
Abstract
Neural stem cells (NSCs) have become promising tools for basic research and regenerative medicine. Intracerebral transplantation studies have suggested that these cells may be able to adopt neuronal phenotypes typical of their engraftment site and to establish appropriate connections in the recipient circuitries. Here, we examined the in vivo neurogenic competence of well-characterized NSC lines subjected to in vitro priming and subsequent implantation into the adult intact mouse brain. Upon implantation into the hippocampus and, less frequently, in the striatum and in the cerebral cortex, numerous green fluorescent protein (GFP)-tagged cells acquired differentiated features indistinguishable from resident neurons. Upon closer examination, however, we found that this outcome resulted from fusion of donor cells with local neuronal elements generating long-term persistent GFP+ neuronal hybrids. This fusogenic behavior of NSCs was unexpected and also observed in coculture with E18 hippocampal immature neural cells, but not with microglia or astrocytes. Similar findings were consistently obtained with different NSC lines, mouse recipients, and donor cell-labeling methods. The frequent and cell type-specific fusion of donor NSCs with host neurons highlights a previously underestimated biological property of the nervous tissue that might prove profitable for basic and therapeutically oriented studies.
Introduction
T
Several studies with immortalized NSC lines [4 –8] have provoked an upsurge of optimism for the ability of grafted NSC lines to generate neuronal subtypes in vivo [9]. Studies using neurospheres and mouse and human embryonic stem cell (ESC) lines have demonstrated variable differentiation, commitment, and long-distance axonal growth competence as well as the host target projection specificity of the donor cells [10 –13]. In most studies, the behavior of the implanted cells has been interpreted according to their ability to acquire a mature phenotype and integrate in the recipient texture. An alternative possibility however is the fusion of the donor cell with a host partner. In the brain, spontaneous cell fusion occurs between bone marrow-derived cells (BMDCs) and Purkinje neurons [14 –20]. Nevertheless, fusion events so far observed in the CNS are extremely rare and slow, and the underlying mechanisms are poorly understood, thus greatly reducing the interest of this phenomenon for possible applicative goals.
Here, we examined the in vivo neurogenic competence of in vitro primed green fluorescent protein (GFP)-labeled NSC lines obtained from different sources, after implantation into the adult mouse brain. We show that within a few days after intracerebral transplantation, GFP-labeled mouse NSCs display differentiated features indistinguishable from resident neurons. This outcome results from in vivo fusion of donor cells with local neurons and is observed also in coculture with hippocampal progenitors or primary neurons, but not with non-neuronal cells. The rather high frequency, cell specificity, and consistency of the results obtained with different cell lines and mouse recipients indicate that fusion of donor NSCs with host neurons is a biologically relevant phenomenon that that might prove therapeutically profitable.
Material and Methods
Animals
Mice were housed 3 to 4 per cage in a 12-h (7:00 A.M. to 7:00 P.M.) light/dark colony room at 22°C and had ad libitum access to food and water. Experimental protocols were approved by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. For transplantation experiments, we have employed the following mouse strains: syngenic 129 s/v, ROSA26, Z/AP, R26R-YFP, CMV-Cre, R6/2, (Charles River, Italy), CD-1 Nude mice (Charles River, Italy), and C57BL/6 (Charles River, Italy).
Cell lines and culture procedures
The following mouse NSC lines have been employed in this study [21 –23] LC1-GFP cells (LC1 ESC-derived cells lentivirally transduced to express GFP); Cor1-GFP cells (cells derived from E16 fetal cortex stably expressing GFP by means of electroporation of a pCAG-GFP vector); NS 14 CX cells (cells derived from E14 fetal cortex of GFP transgenic mice; CX 16 RFP cells (cells derived from E16 fetal cortex of CAG-mRFP1 transgenic mice); NS YFP-floxed cells (cells derived from E16 fetal cortex of R26R-YFP mice); and NS Cre 16 CX cells (cells derived from E16 fetal cortex of CMV-Cre mouse). For the experiments here reported, long-term passaged cells (at least 10–15 in vitro passages) have been employed. NSCs were maintained and passaged as previously described. L14 GFP neurospheres (derived from E14 fetal cortex of GFP transgenic mice) have been generated and passaged as previously reported. Priming procedure was as described in Spiliotopolus et al., (2009) [24]. In particular, here we applied the first step of the differentiation protocol by plating 1.0×105–1.5×105 cells/cm2 on an uncoated plastic cell culture flask dish using the D1 medium consisting of the EUROMED-N medium (Euroclone, Celbio) supplemented with 1% B27 (Invitrogen), 0.5% N2 (Invitrogen), and 10 ng/mL FGF-2 (Peprotech, Tebu-Bio) for 3 days (priming protocol).
When specified, cells have been loaded for 20 min with DiI solution (Vybrant DiI solution; Molecular Probes) 1 h before transplantation, and then washed thrice for a total of 30 min with a warmed medium.
To evaluate the presence of p24 viral protein in the medium of NSCs, a PerkinElmer p24 ELISA Kit Assay has been employed (see manufacturer's instructions).
Primary astrocyte and microglial cultures were prepared from P1-P3 old GFP or RFP mouse pups. Cerebral cortices were isolated and cleaned of meninges in Hank's balanced salt solution (HBSS). After trypsin digestion, the cell suspension was filtered through a 40-μm filter, centrifuged, and resuspended in Dulbecco's modified Eagle medium (DMEM) supplemented with 20% fetal bovine serum. Cells were then plated onto 75-cm2 flasks and cultured in 5% CO2 humidified incubator at 37°C. To obtain microglial cultures, the flasks were shaken at 200 rpm for 2 h after 9–10 days in culture. The medium containing floating microglia was removed, and the cells were centrifuged, resuspended in DMEM supplemented with 10% fetal bovine serum, and plated onto 4-well plates at a density of 2×105 cells/well. The enriched microglial cultures were composed of 95% of microglia as determined by staining using an anti-IBA1 primary antibody and the nuclear staining dye DAPI.
After shaking off the microglia, enriched astrocyte cultures were lifted from the flask with 0.05% trypsin and plated onto 4-well plates at a density of 2×105 cells/well. Cells were cultured in 5% CO2 humidified incubator at 37°C. The enriched astrocyte cultures were composed of more than 95% of astrocytes as determined by staining using an anti-GFAP primary antibody and the nuclear staining dye DAPI.
Primary hippocampal cultures were prepared from E18 GFP+ or RFP+ mouse embryos. Hippocampi were isolated and cleaned in HBSS; pooled tissues were dissociated by digestion with trypsin and deoxyribonuclease for 5 min. The enzymes were removed by washing twice in phosphate-buffered saline (PBS). The cells were passed through a 40-μm cell filter to obtain a single-cell suspension. After centrifugation, the cells were washed and plated onto 4-well plates laminin-coated (3 μg/mL) at a density of 2×105 cells/well. The cultures were maintained in a proliferation medium consisting of a Neurobasal medium, B27 (2%) and N2 (1%) supplement, 20 ng/mL FGF-2, and EGF (human recombinant; Prepotech, TebuBio). Cells were cultured in 5% CO2-humidified incubator at 37°C. Primary neuronal cultures were prepared form E15 RFP+ mouse embryos; the cultures were maintained in a neuronal differentiation medium consisting of a combination of 1:1 DMEM/F12:Neurobasal media, B27 (2%) and N2 (0.5%) supplement, and 20 ng/mL BDNF (human recombinant; Prepotech, TebuBio). Cells were cultured for 10 days in 5% CO2-humidified incubator at 37°C.
Coculture experiments were performed with primary cultures (neural progenitors, microglia, and astrocytes) from RFP+ or GFP+ mice and fetal-derived GFP+ or RFP+ NSCs in self-renewal conditions. Astrocytes and NSCs were plated at a 1:1 proportion in an NS proliferation medium. Microglial cells and NSCs were plated at a 2:1 proportion, in a proliferation medium. For the coculture between NSCs and hippocampal progenitors, cells were plated 1:1 in an NSC proliferative medium. Neurons and primary neurons were plated at a 1:2 proportion in the NS proliferation medium. Alive cells were observed at the microscope at defined time points; subsequently, cells were fixed and stored at 4°C until immunocytochemistry was carried out.
Transplants and tissue processing
All experiments were conducted in accordance with the principles and procedures outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
For each experimental group, at least n=6–12 male mice (6–8 weeks old) were grafted with specific NSCs. In initial experiments, a number of animals received saline. On the whole, more than 300 animals were analyzed.
Animals, anesthetized with Ketamine/Xylazine (100 and 10 mg/kg i.p. respectively), received stereotaxic injection of 1 μL (2,5×105 cells/μL) of NSC suspension in the hippocampus (hp), striatum (st), and neocortex (ctx) (coordinates: hippocampus AP−1,46; ML±1,5; DV−2,3 mm from Bregma striatum AP 0,7; ML±2,0; DV−3,5 mm from Bregma; cortex AP−0,40; ML±1,4; DV−1 from Dura Mater). Transplant recipient animals were killed by overdose of anesthetic and transcardially perfused with 4% paraformaldehyde (PFA) at the indicated time points with ice-cold PBS, followed by ice-cold 4% paraformaldehyde. Animals were transplanted at 4–6 weeks of age. R6/2 transgenic mice were transplanted (n=12) at 9–10 weeks of age to see huntingtin aggregates in cortex.
For in vivo BrdU labeling of hippocampal progenitors of P20 129, s/v mice were treated with 4 consecutive (6-h interval) intraperitoneal BrdU injections (50 mg/kg). The animals (n=18) were grafted 3 weeks after the last injection of BrdU and sacrificed 1 week later.
Immunohistochemistry and microscopy
Perfused brains were removed, postfixed overnight in 4% PFA at 4°C, cryoprotected in 20% sucrose, and stored at 4°C. About 30–40-μm-thick coronal sections were cut on cryostat (Leica Microsystem) and incubated for 1 h at room temperature in a blocking solution (10% normal goat or donkey serum as appropriate and 0.3% TritonX-100), followed by incubation in a blocking solution (1% normal goat or donkey serum as appropriate and 0.1% TritonX-100) with proper primary antibody overnight at room temperature. The next day, the sections were washed thrice in PBS and incubated with the appropriate fluorescence-conjugate secondary antibody (1:500; Alexa, Molecular Probes) for 1 h at room temperature. For BrdU staining, before the blocking step, sections were treated with 2N HCl for 30 min at room temperature, followed by 3 washes in 0.1 M Boric acid, pH 8.8. For the counting of grafted cells in Fig. 1E, we have considered as undifferentiated those cells with no branching and as intermediates those cells carrying either 2 to 4 outgrowth. GFP+ differentiated were those cells (also for graph in Supplementary Fig. S2; Supplementary Data are available online at

Fate of neural stem cells (NSCs) grafted to the adult brain.
Antibodies and dilutions were as follows: mouse anti-BrdU (1:400; Becton Dickinson); rabbit anti-GFP (1:500; Invitrogen; used also for YFP detection); chicken anti-GFP (1:1000; Abcam); rabbit ani-βGal (1:1000; Cappel); rabbit anti-IBA1 (1:1000; Wako); rabbit anti-Prox1 (1:400; Millipore); rabbit anti-NG2 (1:1000; Millipore); rabbit anti-GFAP (1:1000; Dako); rabbit anti-RFP (1:1000; Abcam); goat anti-RFP (1:1000; Santa Cruz); goat anti-Dcx (1:1000; Becton Dickinson); mouse anti-NeuN (1:50; Millipore); mouse anti-Nestin (1:300; Millipore); rabbit anti-Prox1 (1:500; Chemicon), rabbit anti-calbindin (1:1500; Swant); mouse anti-EM48 (1:300; Millipore); and DAPI (1:10000; Molecular Probes).
For immunocytochemistry analysis, fixed cells were incubated for 1 h at room temperature in a blocking solution (5% normal goat serum and 0.5% TritonX-100), followed by incubation in a blocking solution (1% normal goat serum and 0.1% TritonX-100) with the primary antibody overnight at 4°C. The following day, cells were washed thrice in PBS and incubated with the appropriate fluorescence-conjugated secondary antibody (1:500; Alexa, Molecular Probes) for 1 h at room temperature.
Antibodies and dilutions for immunocytochemistry were as follows: rabbit anti-GFP (1:1000; Invitrogen); rabbit anti-IBA1 (1:1000; Wako); rabbit anti-GFAP (1:1000; Dako); goat-anti-RFP (1:1000; Santa Cruz); and DAPI (1:10,000; Molecular Probes).
For confocal analysis of tissue sections, images were acquired with a Leica (TCS-SP2) and Zeiss (LSM-510) confocal laser-scanning microscopes and digitalized by ImageJ software. Cell culture images were acquired with an inverted Leica (DMI6000B) fluorescence microscopy and digitalized by ImageJ software.
Single-cell laser microdissection and single-cell genomic polymerase chain reaction
For single-cell Laser Microdissection (LMD), serial horizontal cryostat sections (thickness: 15 μm) were cut and mounted on sterile polytarthalene (PET) foil stretched on a metal frame (Leica). GFP immunostaining and DAPI staining were directly performed on the mounted section as described above. The sections were then dried at 40°C for 10 min and subjected to LMD. PET foil metal frames were mounted on a Leica AS LMD 6500 system (Leica) with the section facing downward. Using a 40×objective, cutting intensity, aperture, and velocity were adjusted. Then, the pulsed UV laser beam was carefully directed along the borders of single GFP+ cells or DAPI+ nuclei (for GFP− cells). Each cell cut (n=83) was then transferred by gravity alone into a microcentrifuge tube cap placed directly underneath the section. Microcentrifuge tubes were immediately transferred and the collected single cells processed for polymerase chain reaction (PCR) assay. For duplex LacZ/GFP PCR reaction, amplification included initial denaturation at 95°C for 5 min, 50 cycles of denaturation at 95°C for 45 s, annealing at 60°C for 45 s, and extension at 72°C for 30 s. For the single LacZ PCR, amplification included initial denaturation at 95°C for 5 min, 35 cycles of denaturation at 95°C for 45 s, annealing at 60°C for 45 s, and extension at 72°C for 30 s. Single GFP PCR amplification included initial denaturation at 95°C for 5 min, 35 cycles of denaturation at 95°C for 45 s, annealing at 60°C for 45 s, and extension at 72°C for 30 s. Primers' sequences are as follows:
LacZ (Fw): GCACATGGCCCTTTCGCCAGCTGGCGTAAT;
LacZ (Rev): CCCGTCTGGCCTTCCTGTAGCCAGGTTTCA;
GFP (Fw): ACCACATGAAGCAGCACGAC;
GFP (Rev): TTGTAGTTGCCGTCGTCCTT.
Time-lapse experiments
Time-lapse studies have been performed using a Leica AF7000 time-lapse videomicroscopy station. Experiments were performed starting from 1 h after coculture plating. Images were collected in phase contrast and in fluorescence every 10 min during 48–72 h (37°C, 5% CO2). For coculture experiments involving microglia and astrocytes, time-lapse recording was performed also at later time points (7–9–14 days). Movies were visualized with either dedicated Leica software or ImageJ.
Statistical analysis
For the statistical analysis, one-way analysis of variance (one-way ANOVA) has been used.
Results
Intracerebral transplantation of mouse NSC lines, neurospheres, and ESC derivatives has demonstrated variable competence of donor cells for differentiation and incorporation into host circuitries [1,2]. In most studies, the behavior of the implanted cells has been attributed to their ability to acquire mature phenotypes and integrate in the recipient texture.
In this work, GFP+ fetal-derived NSCs (Cor1-GFP cells) were transplanted into adult syngenic wild-type hippocampus either in a self-renewal condition or after 3-day exposure to a neuronal priming procedure (hereafter defined as primed cells) (Supplementary Fig. S1A, B; Supplementary Data are available online at
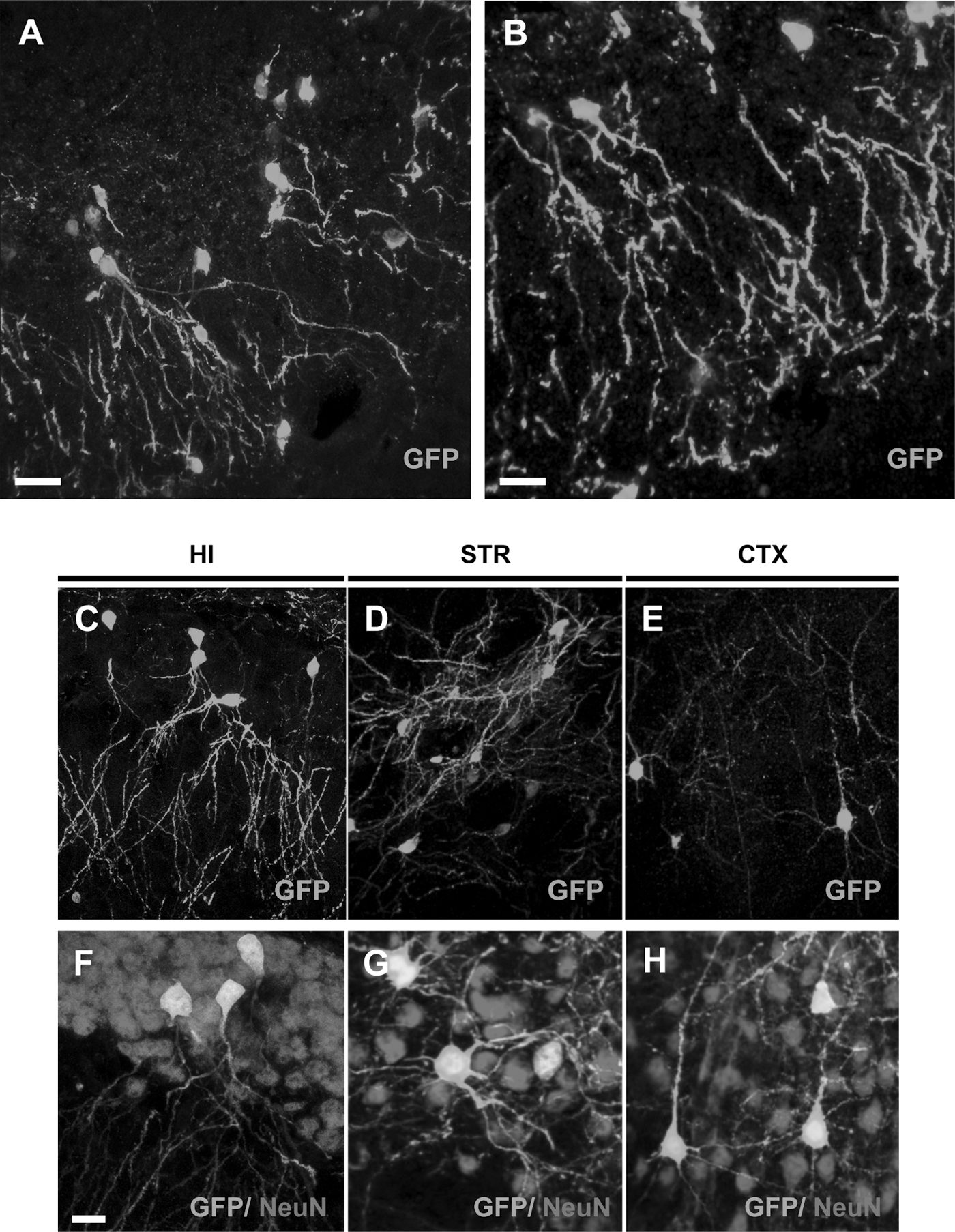
Fate of NSCs grafted to the adult brain.
Importantly, transplantation of Cor1-GFP NSCs into the non-neurogenic adult striatum and the cerebral cortex also led to GFP+/NeuN+ cells displaying the morphology of medium spiny neurons (Fig. 2D, G) and cortical pyramidal neurons (Fig. 2E, H) (n=16). In particular, cells found in the cerebral cortex exhibited large somata and extensive arbours of branching and spine-bearing apical and basal dendrites, which displayed the typical orientation in the cortical architecture. In contrast, in no case, we did find evidence for coexpression of GFP with GFAP (an astrocytic marker) (Supplementary Fig. S2A–D) or IBA1 (a microglial marker) (Supplementary Fig. S2E–H). Analogous results were obtained when performing a colocalization analysis with NG2, an oligodendrocyte precursor marker (Supplementary Fig. S2I, J). Out of 250,000 injected primed cells, 515±193 GFP+ cells were counted per hippocampus (0.21%±0.07%, n, number of animals=15; Supplementary Fig. S3), of which 412±163 (79.3%±8.4%) displayed mature neuronal morphologies. The same outcome was observed in more than 300 animals that have been transplanted in different experimental settings (see below), in the last 5 years, by at least 4 different operators from 2 independent groups. In contrast, only rare (<0.04%) GFP+ cells with neuronal morphologies were found in the nonprimed conditions at the 7-day time point.
Comparable results were obtained with other mouse NSC lines, as well as NSCs grown as neurospheres cultures [22]. Grafting experiments performed with NS CX16 RFP cells (Supplementary Fig. S4A), NS 14CX GFP cells (Supplementary Fig. S4B), and L14 GFP neurospheres cultures (Supplementary Fig. S4C) generated GFP+ cells with hippocampal-specific mature neuronal morphologies. To investigate if the behavior of the grafted NSCs could be different depending on the host immune system activation, primed Cor1 GFP cells were transplanted in an allogenic mouse strain (C57BL/6 wild-type mice) (Supplementary Fig. S4D) and in 2 animal models of immune depression, that is, Cyclosporine A-treated 129 s/v wild-type mice (Supplementary Fig. S4E) and CD-1 nude mice (Supplementary Fig. S4F). In all of the 3 different cases, GFP+ cells with the neuronal morphology were found, thus indicating that the behavior of NSCs was host independent. Furthermore, the generation of GFP+ neurons in the recipient CNS required viable donor cells, since no GFP+ cells could be detected when grafting donor cells subjected to repeated freezing and thawing cycles. Finally, direct transfer of the GFP transgene to host cells due to lentiviral vectors used to tag NS donors was excluded by the absence of p24 viral protein assessed by ELISA assay (Supplementary Fig. S5).
The abrupt appearance of fully mature and properly integrated GFP+ neurons, a few DPGof our NSCs in different forebrain sites suggested fusion of grafted cells with host neurons. We injected Cor1-GFP cells into the hippocampus of ROSA26 transgenic mice in which the β-Galactosidase reporter is ubiquitously expressed [25] (Supplementary Fig. S6A). Confocal analysis of Cor1-GFP cells grafted to the hippocampus of ROSA26 transgenic mice showed that all GFP+ neurons were also positive for β-Gal immunostaining (n=12; Fig. 3A–B; Supplementary Fig. S7A–E). Extensive analysis of the entire specimens revealed that all of the GFP+ cells with mature neuronal morphologies and correct orientation in the host hippocampus were also immunoreactive for β-Gal signal. The possibility that the β-Gal could be released by recipient cells and phagocytosed by donor cells cannot be completely excluded. However, the available literature points to β-Gal as a strictly intracellular cytosolic protein. Accordingly, β-Gal is from ROSA26 mice, and since it has no leader peptide, it should not be secreted. In addition, there are several genetic models that express this gene, and there is no evidence of regulated or constitutive secretion. Therefore, these results indicated that the emergence of native-like GFP+ neuronal elements after transplantation of NSCs in an adult recipient brain was not due to differentiation of donor NSCs, but to fusion events occurring between such cells and host neurons. In addition, transplantation of Cor1-GFP cells in the striatum of adult R6/2 HD transgenic mice carrying a mutant huntingtin fragment [26] also led to GFP+ neurons coexpressing EM48-immunoreactive mutant huntingtin inclusions (Supplementary Fig. S7F, G) (n=6). Further evidence for fusion came from transplantation of NSCs generated from CMV-CRE deleter mice in R26RYFP mice [27]. In this condition, YFP expression can only occur if donor cells fuse with CRE-carrying host cells. As a consequence, the presence of YFP-positive cells in the recipient brain indicates the occurrence of fusion events. To visualize R26R-YFP-grafted cells in the recipient tissue, in these experiments, we loaded donor cells with DiI. Although in these experiments the number of engrafted cells was smaller (0.05%±0.02%, n=10), we consistently observed 4–10 YFP+ hippocampal neurons in each recipient brain that was specifically due to recombination between the 2 genomes (Fig. 3C–E; Supplementary Fig. S7H). Finally, after transplantation of LC1-GFP cells to ROSA26 recipients, we performed laser-capture microdissection of GFP+ mature neurons, GFP+ immature cells, or GFP- host cells (Fig. 4A), followed by single-cell PCR using primers for the GFP and LacZ genes (Supplementary Fig. S7I). This experiment showed that GFP + neurons carry both GFP and LacZ DNA, whereas immature GFP + cells do not have LacZ DNA, and GFP-cells only bear LacZ DNA (Supplementary Fig. S7I). These observations indicate that GFP + mature neurons actually derive from fusion of donor cells with host neurons.

NSCs fuse with endogenous neurons.

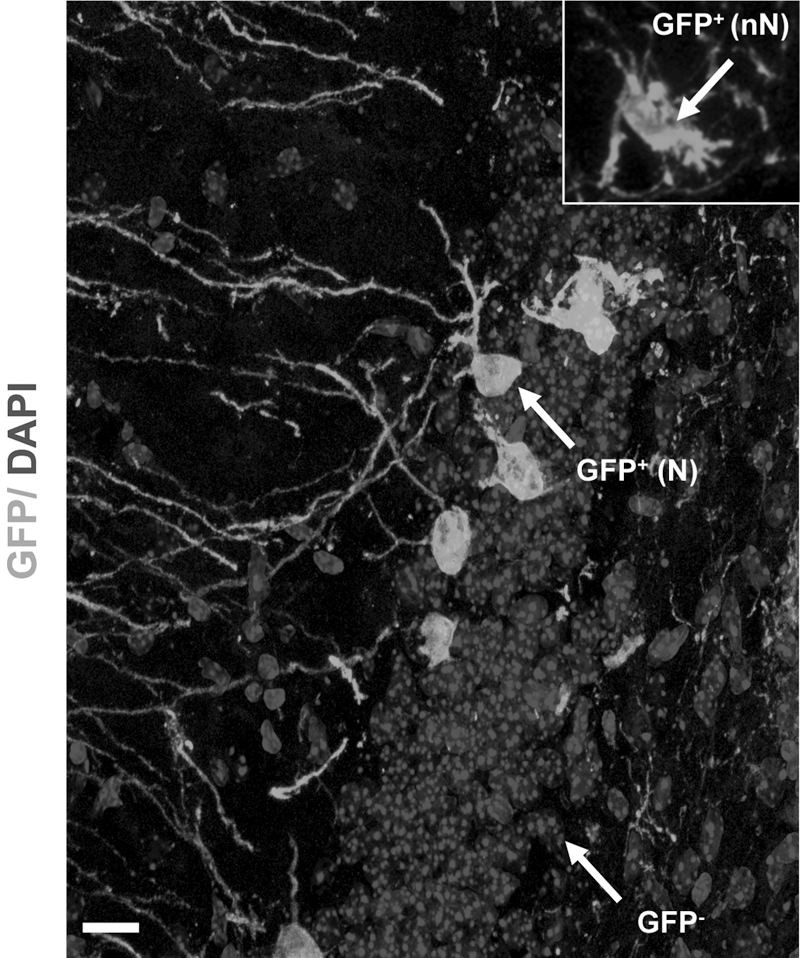
NSCs fuse with endogenous neurons. Arrow points to representative GFP+ mature neurons [GFP+(N)], GFP+ immature cells [GFP+(nN); inset], and GFP- cells sampled for laser microdissection from ROSA26 hippocampi transplanted with Cor1-GFP cells.
To ask whether NSC fusion is related to the age and renewing capacity of hippocampal neurons, Cor1-GFP cells were grafted to wild-type mice 3 weeks after BrdU injection. At 7 DPGa, a small, but consistent, number of mature granule neurons double labeled for BrdU and GFP were present in the recipient hippocampus (4–6 cells/animal, n=18; Fig. 5A–D and Supplementary Fig. S8A–D). Therefore, these results indicated that NSCs may fuse with all granule cells, including both recently generated neurons and more aged elements. Further demonstration of the fusogenic abilities of NSCs was obtained by coculturing primary E18 hippocampal progenitors from RFP+ mice with neuronally primed Cor1-GFP cells. In living-cell imaging, fused GFP+/RFP+ cells were visible at 2 h (Fig. 6A, arrow) and up to 48 h (Fig. 6B–D; Supplementary Fig. S9A–E and Supplementary Movie M1), but most of them disappeared by 72 h. Immunocytochemical analysis also showed increasing numbers of GFP+/RFP+ double-labeled cells until 48 h (Fig. 6E–H, arrows). At this time point, only 5.3% of the cells were double immunoreactive for GFP and RFP. During time, the percentage of GFP+/RFP+ double-immunoreactive cells increases (number of GFP+/RFP+ double-immunoreactive cells: 14.3%±0.9%, 31.6%±3.6% and 22.1%±2.8% at 6, 24 and 48 h, respectively). At later time points, GFP+/RFP+ double-immunoreactive cells do not seem to be visible (Supplementary Fig. S9F). Similar outcomes were observed in co-ultures of GFP+ NSCs with RFP+ primary neurons, with fused binucleated cells (Fig. 7A–T), but never with non-neuronal cells (Supplementary Fig. S10).

Fusion of NSCs with newborn DG neurons in vivo and in hippocampal cultures.

Coculture of Cor1-GFP cells with primary hippocampal progenitors from E18 RFP mice

Spontaneous fusion of primed GFP NSCs cocultured with RFP+ primary neurons.
Discussion
Evidence over the last few years has shown the unexpected capacity of donor cells to fuse with native cellular elements. So far, however, cell fusion has been considered a relatively rare phenomenon, physiologically occurring in vivo in different non-neural tissues of normal individuals, and possibly contributing to the regeneration of damaged tissue [19,28]. In the brain, only Purkinje neurons of the cerebellum have been shown to fuse spontaneously with injected BMDCs, giving rise to binucleated Purkinje-like neuronal hybrids [15 –17,28 –30].
Here we demonstrate an unprecedented ability of NSCs for fusing with mature neurons in the adult CNS. Fusion events occur with high frequencies, within a few days after transplantation and specifically involve neurons. The evidence pointing to the occurrence of fusion events can be summarized as follows: (i) in chimeric experiments in which GFP+ NSCs were transplanted into the ROSA26 or R6/2 mice, GFP signal was found to colocalize with the β-Gal immunoreactivity or with the EM48 staining, respectively (Fig. 3A, B and Supplementary Fig. S7F, G); (ii) the GFP+ neurons observed in the hippocampus and cortex appeared within 7 days of fetal-derived NSC injection, while endogenous neurons would be expected to mature over a much longer period of time; (iii) experiments aiming at detecting any transfer of the GFP gene/viral construct or protein gave negative results; (iv) grafting of 6 mouse NSC lines—which were derived from either ESC or fetal brain—gave similar results (though with different efficiencies); (v) different genetic labeling approaches (infection or transfection of the NSC with the GFP or RFP transgenes or generation of NSC lines from RFP+, GFP+ mice, and CRE+ mice) and different host mouse strains (129 S/V, CD1, C57BL6/J, and mixed strains) were used leading to similar results (although some donor cell lines displayed lesser efficiency); (vi) highly branched GFP+ neurons persisted in the hippocampus for over 18 months, further supporting the evidence of a permanent contribution of donor cells. Also, neural progenitors from neurospheres appeared to fuse in vivo, although to a lower efficiency. We also show that donor cells preferentially fuse with resident neuronal elements and not with glial and microglial cells, suggesting that this event is cell specific. Fusion events are also observed outside the hippocampus (although more rare) and never detected when the same NSCs were transplanted in the cerebellum. Therefore, the frequency of cell-to-cell fusion might be different depending on the neuronal subtype.
The functional effects of most CNS replacement strategies are thought to largely depend on the establishment of graft–host connection [8,31]. Our study however prompts a caution against the risk of misinterpreting the results of transplantation experiments. In the literature, other groups have reported the generation of perfectly integrated native-like neurons after intracerebral grafting of ex vivo expanded NSCs. One of the most striking examples came from transplantation of the immortalized RN33B neural progenitor cell line [4,5]. Grafts with similar behaviors have been described for human neurospheres exposed to a 6-day neuronal priming procedure that gave rise almost to several exclusively neuronal subtypes [32] and also in most recent studies that employed cells derived from ESCs [11,13] or from leptomeninges [33]. In these studies, the possibility of cell fusion was not considered or poorly investigated.
Our observations reveal an unexpected capacity of mature neurons to acquire the donor genetic material from NSCs. While this raises the possibility that fusion may play a physiological role in the development and maintenance of neogenerated neurons in the adult hippocampus, the frequency and specificity of the observed phenomenon suggest for the first time the fusogenic properties of NSCs as a new route for delivering therapeutic purposes in genetic diseases caused by loss of function.
Footnotes
Acknowledgments
We thank Alessandro Vercelli for the RFP mice, Manuela Marullo and Gaetano Finocchiaro for help with ELISA assay, Giulio Simonutti for imaging, Alessia Moiana and Roberta Azzarelli for cell culture, and Ketty Leto for help with the immunocytochemistry. We are also grateful to Mauro Baron from Leica Microsystems for assistance with the single-cell laser microdissection procedure and to Lucia Colombo for the use of the Leica Laser microdissection workstation. The work is supported by StemStroke (FP6, European Union Health 2007-037526), EuroStemCell (FP6, European Union Health 2006-503005), and NeuroStemCell (FP7, European Union Health-2008-B-222943) to E.C., Progetto Piattaforma Cariplo Nobel (Fondazione Cariplo, Italy; 20052042/104878) to E.C. and G.C. and PRIN (MIUR, Italy; #20074MW29N) to L.C.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.