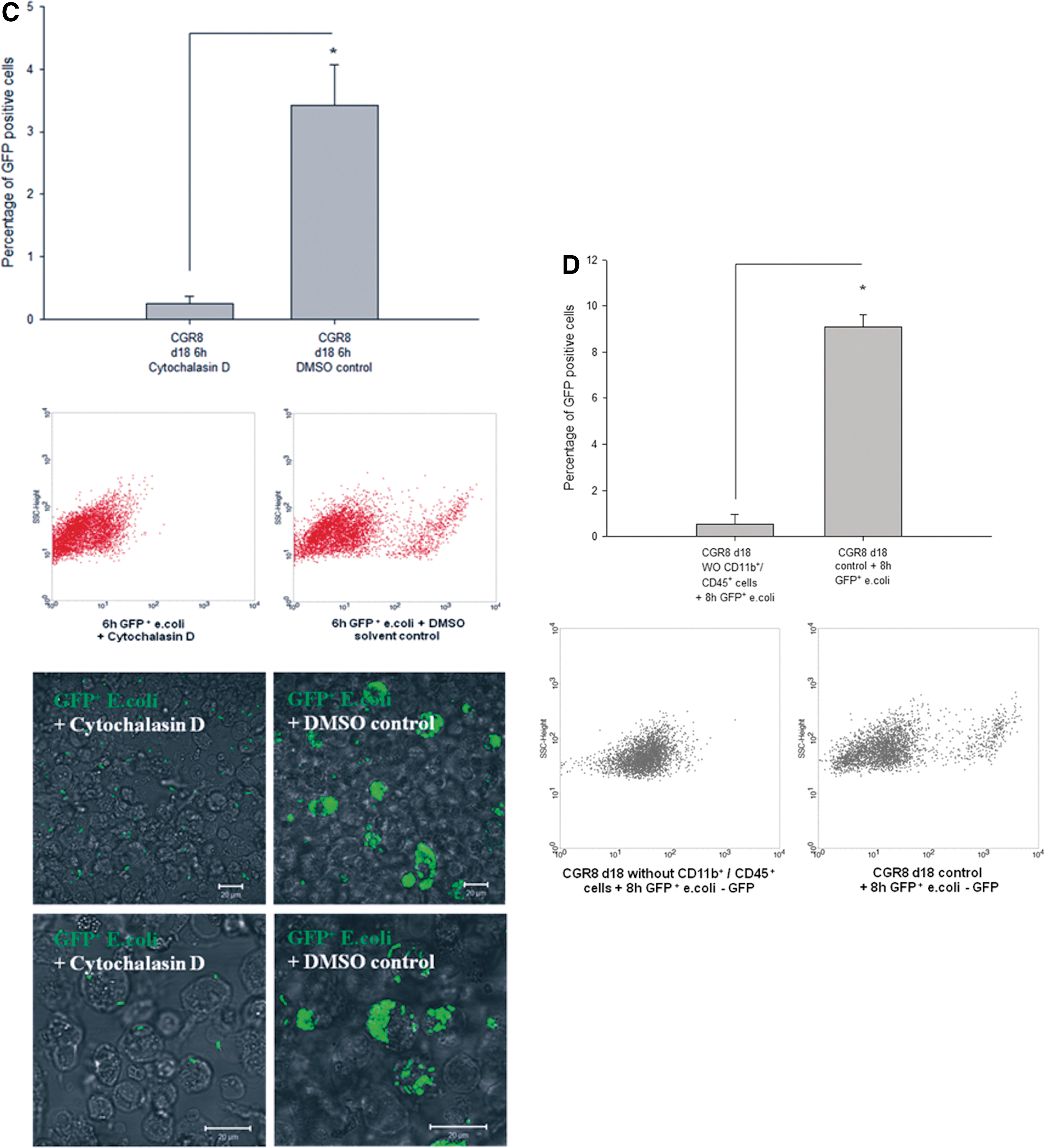
To further elucidate which type of cells within the differentiated CGR8 EBs was mainly responsible for the initially observed reduction of bacterial CFUs in the presence of EBs, we analyzed GFP+ cells containing E. coli by ICC and FCM analysis and costained them with the markers of cells of the innate immune response. Using FCM, we detected that after 6 h of coculture with 18-day-old EBs, cells containing GFP+ E. coli or even bacterial fragments were present in CD45+ as well as CD68+ cells (Fig. 4A, B). In comparison to an appropriate control, ∼85% of all GFP+ cells carried in parallel the CD45 surface antigen after 6 h of coincubation with the living E. coli (Fig. 4A). Further, ∼58% of GFP+ cells carried in parallel the CD68 surface antigen in the same context (Fig. 4B). To visualize colocalization, ICC in combination with confocal microscopy was carried out. After coincubation with GFP+ E. coli, a number of cells carried GFP+ E. coli on the cell surface as well as within the intracellular compartment. These cells with absorbed GFP+ or inactivated RFP+ bacteria carried the neutrophil antigen (Fig. 5A), CD45 (Fig. 6A), CD68, CD11b, and F4/80 (Fig. 5B, C). Using an additional LysoTracker-Red® dye, we observed that some of the intracellular localized GFP bacterial particles were distributed into typical acid granular lysosomes (Fig. 6A, B). These findings were completed by preincubating differentiated CGR8 EBs with cytochalasin D (5 μM for 30 min), leading to a significantly reduced uptake of GFP+ E. coli (Fig. 6C). These results were additionally validated by counting the number of CFUs, which did not show a significant difference between the cytochalasin D-treated EBs and the control population (data not shown). Interestingly, it was further possible to abolish the observed uptake of GFP+ E. coli by removing the CD11b+ and CD45+ cells together from the population of differentiated CGR8 EBs using MACS before exposing the negative cell fraction (without CD11b/CD45 cells) to GFP+ E. coli (Fig. 6D).
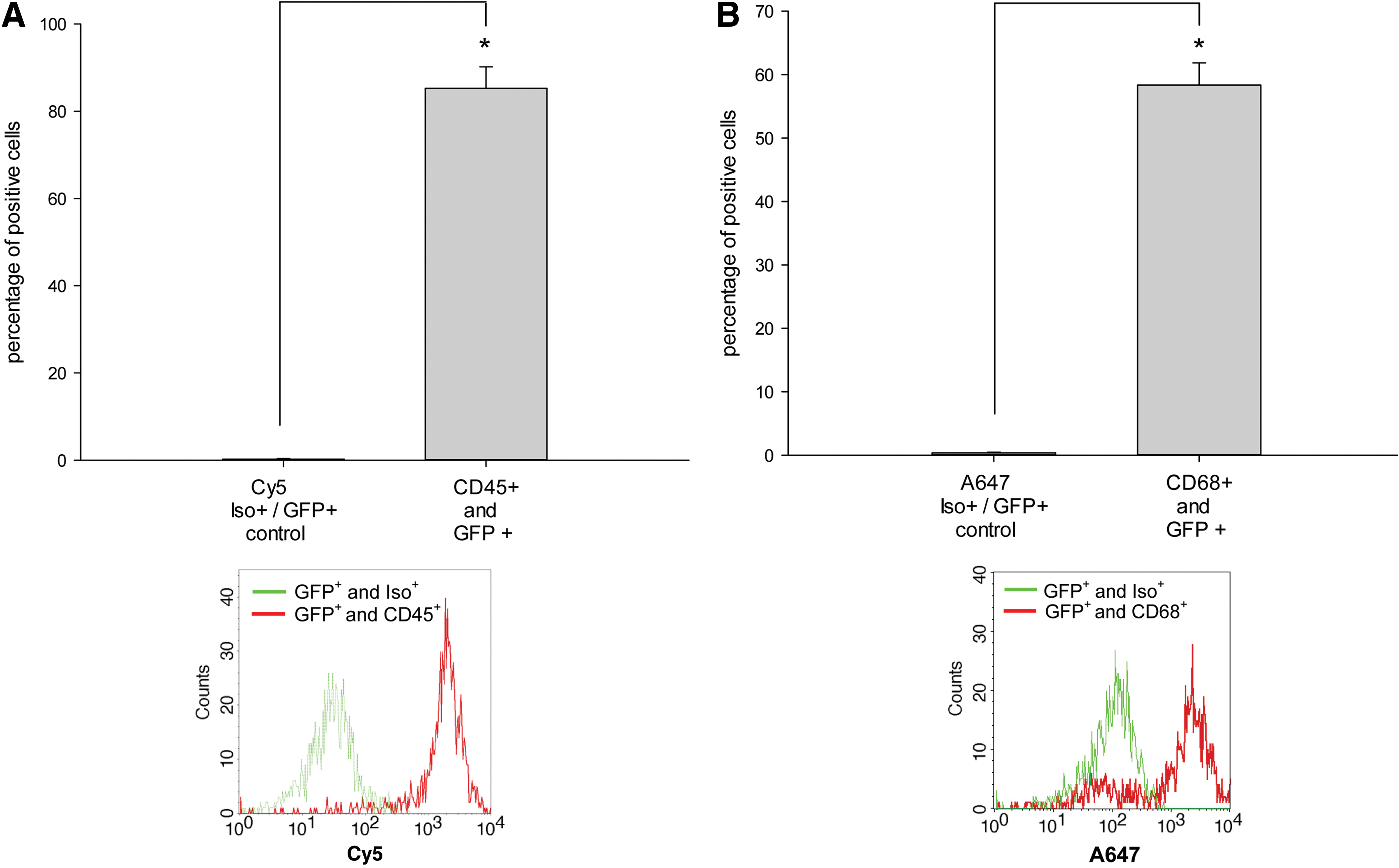
Fingerprinting of phagocytic active cells that endocytosed GFP+ E. coli. To characterize phagocytic active cells, the percentage of GFP+/CD45+

Representative immunocytochemistry (ICC) images of two different magnifications of neutrophil antigen+ cells (red) and GFP+ particles (green) after coincubation of living GFP+ E. coli with 18-day-old CGR8 EBs for a period of 8 h
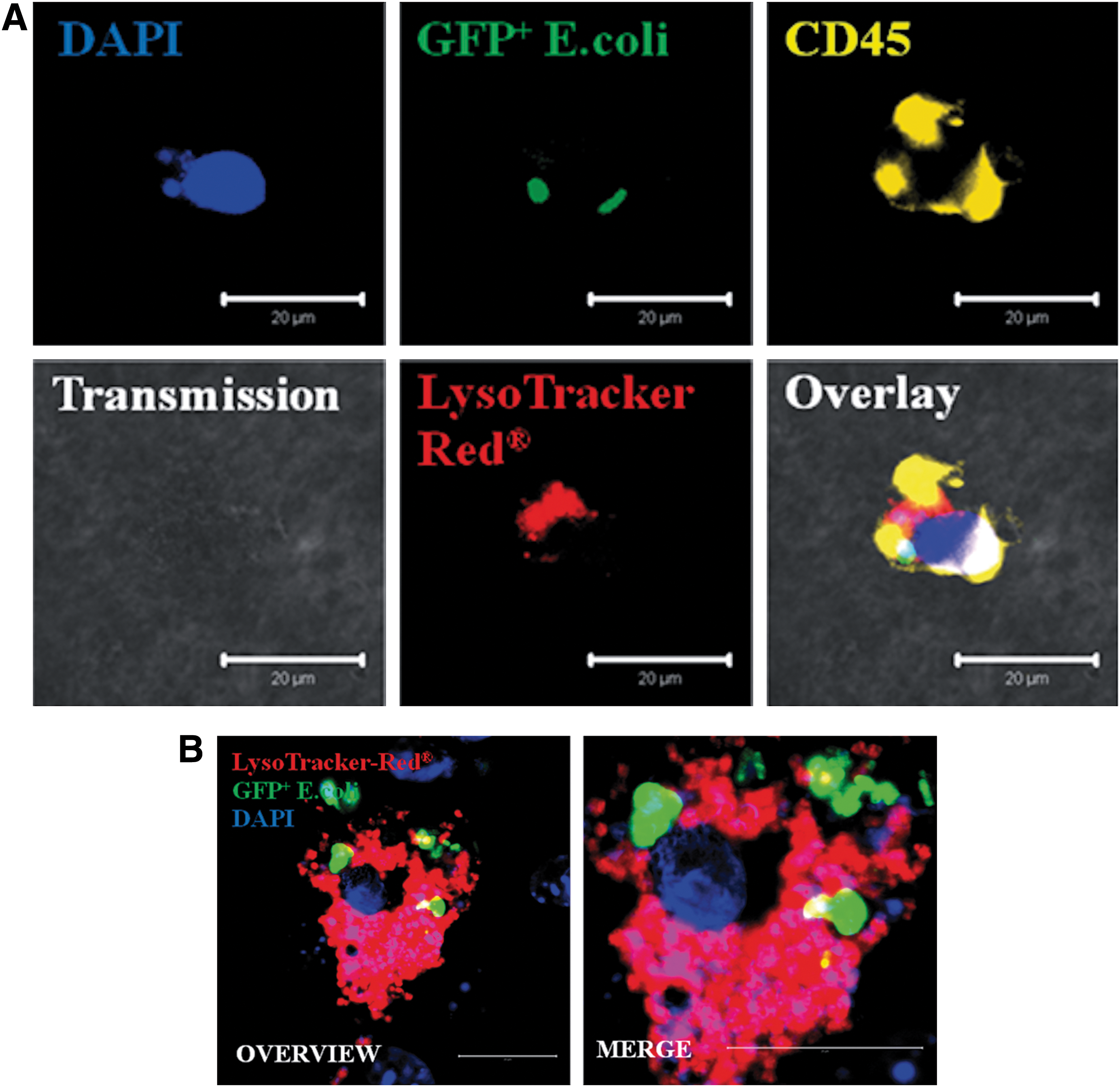
Representative ICC of a CD45+ cell (yellow)
Coincubation of differentiated 18-day-old CGR8 EBs with E. coli led to an upregulation of the monocyte differentiation antigen CD14 as well as NOX2
The monocyte differentiation antigen CD14, which is known to play an important role during the initiate immune response to bacterial LPS, was only slightly expressed in differentiated CGR8 EBs. Interestingly, a significant upregulation of the CD14 protein was detected in 18-day-old EBs within 10 h of coincubation with E. coli using western blot analysis (Fig. 7A, B). This could indicate that coincubation with E. coli boosts differentiation of monocytes to functional macrophages, which efficiently phagocyte bacteria. Alternatively, the density of CD14 antigen per cell surface may increase. A further characteristic for an efficient primary immune response is an increased generation of ROS-generating enzymes, for example, NOX2. To verify this point, western blot analysis of NOX2 was performed with samples of 18-day-old differentiated CGR8 EBs coincubated with E. coli. Notably, NOX2 protein was also upregulated within 6 h of coincubation (Fig. 8A). NOX2-positive cells derived from differentiated CGR8 were visualized by ICC (Fig. 8B).
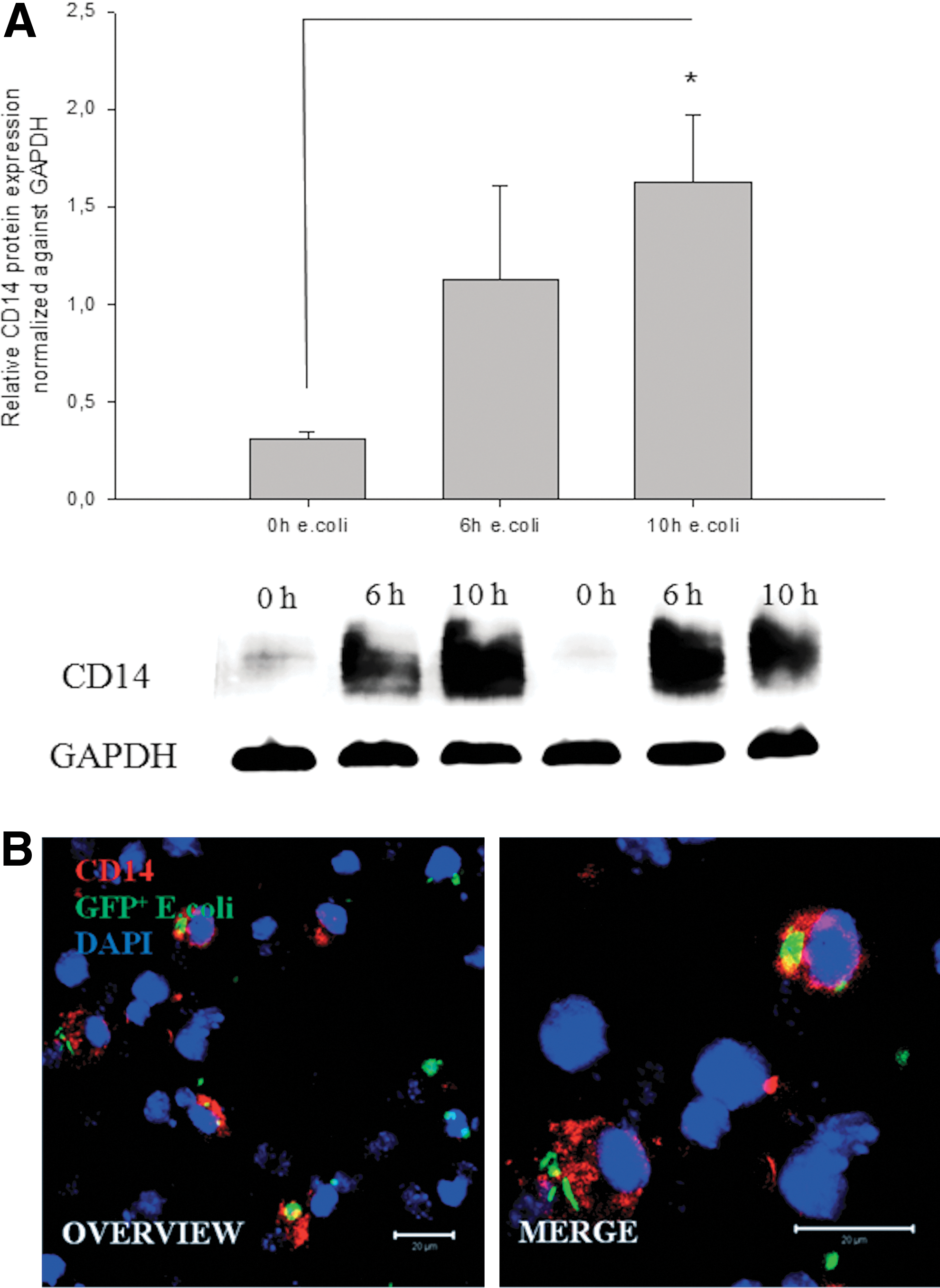
Representative western blot analysis and quantification of the CD14 antigen
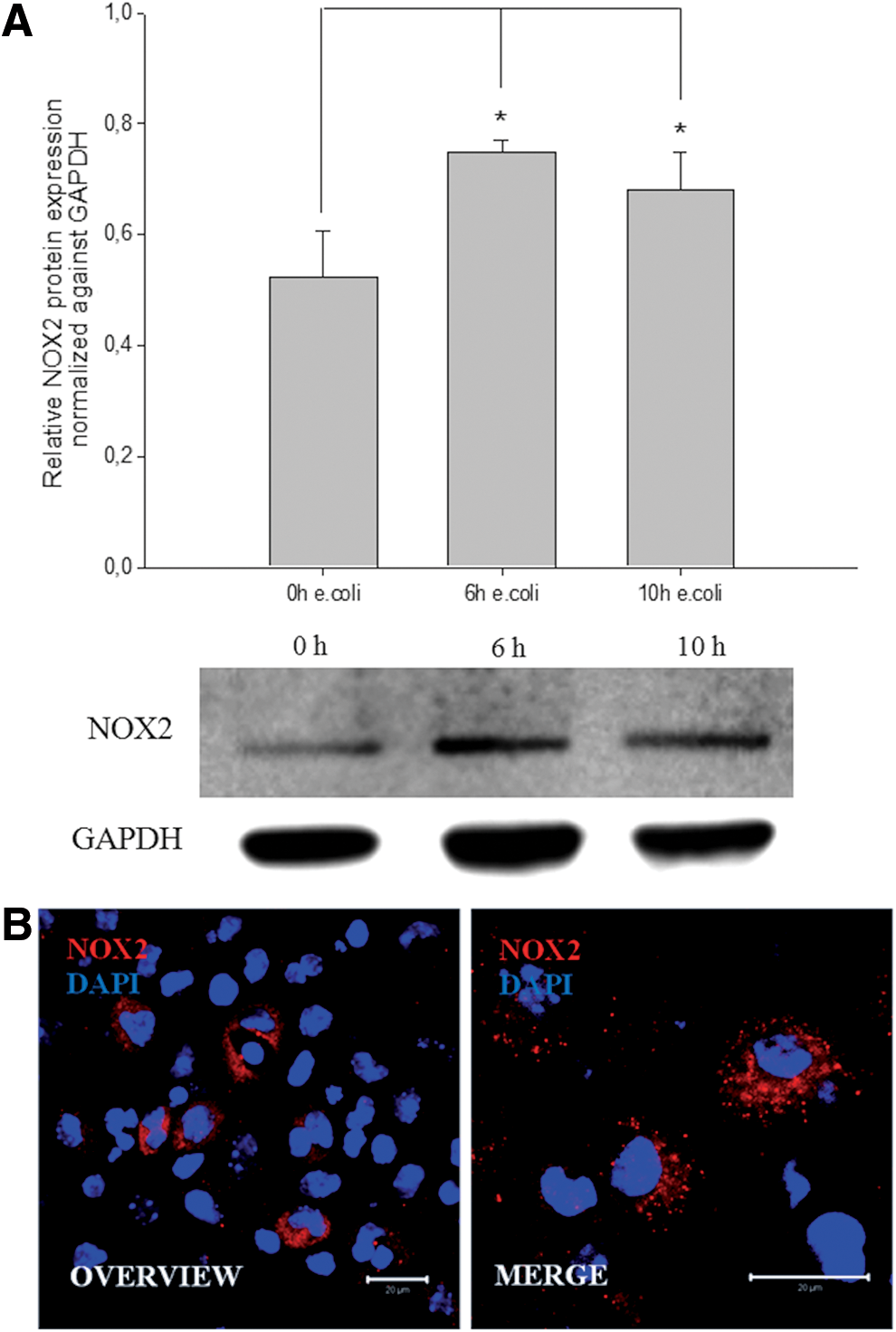
Representative western blot analysis and quantification of NOX2
Differentiated CGR8 EBs in coincubation with E. coli initiate a primitive immune response
To verify a potential cytokine production within the differentiated EBs during coincubation with E. coli, typical inflammatory cytokines, that is, TNF-α, IL-1β, and IFN-γ, were analyzed using the enzyme linked immunosorbent assay technique. Depending on the initial bacterial dose, a significant increase of TNF-α was observed in the supernatant of 18-day-old CGR8 EBs after 6 and 10 h, respectively, of coincubation with E. coli (Fig. 9A). IL-1β showed a significant upregulation after exposure for 10 h toward a high bacterial load (Fig. 9B). Further, the IFN-γ concentration was significantly elevated after 6-h exposure toward a low dose of E. coli (Fig. 9C).
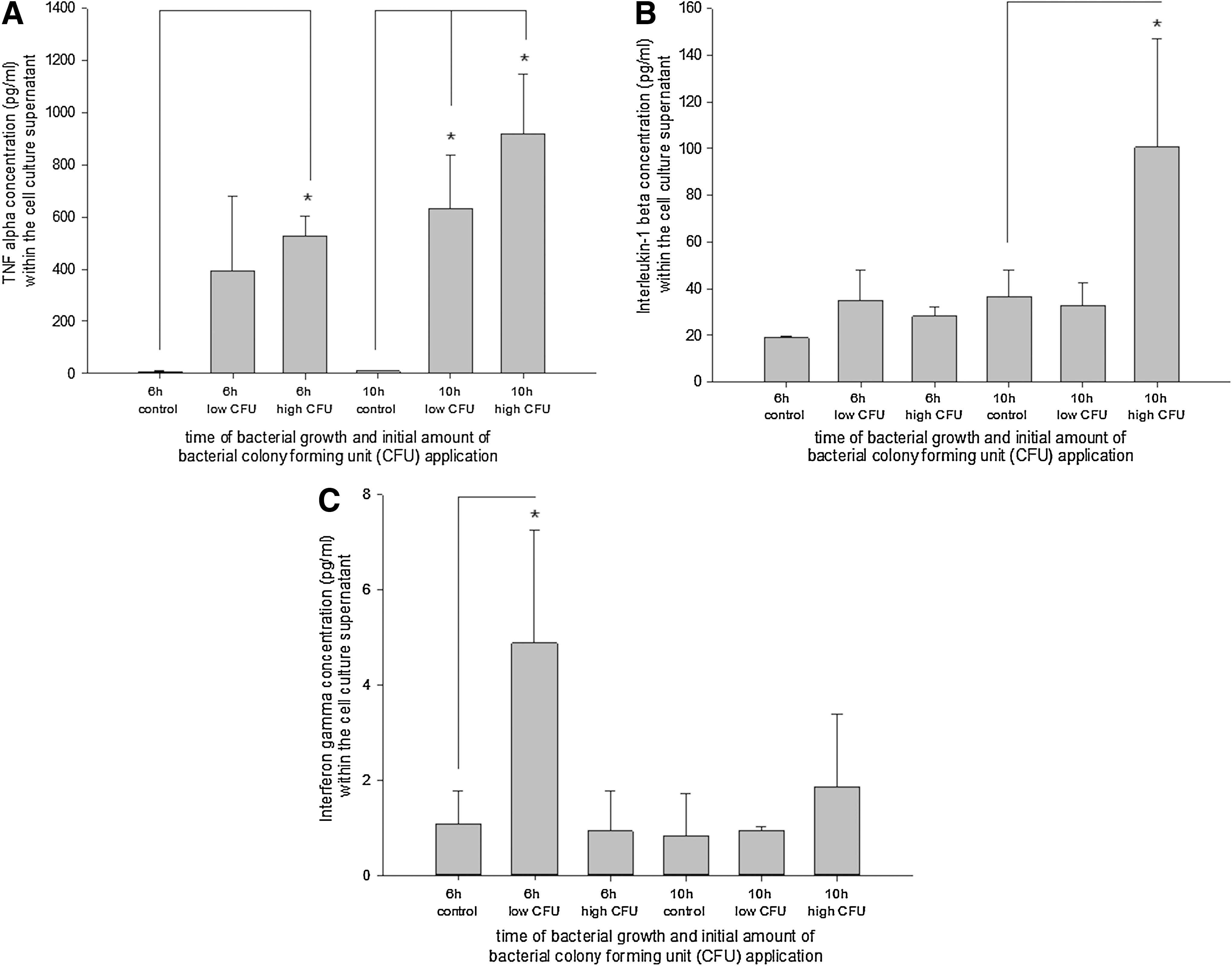
Secretion profile of TNF-α
Sorting of phagocytic cells derived from differentiated CGR8 EBs after coincubation with living GFP+ E. coli
Common cell-sorting strategies for immune cells are mostly based on antigen–antibody binding. This method blocks at least temporarily some cell functions and signaling events of the respective cells. To purify phagocytic active cells from the population of differentiated 18-day-old CGR8 EBs after coincubation with GFP+ E. coli, a cell sorter-based strategy for selection and purification of the GFP+ cell population was established (Fig. 10A). After cell sorting, the cells were cultured again and characterized for the expression of immune cell markers (Fig. 10B). The applied cell-sorting process yielded purities >95%, depending on the individual sorting conditions. Especially, macrophages and dendritic-like cells attached to gelatin-coated surfaces after sorting and survived several days in the presence of antibiotics. Isolated white blood cells were analyzed using a cytospin technique. To demonstrate that these sorted and further cultured cells still possess their phagocytic activity, we performed a subsequent coincubation experiment with RFP+ E. coli. Using ICC, it was again possible to demonstrate a still-present phagocytic activity. To visualize this observation, we colocalized the F4/80 antigen with living RFP+ E. coli (Fig. 10C). Additionally, it was possible to colocalize iNOS as well as the TLR4 by ICC on cells that previously had phagocytosed RFP E. coli (Supplementary Fig. S6). To further generalize the technical feasibility of cell sorting based on the phagocytic uptake of GFP+ E. coli, bone marrow cells were obtained from mice that were previously stimulated with LPS for 10 days and subsequently coincubated for 6 h with GFP+ E. coli. After cell sorting, the phagocytic active cells could be clearly identified within the whole population of bone marrow-derived cells (Supplementary Fig. S7).
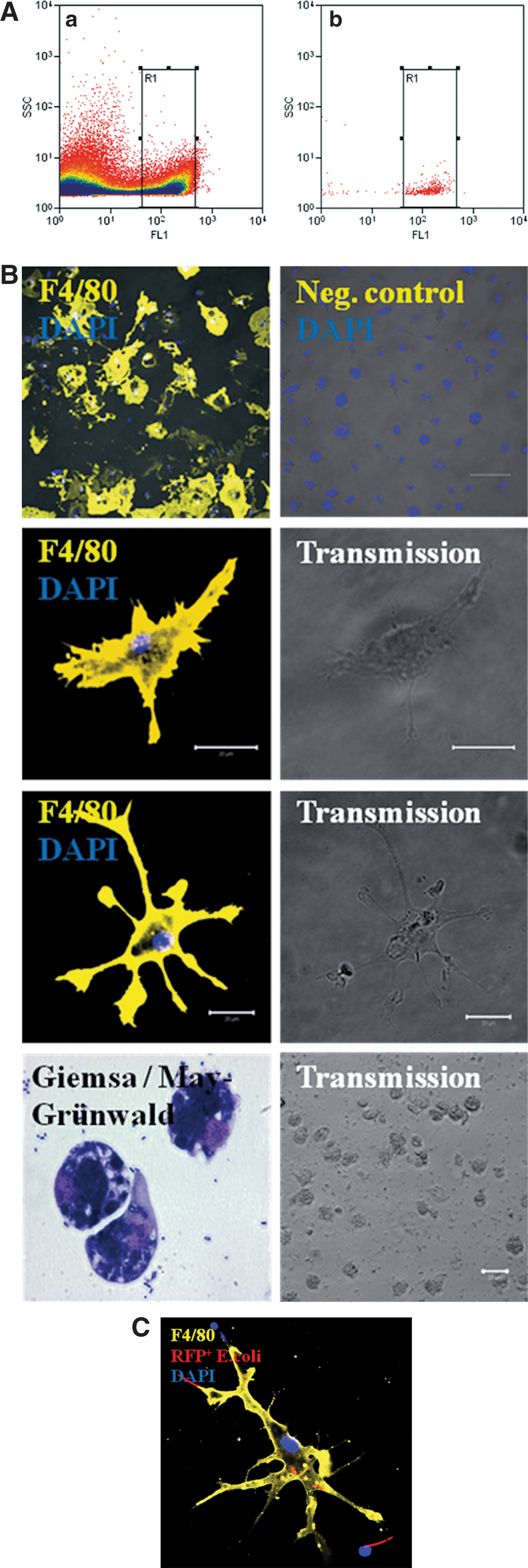
Representative scatter plot used for selective cell sorting of GFP+ immune cells
Silencing NOX2 using shRNA technique within ES cell-derived EBs decreases the antibacterial capacity upon confrontation with E. coli
To investigate the impact of NOX2 and a potential ROS production on the antibacterial capacity of EBs, NOX2 was silenced using the shRNA technique. Subsequently, the antibacterial capacity of cells was analyzed in confrontation with E. coli using low- and high-infection-dose CFUs (Supplementary Fig. S8). Silencing of NOX2 led to a significant reduction of the antibacterial capacity of EBs within 10 h of coincubation (Fig. 11). This may be due to absence of oxidative burst upon genetic inactivation of NOX2. Further, the differentiation of F4/80+ cells in NOX2-silenced EBs may be impaired. Indeed, FCM analysis of the shRNA-silenced EBs indicated a reduction of F4/80+ cells within the population of analyzing cells, suggesting a potential role of NOX2 during the differentiation of this specific type of immune cells within the EB system (Fig. 12A, B).

Antibacterial capacity of 18-day-old differentiated EBs derived from the murine ES cell line CGR8 in comparison to EBs that were silenced for NOX2 using the shRNA technique. The time-dependent number of CFUs at the time of 0, 6, and 10 h after starting the infection was analyzed. Respective EBs were incubated with a low and high infection dose of E. coli. Data are presented as mean±SEM. Asterisk indicates significance (p<0.05/n=4).
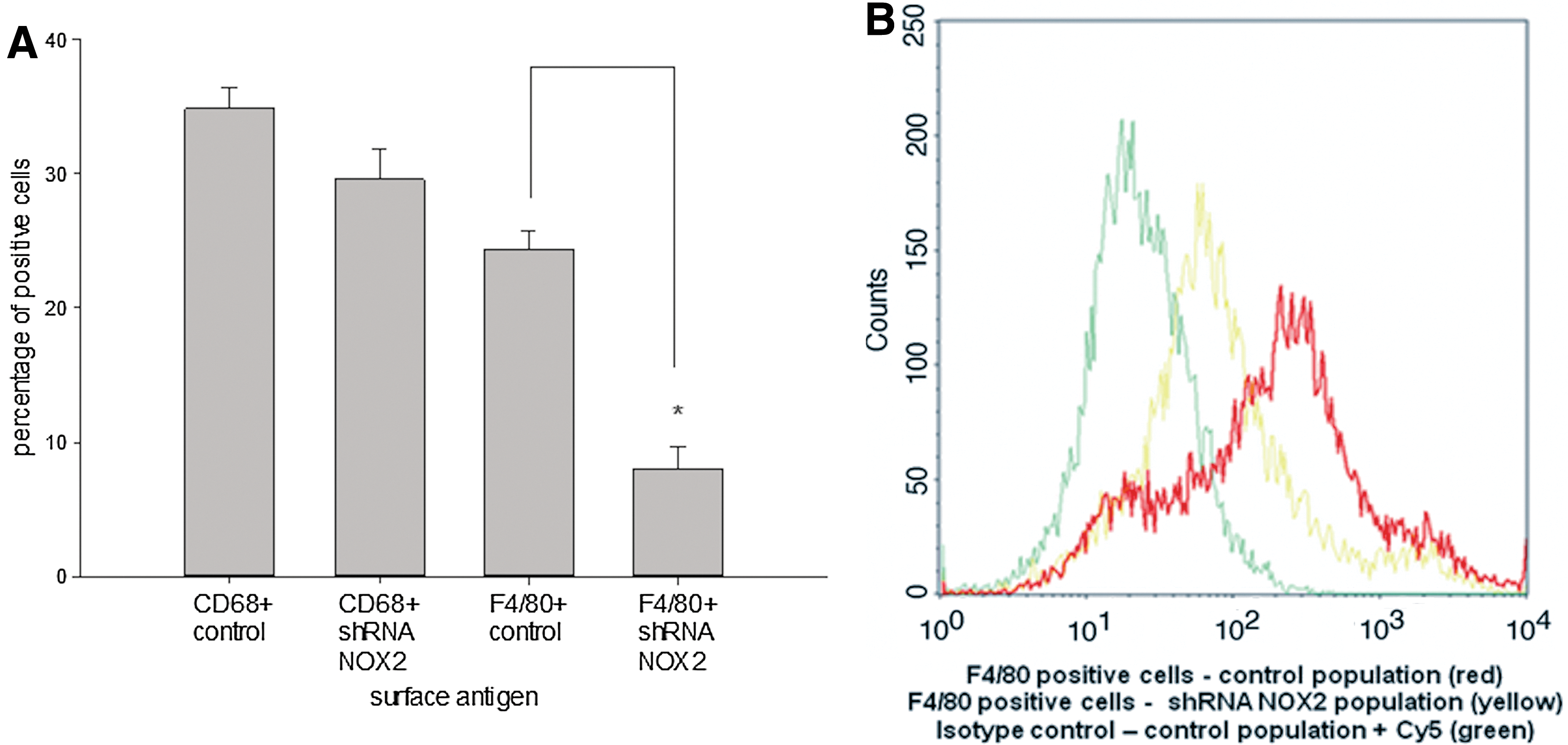
Quantitative FCM analysis
Discussion
Although the relation between differentiation of ES cells and the chronological appearance of immune cells has been previously characterized [2 –6,11], less is known about the antibacterial capacity and activation of these cells in the differentiated EB system. Our data show that a certain percentage of murine ES cells spontaneously differentiate into immune cells. These cells can be characterized as typical leukocytes, for example, neutrophils, macrophages, and cells of the B-cell lineage [1 –4]. ES cells and early immune cells show a defined profile of PRRS that differs from cells that have been previously exposed to distinct antigens and passes through an immune response [7]. To assess the potential functionality of ES cell-derived immune cells, differentiated 18-day-old EBs were directly exposed to living E. coli. This technique was used because previous approaches using pathogens or PAMPs such as LPS, flagellin, and others sometimes failed to fully and sufficiently activate a significant immune response within the population of differentiated ES cells [7]. Our findings indicate that by coincubation of differentiated 18-day-old EBs derived from murine ES cells with living E. coli, a primitive immune response was induced. The point in the time of the analysis was chosen because of its closed relation to the gestation time of mice, which is around 18–23 days. The observed immune response was first of all characterized by a significant reduction of bacterial growth documented by a less number of CFUs in the medium during coincubation with 18-day-old EBs. By using FCM analysis and additional ICC with GFP+ E. coli, it was possible to identify and quantify a significant number of phagocytic active cells within EBs. The technique of using GFP+ bacteria was recently described to study the distinct phagocytosis processes [14]. In consistence with these results, it was possible to identify GFP+ E. coli or their digested deposits within the lysosomes of immune cells. Further analysis identified the phagocytic active cells within the EB system mainly as leukocytes, which belong to the class of monocytes/macrophages as well as neutrophils. The kinetics of the observed phagocytic processes was mainly dependent on the coculture time and bacterial dose.
To further assess the functionality of the observed immune reaction, the supernatant of 18-day-old differentiated EBs after coincubation with E. coli was analyzed, and the secretion of typical inflammatory cytokines was determined and referenced to the serum level of cytokines from septic mice [1,20]. With this method, it was possible—depending on the initial applied bacterial dose—to detect the major cytokines TNF-α and Il-1β, which are elevated during sepsis or severe infection [21,22]. A further dynamic sign of the antibacterial capacity of differentiated EBs was a significant upregulation of the CD14 antigen, which mediates the recognition of LPS [23,24].
Another known key regulator of unspecific host defense and several bacterial killing processes is the activity of NOX2, which interestingly showed a significant upregulation during the coincubation of E. coli with EBs [14]. Moreover, we demonstrated an important role of NOX2 in the antibacterial effect of EBs, since shRNA silencing of NOX2 led to a reduced antibacterial capacity. This may be due to the absence of oxidative burst under these experimental conditions. Notably, in ES cells lacking NOX2, the differentiation of F4/80-positive cells was impaired.
As of yet, there is still an insufficiency to purify monocytes/macrophages from mice or even differentiated ES or induced pluripotent stem cells. Mainly, there are two options that are on the one hand based on a negative selection of monocytes/macrophages from peripheral blood of mice [25]; on the other hand, macrophages can be isolated from the peritoneal cavity of mice [26]. These strategies are using selection procedures based on antibody–antigen recognition with the disadvantage of impairment of cell function. In the present study, we introduced a method that allows to specifically isolate immune-competent cells based on their phagocytic capacity. This selection procedure appeared to be selective and yielded leukocytes, for example, macrophages, in high purity and vitality. Cells sorted by the use of endocytosed GFP+ E. coli may be exploited in antisepsis therapies for patients with a compromised immune response in the future. Recent publications suggest a beneficial effect on lethality of sepsis if murine CD34+ stem cells were injected intraperitoneally in mice, apparently through improved neutrophil and macrophage phagocytosis [27]. These observations and the data of the present study on the antibacterial capacity of differentiated ES cells and the growth reduction of multiresistant bacteria may therefore potentially open up new strategies in antisepsis therapies. Further, the presence of CD19+ cells in an environment of antigen-presenting and cytokine-secreting cells within the differentiated EB system potential rises the opportunity to achieve a transfer of immature B-cells to the antigen-presenting cells and finally antibody-producing plasma cells.
Footnotes
Acknowledgments
We thank PD Dr. J. Roedel (Institute for Microbiology, University Hospital Jena) for providing us his support performing the experiment regarding the ESBL E. coli. We further thank Dr. M. Foerster for providing us his support during the FCM and cell-sorting procedures.
This work was supported by the Federal Ministry of Education and Research (BMBF), Germany, FKZ: 01EO1002.
Author Disclosure Statement
The authors declare that there are no commercial associations to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.