
Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has been used extensively in cancer therapy. However, more than half of glioblastoma multiforme are insensitive to the apoptotic effect of TRAIL. Improvement in therapeutic modalities that enhances the efficacy of TRAIL in glioma is much sought after. In this study, we combined the tumor selectivity of TRAIL and tumor-homing properties of mesenchymal stem cells (MSC) with gap junction (GJ) inhibitory effect of carbenoxolone (CBX) to target orthotopic glioma. MSC were engineered to express TRAIL (MSC-TRAIL) by incorporating the secretable trimeric form of TRAIL into a Herpes Simplex Virus (HSV) type I amplicon vector. Our results showed that combined treatment of MSC-TRAIL and CBX enhanced glioma cell death, especially in three primary human glioma isolates, of which two of those are marginally sensitive to TRAIL. CBX enhanced TRAIL-induced apoptosis through upregulation of death receptor 5, blockade of GJ intercellular communication, and downregulation of connexin 43. Dual arm therapy using TRAIL and CBX prolonged the survival of treated mice by ∼27% when compared with the controls in an intracranial glioma model. The enhanced efficacy of TRAIL in combination with CBX coupled with the minimal cytotoxic nature of CBX suggested a favorable clinical usage of this treatment regimen.
Introduction
G
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), also known as Apo2 ligand, is a member of the tumor necrosis factor (TNF) family and a potent apoptotic inducer. TRAIL signals through death receptor (DR) 4 (TRAIL-R1) and DR5 (KILLER, TRAIL-R2) to mediate apoptosis. Three other members of the TRAIL receptor family, decoy receptor (DcR) 1 (TRID), DcR2 (TRUNDD), and osteoprotegerin, act as negative regulators of TRAIL signaling [2]. Engagement of DR4 and DR5 lead to downstream activation of caspase 8 and subsequently caspase 3. TRAIL has been shown to induce cell death specifically in tumor cells while sparing normal cells in nonhuman primates [3,4]. The tumor selectivity of TRAIL thus raised great interest in the use of TRAIL for cancer treatment. However, clinical application of TRAIL was hampered when hepatotoxicity was observed [5]. This was found to be caused by the zinc content within the poly-Histidine tagged recombinant TRAIL, which was later resolved by the use of untagged TRAIL [6]. However, the improved version of recombinant TRAIL did not generate complete clinical responses. This may be attributed to TRAIL-insensitive tumor cells resulting from the lack of TRAIL death receptors expression [7] and aberrant expression of pro-apoptotic or pro-survival molecules [8,9]. Accordingly, improved treatment strategies, such as the use of TRAIL coupled with other conventional or novel drugs for instance temozolomide (TMZ) [10], paclitaxel, carboplatin and bevacizumab [11], phosphoinositide-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor PI-103 [12], Bortezomib [13], or histone deacetylase inhibitor Entinostat [14], have been explored to enhance the therapeutic efficacy mediated by TRAIL. In gliomas, a study has shown that the use of a gap junction (GJ) inhibitor, carbenoxolone (CBX), was capable of enhancing TRAIL, FasL, and etoposide-induced apoptosis through the disruption of cell–cell interaction [15].
GJ are membrane specializations containing clusters of intercellular channels that consist of six connexin (Cx) monomers on adjacent cells. Transfer of signaling molecules between these channels, also known as GJ intercellular communication (GJIC), enables cells to share second messengers, ions, metabolites, and solutes of less than ∼1000 kDa that assist in cell survival [16]. Inhibition of GJIC will thus block the transfer of these metabolites and signaling proteins, hence leading to breakdown in cell–cell communication [17], tumor cell invasion [18], and apoptosis [15]. CBX, a derivative of 18-glycyrrhetinic acid, is a mineralocorticoid agonist that inhibits the action of 11β-hydroxysteroid dehydrogenase. It has been clinically approved for the treatment of esophageal ulceration [19]. In addition to its anti-inflammatory role, CBX is also widely used as a GJ inhibitor [20]. Thus, this led us to hypothesize that combination treatment of mesenchymal stem cell (MSC)-delivered TRAIL with CBX would augment TRAIL-induced apoptosis in human gliomas. Human bone marrow-derived MSC are multipotent stem cells that are able to differentiate into adipogenic, osteogenic, and chondrogenic lineages [21]. They are easily isolated and expanded; they are able to self-renew, proliferate, and migrate to regions of neoplastic lesions and inflammation [22,23]. Because of their tumor-tropism, MSC have generated a great deal of excitement as a potential source of cells for cell-based therapeutic strategies. Genetic modification of MSC using viral vectors that carry therapeutic gene(s) has been shown to potentially inhibit tumor growth [10,12,24].
In the present study, we investigated whether CBX would enhance MSC-TRAIL-induced apoptosis in orthotopic GBM. To this end, a herpes simplex virus-1 (HSV-1) amplicon viral vector-based TRAIL construct was synthesized. The TRAIL construct, pHGCX-FFZT, contains the extracellular domain of human FMS-like tyrosine kinase 3 ligand (hFlt3) gene, furin recognition sequence, isoleucine zipper domain, and the recombinant TRAIL. The extracellular domain of the hFlt3 (hFlex) allows the secretion of the fusion protein, while the furin recognition sequence between the hFlex and TRAIL allows the cleavage of TRAIL. Trimerization of TRAIL is achieved by the inclusion of an isoleucine zipper domain. Our results showed that concurrent treatment of human glioma cells with CBX and MSC-TRAIL markedly increased TRAIL-induced apoptosis in vitro and prolonged the survival of glioma-bearing mice in vivo. The enhanced efficacy of TRAIL correlated with upregulation of DR5 expression, blockade of GJIC, and downregulation of Cx43 expression. Because CBX is clinically approved for the treatment of inflammatory diseases [19], the combined usage of CBX and TRAIL may offer an attractive alternative for the treatment of glioma.
Materials and Methods
Cell culture
This study has been approved by the SingHealth Centralized Institutional Review Board, Singapore. ΔGli36 cells (kindly provided by Dr. M. Esteves, University of Massachusetts) and 2-2 cells (kindly provided by Dr. R. Sandri-Goldin, University of California Los Angeles) were cultured as previously described [25]. Human glioma cell line, U87MG, was purchased from American Type Culture Collection (Rockville, MD) and was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Grand Island, NY). The identity of the cells was authenticated by short tandem repeats profiling.
Procedures on isolation of MSC were performed as follows. Bone marrow cells were isolated from the femoral head of patient undergoing hip-replacement surgery following informed consent (Age: 68, Sex: M). Culture medium DMEM/F12 with 10% FBS and ascorbic acid (Sigma-Aldrich, St. Louis, MO) was added into the marrow isolates and subjected to Ficoll-Hypaque density gradient centrifugation. The cells were then plated into tissue culture flasks for 2–3 days and subsequently subjected to repeat culture medium changes to remove the hematopoietic cells from the culture. A confluent monolayer culture was observed 7–10 days following initial plating. MSC were then characterized based on the presence of cellular markers CD13, CD44, CD73, CD90, and CD105, as well as its ability to differentiate to osteogenic and adipogenic lineage as previously described [25].
Isolation of primary glioma cells NNI23 (age: 60, sex: F) and NNI24 (age: 49, sex: M) from local GBM patients were performed, after informed consent, as follows. In brief, brain tumor specimens, from patients undergoing tumor resection surgery, were cut into smaller pieces and washed thoroughly with phosphate-buffered saline (PBS) before digestion with 0.25% trypsin at 37°C for 30 min with constant stirring. Equal volume of astrocyte growth medium (AGM; Lonza, Basel, Switzerland) was then added to the suspension. Tumor pieces were allowed to settle before collecting the supernatant and filtering through a 70-μm membrane filter (BD Biosciences, Franklin Lakes, NJ). Filtered supernatant was centrifuged at 1000 rpm for 5 min at room temperature. Cell pellet was then resuspended in fresh media and plated per usual. Primary GBM line, GBM6, was purchased from Mayo Clinic (Rochester, MN) and maintained as subcutaneous xenografts as previously described [26]. CBX and glycyrrhizic acid (GZA) were purchased from Sigma-Aldrich. To test the sensitivity of GBM6 to TRAIL and CBX, short-term explant cultures were established in tissue culture dishes containing DMEM with 2.5% FBS and 1× penicillin/streptomycin.
Cloning of pHGCX-TRAIL HSV-1 amplicon viral vector
The pHGCX HSV-1 amplicon vector containing the enhanced green fluorescent protein (eGFP) gene under the control of the viral immediate early promoter (IE4/5) was obtained from Dr. EA Chiocca (Ohio State University Medical Center, Columbus, OH). The cassette consisting the gene encoding hFlex—Furin-isoleucine zipper—TRAIL was inserted into the HindIII and XbaI site located downstream of the strong cytomegalovirus (CMV) promoter. All sequences were verified by DNA sequencing (1st BASE, Singapore). Purification of HSV-1 amplicon viral vectors and amplicon viral transduction were performed as previously described [25].
In vitro migration assay
In vitro migration assay was performed with a modified Boyden chamber as previously described [25]. To determine the effect of CBX on ΔGli36 capacity in secreting chemotrophic factors, ΔGli36 cells were seeded at 1×106 cells in T75 flask and treated with either 100 μM of CBX or GZA the following day. Conditioned media (CM) was collected 48 h post-addition and was used in the migration assay as described above.
Quantification of TRAIL receptors by flow cytometry
The cell surface expression profile of TRAIL receptors was analyzed by flow cytometry using antibodies against DR4, DR5 (Santa Cruz Biotechnology, Santa Cruz, CA), DcR1, and DcR2 (R & D Systems, Minneapolis, MN). In brief, upon harvesting cells with StemPro®Accutase® (Invitrogen, Grand Island, NY), cells were subjected to repeated washing with cold FACS buffer (PBS supplemented with 2% FBS) and subsequently incubated with antibodies for 30 min at 4°C. Following incubation, cells were subjected to washing steps and finally resuspended in cold FACS buffer for analysis.
Caspase-3 activity assay
Caspase-3 activity was determined using ApoAlert Caspase-3 Colorimetric Assay Kit (Clontech, Mountain View, CA) according to the manufacturer's protocol. In brief, ΔGli36 cells were seeded at 2×106 cells in a 100-mm dish (Corning Life Sciences, Tewksbury, MA). The following day, MSC-TRAIL-CM along with 100 μM of CBX or GZA was added to ΔGli36 cells. Cells were harvested 24 h post-treatment and further subjected to the assay protocol.
Small interfering RNAi transfection
Introduction of RNAi against DR5 and Cx43 was performed on ΔGli36 cells with Lipofectamine RNAi Max (Invitrogen) by reverse transfection method according to the manufacturer's protocol. In brief, a final concentration of 20 nM pooled DR5-RNAi and Cx43-RNAi constructs was transfected into 2×104 ΔGli36 cells in 48-well plate. Cells were exposed to MSC-TRAIL-CM and 100 μM CBX or GZA 24 h after transfection. Cell death assay was performed 48 and 24 h post-treatment for DR5-RNAi and Cx43-RNAi transfected cells, respectively.
Dye transfer assay
Acceptor cells (ΔGli36 and U87MG) labeled with 1 μM of carboxymethyl-diI (CM-DiI) (red; Invitrogen) were pretreated with either 100 μM CBX or GZA for 3 h. Donor cells (ΔGli36 and U87MG) were labeled with 1 μM of Calcein AM (green; Invitrogen) at 37°C for 1 h. After 3 h, Calcein AM-labeled donor cells were overlaid onto their respective CM-DiI-labeled acceptor cells in a ratio of 1:5 in the presence of either 100 μM CBX or GZA to allow dye transfer to occur. Subsequently, the cells were mounted onto microscope slides and images were captured using a Plan Neofluor 40×/Numerical Aperture (N.A.) 0.75 objective mounted on the LSM 510 Meta Confocal Microscope system (Carl Zeiss, Göttingen, Germany).
Immunoblotting
Equal amounts of protein extracted from ΔGli36, U87MG, and GBM6 or CBX and GZA-treated ΔGli36 cells were resolved by 10% SDS-PAGE and electroblotted onto polyvinylidine difluoride membrane (Trans-blot Transfer Medium; Bio-Rad Laboratories, Hercules, CA). Membranes were blotted against Cx43 (1:500; Sigma-Aldrich), β-tubulin (1:5000; Sigma-Aldrich) and actin (1:10,000; NeoMarkers, Fremont, CA).
In vivo intracranial mouse tumor model
All animal experiments were performed in accordance to the guidelines and protocols approved by the Institutional Animal Care and Use Committee at the Singapore General Hospital, Singapore. Inoculation of tumor cells in immunodeficient nude mice (female, 4–6 weeks; Animal Resource Centre, Canningvale, Western Australia) was performed as previously described [23]. MSC were preinfected with MOI of 2.0 of either HGCX or HGCX-TRAIL to increase the amount of TRAIL production. Seven days post-implantation, MSC-TRAIL or MSC-HGCX (1×105) suspended in 2 μL of PBS was injected at the ipsilateral hemisphere at 2.5 mm lateral and 2.8 mm depth from the bregma. Mice group receiving CBX treatment was injected with 2 μL of 100 μM CBX at the same site of ΔGli36 implantation on the same day as MSC injection. The experiment was terminated when the body weight of the control group (ΔGli36) decreased by 30%.
TUNEL staining
Brain sections were subjected to TUNEL assay using DeadEnd Colorimetric TUNEL System (Promega, Madison, WI) according to the manufacturer's protocol. In brief, sections were fixed with 4% paraformaldehyde (PFA) and repeatedly washed with PBS. Sections were then incubated with 20 μg/mL Proteinase K for 15 min at room temperature and subjected to repeated washing in PBS. Following which, sections were incubated in terminal deoxynucleotidyl transferase (TdT) and biotinylated nucleotide mix for 60 min at 37°C. End-labeling reaction was then terminated by incubating the sections in 2× SSC solutions for 15 min at room temperature followed by repeated washing by PBS. Streptavidin horseradish peroxidase (HRP) was then added to the sections and incubated for 30 min at room temperature. Unbound streptavidin HRP was removed by repeated washing in PBS. 3,3′-Diaminobenzidine solution was then applied for approximately 10 min to allow for brown color to develop. Sections were then rinsed with distilled water and subsequently counterstained with hematoxylin. TUNEL-positive stained area was determined using Nikon Eclipse 90i (Nikon, Singapore). Images were captured using a 10×/NA 0.45 Plan Apochromat objectives.
Statistical analysis
Statistical analysis was performed using Prism 3.0 (Graphpad Software, Inc., San Diego, CA). One-way analysis of variance followed by Tukey-Kramer multiple comparisons test were used for comparing statistical significance for more than two groups. P-value<0.05 was considered statistically significant.
Results
CBX does not affect the viability and migratory properties of MSC
The aim of the current study was to investigate whether blockade of GJIC could enhance MSC-delivered, TRAIL-induced apoptosis in human glioma cells. To this end, the optimal concentration of CBX that does not affect the viability of human bone marrow-derived MSC was first tested. These multipotent MSC expressed CD44, CD105, CD90, CD13, and CD73 but not CD34 and CD45; these cells also possessed the capacity to differentiate to adipogenic and osteogenic lineage (Supplementary Fig. S1; Supplementary Data are available online at
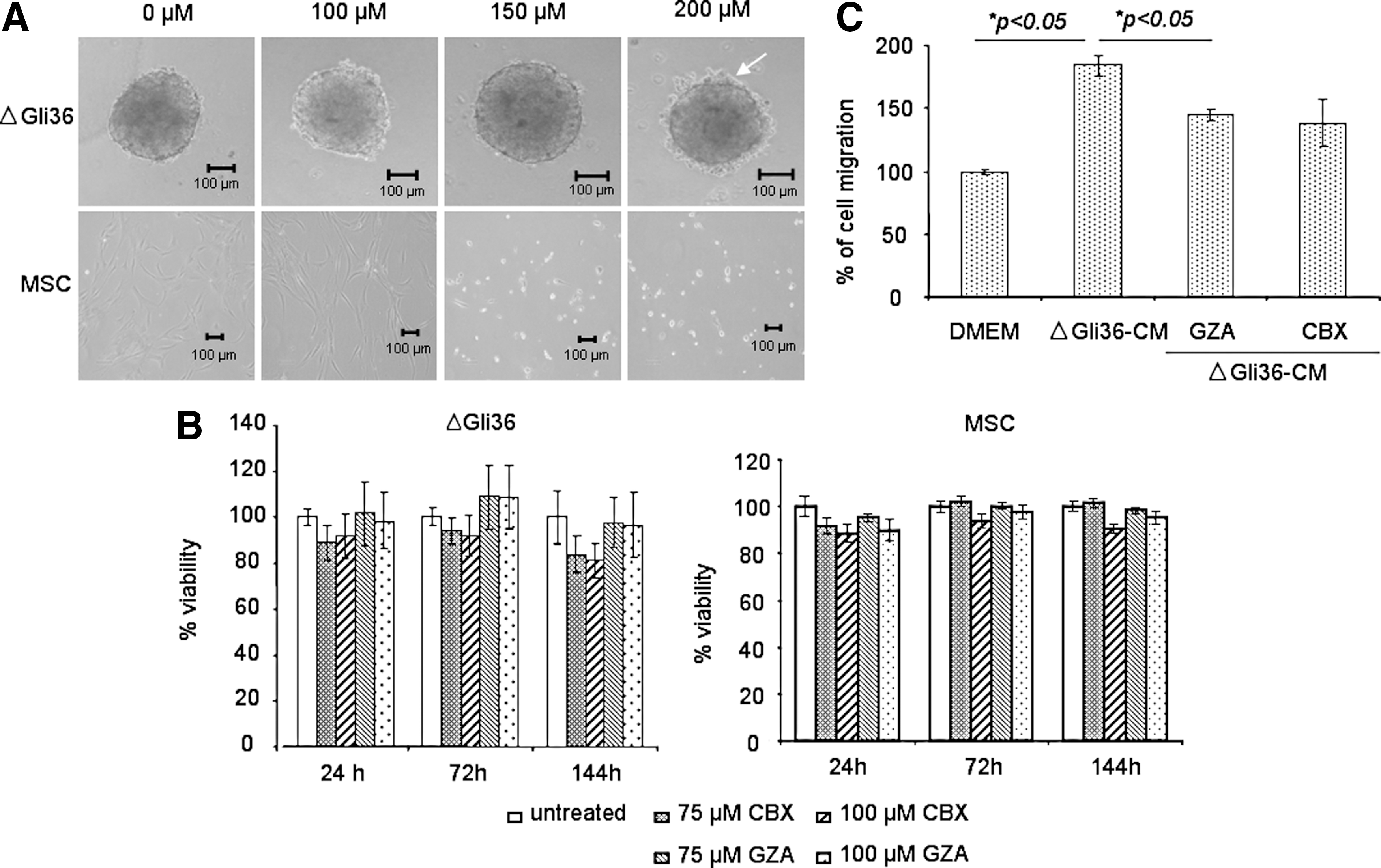
Carbenoxolone (CBX) is noncytotoxic on tumor cells.
Constitutive expression of secretable form of TRAIL, mediated by MSC, increases glioma cell death in the presence of CBX without affecting the viability of MSC
To achieve sustained expression of TRAIL, the gene encoding for hFlex-furin-isoleucine zipper-TRAIL (FFZT) was incorporated into the pHGCX HSV-1 amplicon viral plasmid (Fig. 2A; refer Materials and Methods for details). As shown in Fig. 2B, MSC could easily be transduced by HGCX and HGCX-TRAIL HSV-1 amplicon viral vectors with efficiency of ∼33% without changes to their innate properties [25]. Gene modification of these MSC allowed the expression of TRAIL up to ∼130 pg/mL at MOI of 1.0 (Fig. 2C). To assess the cytotoxic effect of TRAIL on MSC and glioma cells, MSC and ΔGli36 cells were infected with MOI of 1.0 of HGCX-TRAIL and HGCX as control. Transduction efficiency of both MSC and ΔGli36 cells was ∼40% and 70%, respectively (Fig. 2D). Cell death was not observed in HGCX-TRAIL-transduced MSC when compared with the vector-transduced cells. By contrast, 80% cell death was detected in HGCX-TRAIL-transduced ΔGli36 cells, thus confirming the tumor-specific effect of TRAIL (Fig. 2D). FACS analysis showed that MSC lacked DR4 expression but expressed DR5 (39.1%), DcR1 (22.8%), and DcR2 (37.9%), whereas ΔGli36 expressed low level of DR4 (0.5%) and DcR1 (1.8%) but expressed high DR5 (75.3%) and moderate DcR2 (30.4%) (Fig. 2E). Both DcR1 and DcR2 have been shown to differentially inhibit the death-inducing signaling complex (DISC) formation between TRAIL and DR5 [28], possibly contributing to the insensitivity of MSC to TRAIL-induced apoptosis. Thus, these results indicated that MSC are refractory to TRAIL and could be employed for cancer gene therapy purpose.
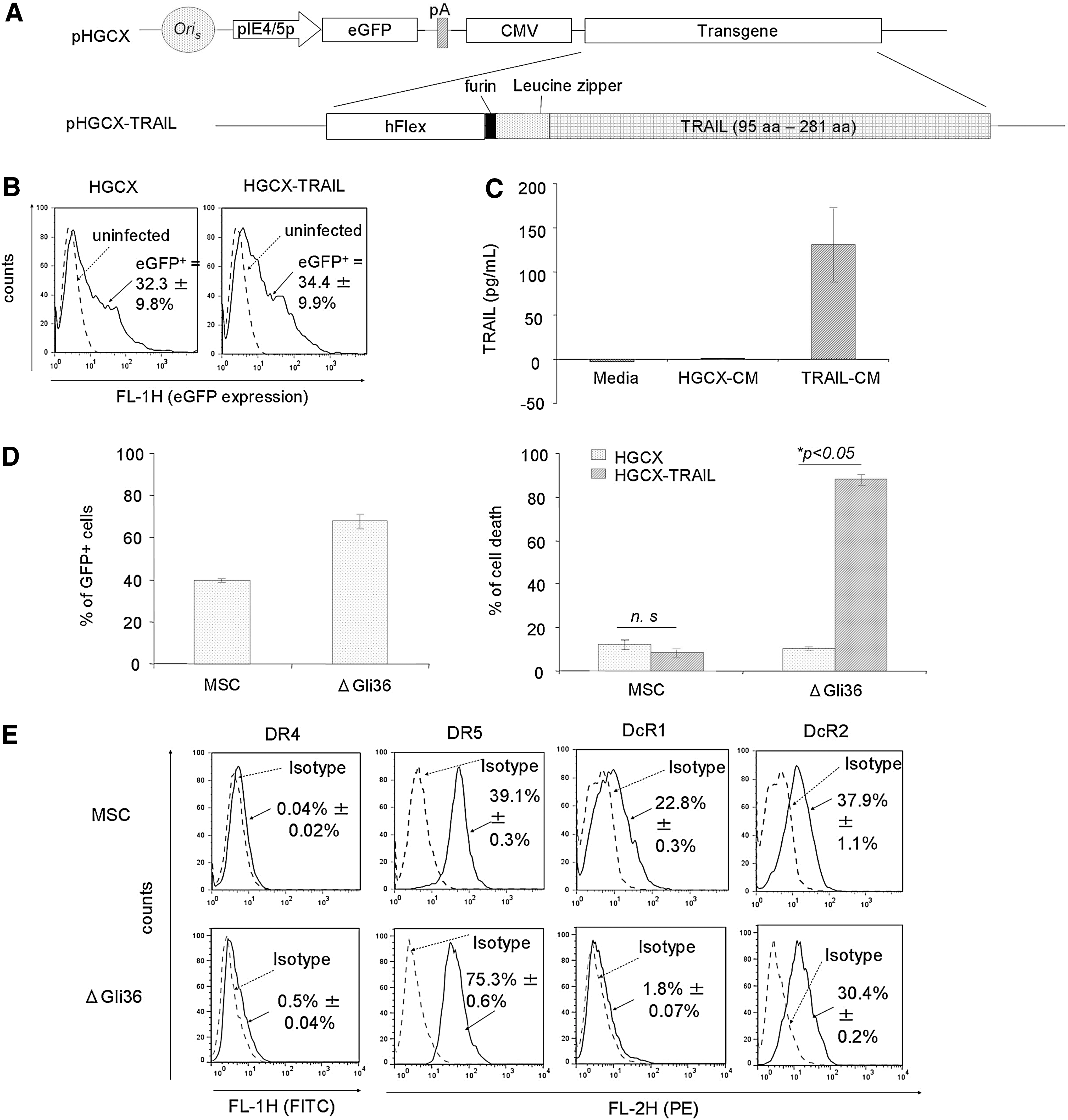
MSC can be easily transduced by herpes simplex virus-1 (HSV-1) amplicon virus and are resistant to TRAIL-induced apoptosis.
Next, the effect of TRAIL and CBX in ΔGli36 and U87MG human glioma cells, which exhibit different degree of sensitivity toward TRAIL, was evaluated using secretable TRAIL produced by genetically modified MSC. MSC were first transduced with HGCX-TRAIL to generate CM containing TRAIL, which is denoted as MSC-TRAIL-CM henceforth. Co-incubation of MSC-TRAIL-CM with CBX markedly augmented the percentage of cell death in ΔGli36 and U87MG cells by 27.1% and 19.1%, respectively, when compared with MSC-TRAIL-CM with GZA (Fig. 3A). On the other hand, significant difference was not observed between cells incubated with MSC-TRAIL-CM with or without GZA. Similarly, cell death was not observed in MSC even in the presence of 100 μM CBX (Fig. 3B), thus indicating that these cells are resistant to TRAIL-induced apoptosis. As many of the glioma cell lines have been propagated extensively in the laboratory and might not accurately represent the heterogeneous primary brain tumors, we further challenged the efficacy of CBX in enhancing TRAIL-induced apoptosis in three primary cultures of patient-derived glioma cells. NNI23 and NNI24 are short-term cultures of primary glioma derived from local patients and histologically classified as GBM and oligoastrocytoma, respectively. GBM6 were originally obtained from Mayo Clinic (refer Materials and Methods section). These primary glioma cells expressed glial fibrillary acidic protein indicating that they were of glial origins (Fig. 3D). As shown in Fig. 3C, both NNI23 and NNI24 are marginally sensitive to TRAIL-induced apoptosis (12% and 14% respectively). Co-incubation of MSC-TRAIL-CM with CBX in NNI23 and NNI24 markedly augmented the percentage of cell death by 14.3% and 18.1%, respectively, when compared with that of MSC-TRAIL-CM with GZA (Fig. 3C, respectively), while minimal cell death was observed in either GZA or CBX-treated cells. Similar enhancement in TRAIL-induced cell death was also observed in GBM6 treated with CBX and MSC-TRAIL-CM (Fig. 3C). By contrast, the difference in the percentage of cell death detected between MSC-TRAIL-CM with GZA and MSC-TRAIL-CM was not statistically significant. The increased in cell death observed in MSC-TRAIL-CM and CBX-treated ΔGli36 cells correlated with a marked increase in caspase-3 activities when compared with that of MSC-TRAIL and GZA or MSC-TRAIL alone (Fig. 4A). Collectively, these results showed that CBX is able to enhance TRAIL-induced apoptosis in human glioma cell lines and patient-derived glioma cells.
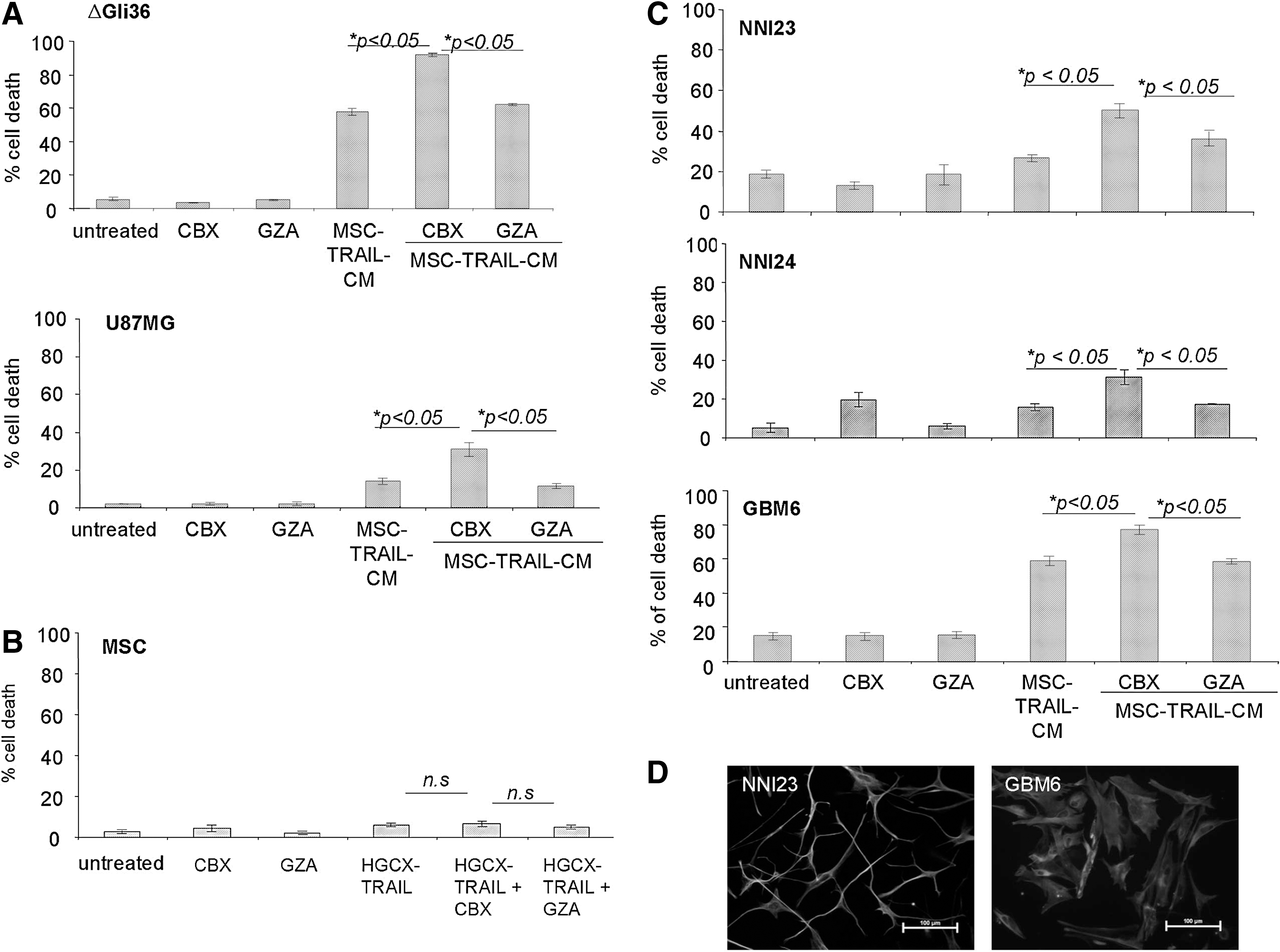
CBX augments MSC-mediated TRAIL-induced apoptosis in human glioma cell lines and in patient-derived primary glioma cells. MSC-TRAIL-CM was collected 24 h post-transduction of MSC at MOI of 2.0. MSC-TRAIL-CM along with 100 μM CBX or GZA was then added to glioma cells,

Enhanced TRAIL-apoptosis by CBX is correlated with increased in caspase activities and is partially mediated by DR5 upregulation.
DR5 activation and Cx43 downregulation are essential for CBX-enhanced TRAIL-induced apoptosis
To explore the underlying mechanism on how CBX enhances TRAIL-induced apoptosis, we investigated the effect of CBX on the cell surface expression of DR4 and DR5. Upon treatment with CBX, upregulation of DR5 cell surface expression was observed in CBX-treated ΔGli36 and U87MG (Fig. 4B), while no effect was observed for DR4 (Fig. 4C), suggesting that the induction of apoptosis by CBX and TRAIL was mediated through DR5. To confirm the functional role of DR5 in CBX-enhanced TRAIL-induced apoptosis, three DR5-RNAi that target different regions on the DR5 transcript were synthesized. These RNAi showed at least 90% reduction in the %DR5 expression (Supplementary Fig. S2A) when compared with the control-RNAi (ctrl-RNAi)-transfected cells, while no effect was observed on DR4 expression (Supplementary Fig. S2B). Upon transfection into CBX-treated ΔGli36 cells, targeted knockdown of DR5 reversed the action of CBX; increased cell death was not observed in DR5-RNAi-transfected cells that were treated with MSC-TRAIL-CM and CBX (Fig. 4D). On the contrary, combined treatment of MSC-TRAIL-CM and CBX increased cell death in ctrl-RNAi-transfected cells in comparison to that of TRAIL alone (Fig. 4D). These results suggested that upregulation of DR5 is, in part, essential for CBX-enhanced TRAIL-induced apoptosis.
CBX has been shown to inhibit GJ communication through its effect on Cx43 [29], which is the most abundant connexin molecules in the brain. Phosphorylation of Cx43 was shown to associate with closure of GJ channel [30]. Immunoblot performed on total cell lysate harvested from ΔGli36, U87MG, and GBM6 showed the expression of Cx43 (Fig. 5A). Two distinct bands were visible in ΔGli36, namely, the nonphosphorylated and phosphorylated isoform of Cx43 (Fig. 5A), whereas U87MG and GBM6 expressed multiple isoforms of phosphorylated Cx43. Using dye-transfer assay, we demonstrated the transfer of the green fluorescence Calcein AM from the donor to the CM-DiI-labeled ΔGli36 and U87MG recipient in GZA-treated cells (Fig. 5B arrow). By contrast, dye transfer was absent in CBX-treated ΔGli36 and U87MG (Fig. 5B arrowhead), demonstrating that CBX inhibited GJ communication in both cells. As shown in Fig. 5C, CBX significantly downregulated Cx43 mRNA expression in ΔGli36 and U87MG cells after 72 h, whereas no difference in Cx43 gene expression was observed in cells treated with GZA. These results were also evident in the immunofluorescence staining of Cx43 expression in CBX-treated ΔGli36, U87MG, and primary glioma GBM6 (Fig. 5Di). Cx43 staining was observed at cell–cell apposition, plasma membrane, and cytoplasm (Fig. 5Di arrow). Upon treatment with CBX, Cx43 expression was virtually undetectable from these cells (Fig. 5Di and Supplementary Fig. S3). Immunoblot analysis of CBX-treated ΔGli36 cells confirmed the downregulation of Cx43 protein expression compared to GZA-treated cells (Fig. 5Dii). To confirm the role of Cx43 in CBX-enhanced TRAIL-induced apoptosis, a combination of three different Cx43-RNAi was used to knockdown Cx43 expression in ΔGli36. As shown in Fig. 5E, these RNAi could effectively reduce Cx43 expression compared to ctrl-RNAi-transfected cells. Targeted knockdown of Cx43 resulted in similar observation as CBX treatment; increased cell death was observed in Cx43-RNAi-transfected cells upon treatment with MSC-TRAIL-CM but not in ctrl-RNAi-transfected cells (Fig. 5F). Taken together, these results suggested that Cx43 is essential for enhanced TRAIL-induced apoptosis by CBX.

CBX disrupts GJIC and downregulates Cx43.
Double arm therapy using MSC-TRAIL with CBX prolonged the survival of glioma-bearing mice
The efficacy of TRAIL and CBX was further investigated in an intracranial mouse tumor model system. To mimic the clinical scenario, ΔGli36 human glioma cells were first implanted into the right hemisphere of immunodeficient nude mice followed by administration of either MSC-HGCX or MSC-TRAIL with or without CBX (refer to Materials and Methods section for details) 1 week later. As shown in Fig. 6A, concurrent treatment with MSC-TRAIL and CBX significantly prolonged the survival of tumor-bearing mice by 26.7% in comparison to the group of mice receiving either MSC-HGCX with CBX or CBX alone. By contrast, MSC-TRAIL, as a single agent, was not as effective as MSC-TRAIL with CBX in extending the survival of the glioma-bearing mice. The prolonged survival observed in MSC-TRAIL and CBX group correlated with enhanced apoptosis (Fig. 6B) as observed in the higher percentage of TUNEL-positive cells. Furthermore, marked reduction of Cx43 expression was also detected in MSC-TRAIL with CBX-treated group in comparison to MSC-TRAIL-treated group as shown by immunofluorescence staining (Fig. 6C and Supplementary Fig. S4). Taken together, our results showed that CBX, when used in combination with MSC-TRAIL, improves the therapeutic efficacy of TRAIL.

CBX synergizes with MSC-TRAIL to prolong the survival of glioma-bearing mice.
Discussion
TRAIL-based therapeutic approaches have been particularly attractive due to its low toxicity that are well tolerated in clinical trials [2,3]. In a first-in-human trial of patients with advanced cancer, 46% of patients had stable disease at the end of the treatment cycle and two of five patients with chondrosarcoma had confirmed partial response after treatment with recombinant TRAIL, Dulanermin [31]. Although this trial has demonstrated safety and efficacy, further clinical development has been hampered by the minimal anticancer activity when Dulanermin was administered as a single agent [3], suggesting that some cancer cells may be resistant to TRAIL or that the treatment failed to reach majority of the cancer cells [4]. The latter becomes a pressing issue considering that glioma cells are notorious for its diffuse infiltrative nature. In the current study, we examined the feasibility of using MSC as carriers of TRAIL in the presence of a GJ inhibitor. We reasoned that the therapeutic efficacy of TRAIL would be enhanced with the tumor homing potential of MSC, and reinforced by inhibiting the astrocytic intercellular communication.
Cell-to-cell communication occurs through transfer of signaling molecule via GJ, which not only contributes to cell death through the transfer of apoptotic messengers, it also functions as a “good samaritan” by passing cell survival factors between cells [32]. Consequently, disruption of GJIC and hemichannels will interfere with cancer cells survival and thus amplify the death signal. Indeed, enhanced apoptosis was observed in ΔGli36 and U87MG human glioma cells that were treated with CBX and TRAIL (Fig. 3) in comparison to TRAIL alone, while no difference was observed in GZA and TRAIL-treated cells. This increased in cell death may be due to blockage in GJIC, which is evident in the inhibition of Calcein AM dye transfer in CBX-treated ΔGli36 and U87MG cells (Fig. 5B), indicating a GJ-mediated effect of CBX. It has been suggested that only membrane localized Cx43 forms functional GJ [33]; however, this does not seem to be the case. Although Cx43 was predominantly cytoplasmic in ΔGli36 and U87MG, dye transfer could still occur (Fig. 5B; GZA-treated cells). In fact, cytoplasmic-Cx43 is frequently observed in GBM [34]. In a study examining the localization and functionality of GJ in 74 human GBM samples, 77% of the GBM tissues expressed Cx43, and four out of eight of the short-term culture established from these GBM tissues have cytoplasm-localized Cx43 [34]. Furthermore, the short-term cultures that have cytoplasmic-Cx43 retained functional GJ assessed via dye transfer assay. The link between cytoplasmic-Cx43 expression and functional GJIC was also observed in a study by Cottin et al. in U87MG and SKI-1 human glioma cells [35]. Other studies performed on cardiomyocytes showed functional hemichannels formed by mitochondrial localized Cx43 [36 –38]. Thus, it is plausible that the remaining GJs present in our glioma cells are highly functional.
The role of Cx43 in apoptosis is controversial; Cx43 was reported to mediate cell death through GJ-dependent and GJ-independent manner. Dysfunctional GJ through the silencing of Cx43 has been shown to render thyroid cancer cells susceptible to anoikis and inhibit Akt activation [39]. Furthermore, downregulation of Cx43 expression was observed in cell death induced by serum deprivation or etoposide treatment in various cell types, including osteosarcoma cells [40], human corneal fibroblast [41], and primary bovine lens epithelial cells [42]. Taken together, our results on the inhibition of dye transfer and downregulation of Cx43 in CBX-treated cells suggested that the enhancement in cell death observed upon treatment with CBX and TRAIL occurred through a GJ-dependent manner. However, it is difficult to rule out GJ-independent cell death because CBX was also shown to induce mitochondrial depolarization and oxidative stress through the production of reactive oxygen species (ROS) in neurons [43]. Generation of ROS subsequently activates mitogen-activated protein kinase (MAPK) and DR expression to initiate downstream death signals [44]. Indeed, our results showed that CBX upregulated DR5 expression in ΔGli36 and U87MG by 18% and 15.7%, respectively (Fig. 4B). These results are in agreement with published reports whereby upregulation of DR5 expression was observed in studies that used therapeutic agents such as zerumbone [45] and garcinol [46] to augment TRAIL activities. Most of the studies that showed GJ-independent aspect of Cx43 in cell death are based on manipulation of Cx43 expression in cells. For example, the mutant form of Cx43 that contains point mutation in the second extracellular region of the Cx43 gene failed to form functional GJ, but retained its ability to suppress HeLa cells growth [47]. Likewise, interaction of Cx43 with other growth regulators such as c-Src through its nonchannel forming carboxyl tail was shown to inhibit glioma cells tumorigenesis [48]. Furthermore, interaction between Cx43 and apoptosis-signal-inducing kinase 1 (ASK1) was also shown to protect against hydrogen peroxide-induced apoptosis in C6 glioma cells without affecting its channel-forming ability [49]. Collectively, our results suggested that CBX-enhanced TRAIL-induced apoptosis is Cx43- and DR5-dependent (Fig. 7). It is possible that CBX treatment increased the production of ROS [43], which induced phosphorylation of Cx43 and the subsequent inhibition of GJ through MAPK signaling [50]. Along the same line, ROS-induced DR5 upregulation most probably occurs through activation of c-Jun N-terminal kinase (JNK) [51], which increased the expression of DR5 and the subsequent recruitment of Fas-associated death domain to form the DISC [52] and activation of downstream active caspases.
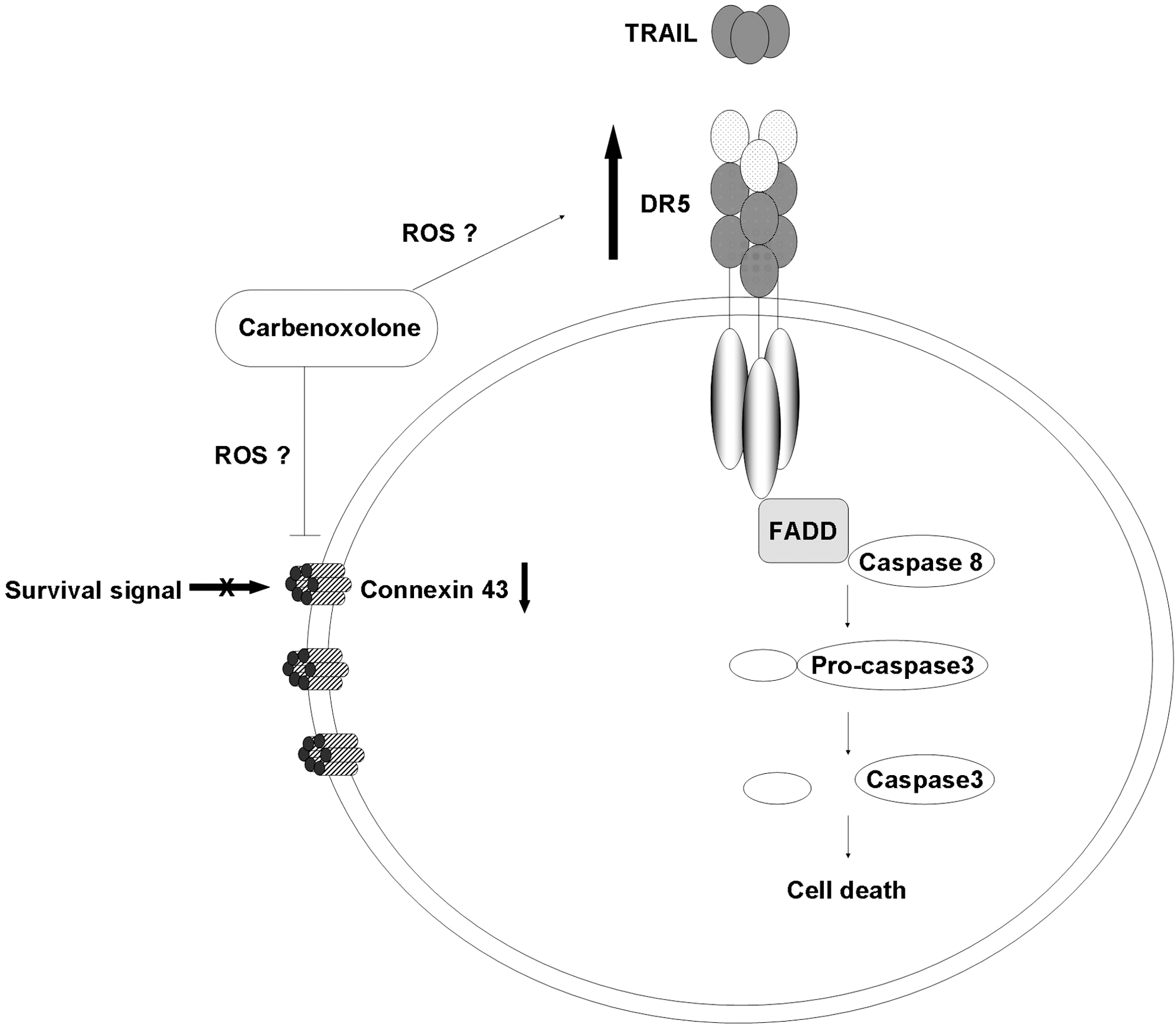
Synergistic mechanism of TRAIL-enhanced apoptosis mediated by CBX. In the presence of CBX, DR5 is upregulated, thereby increasing the apoptotic signals and downstream active caspases. Additionally, GJIC is disrupted and Cx43 is downregulated. CBX might also possibly increase reactive oxygen species (ROS) production, which may subsequently phosphorylate Cx43, leading to disruption in GJIC, and upregulates the expression of DR5.
Although our in vivo results demonstrated enhanced survival in the presence of MSC-TRAIL and CBX, the animals, nonetheless, succumbed to glioma. This is because the transgene expression can only be sustained up to a maximum of 14 days due to the nonintegrating nature of the HSV-1 virions [25]. We have recently generated a HSV/Epstein Barr virus (EBV) hybrid vector and showed stable luciferase gene expression in the mouse brain for up to 1 year [53]. Thus, to improve the stability, the pHGCX-TRAIL construct can be subcloned into the HSV/EBV hybrid vector for stable gene expression. Besides this, the half-life of both CBX and TRAIL in rodents are rather short; the half-life of CBX is less than 1 h [54], while that of TRAIL is 3–5 min [55]. Recently, Rozanov and colleagues generated a leucine zipper-TRAIL (LZ-TRAIL) chimera with extended half-life of more than 1 h [56]. This chimeric construct contains the optimized sequence of the GAP promoter, α-factor signal peptide, the SRKKRS-modified Kex2 cleavage site, and the modified yeast GCN4-pII LZ motif linked via the GSG linker to the TRAIL 120–281 fragment [56]. Thus, to improve stability, the LZ-TRAIL gene can be subcloned into the HSV/EBV hybrid vector for stable transgene expression. TMZ in combination with irradiation is currently the first-line treatment strategy for recurrent GBM and, when used concurrently, has been shown to improve the median survival time of glioma patients for up to 5 years of follow-up [57]. Accordingly, pretreatment of glioma xenograft with CBX before administration of TRAIL transduced MSC, or in combination with TMZ might improve the efficacy and further prolong survival. Further manipulation of the in vivo administration will need to be performed to achieve a substantial therapeutic outcome.
In summary, we showed that CBX increased the efficacy of TRAIL in human glioma cell lines and patient-derived primary glioma cells in vitro, and prolonged the survival of glioma-bearing mice in vivo through the inhibition of GJIC, downregulation of Cx43, and upregulation of DR5. More importantly, no obvious physiological or neurological effect was observed in mice administered with CBX, thus supporting the in vitro results that CBX is noncytotoxic. Considering that CBX is approved clinically as an anti-inflammatory agent, our results on the enhanced anti-tumor efficacy in combination with TRAIL suggested a clinical usage for potential cancer treatment.
Footnotes
Acknowledgments
We thank Dr. E.A. Chiocca (Ohio State University Medical Center, Columbus) for providing the pHGCX HSV-1 amplicon vector and the fHSVΔpac27 0+ and Mr. K.C. Sia for assistance in packaging the HSV-1 amplicon viral vectors. GBM6 was kindly provided by Jann Sarkaria (Mayo Clinic, Rochester, MN). This research was supported by grants from the SingHealth Foundation, National Medical Research Council, Singapore, and the Community Cancer Fund, National Cancer Center of Singapore.
Author Disclosure Statement
The authors declare no potential conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.