
Abstract
Adipose-tissue-derived stem cells (ASCs) have received considerable attention due to their easy access, expansion potential, and differentiation capacity. ASCs are believed to have the potential to differentiate into neurons. However, the mechanisms by which this may occur remain largely unknown. Here, we show that culturing ASCs under active proliferation conditions greatly improves their propensity to differentiate toward osteogenic, adipogenic, and neurogenic lineages. Neurogenic-induced ASCs express early neurogenic genes as well as markers of mature neurons, including voltage-gated ion channels. Nestin, highly expressed in neural progenitors, is upregulated by mitogenic stimulation of ASCs, and as in neural progenitors, then repressed during neurogenic differentiation. Nestin gene (NES) expression under these conditions appears to be regulated by epigenetic mechanisms. The neural-specific, but not muscle-specific, enhancer regions of NES are DNA demethylated by mitogenic stimulation, and remethylated upon neurogenic differentiation. We observe dynamic changes in histone H3K4, H3K9, and H3K27 methylation on the NES locus before and during neurogenic differentiation that are consistent with epigenetic processes involved in the regulation of NES expression. We suggest that ASCs are epigenetically prepatterned to differentiate toward a neural lineage and that this prepatterning is enhanced by demethylation of critical NES enhancer elements upon mitogenic stimulation preceding neurogenic differentiation. Our findings provide molecular evidence that the differentiation repertoire of ASCs may extend beyond mesodermal lineages.
Introduction
Despite their natural propensity to differentiate into mesodermal derivatives, there is also evidence for the differentiation of MSCs, including ASCs, toward ectodermal and endodermal lineages [3 –7]. A subpopulation of MSCs termed multilineage-differentiating stress-enduring (Muse) cells, recently isolated from several tissues, including adipose tissue, expresses the pluripotency cell surface marker SSEA3 (stage-specific embryonic antigen 3) and can self-renew and differentiate into endodermal, ectodermal, and mesodermal cell types [8].
On the basis of morphological and immunohistochemical criteria, a number of studies have reported a neurogenic differentiation of ASCs [4,9 –25]. Electrophysiological recordings from neuron-like cells derived from ASCs support the idea that these have differentiated into functional neurons [10,15 –17]. Moreover, transplantation of ASCs into a variety of animal models for stroke, spinal cord injury, or multiple sclerosis, suggests a substantial functional benefit [18]. However, the mechanisms by which these improvements occur remain unclear, and evidence that ASCs can generate neurons capable of transmitting information and integrating into functional neuronal networks remains to be provided. One particular shortcoming lies in the molecular characterization of the commitment of ASCs to the neurogenic pathway.
Neurogenic differentiation is a complex process that involves fine-tuned orchestration of a series of transcription factors (TFs) and regulatory mechanisms [26 –28]. Nestin is a type-VI intermediate filament that has been extensively used as a marker for neurogenesis [29] and proliferation of neural progenitors [30,31]. Nestin is also expressed in developing skeletal muscle [32] and in several other cell types during development, such as migrating and proliferating cells with a potential to differentiate into neuroectodermal, ectodermal, and mesodermal lineages [33]. During neurogenic differentiation, downregulation of nestin is associated with entry into a postmitotic state [32]. Organization of the nestin gene (NES) is well conserved in mammals and reveals 3 introns and 4 exons [34]. Interestingly, tissue-specific expression of Nes in murine muscle and neural progenitor cells is regulated by 2 tissue-specific enhancers in the first and second introns, respectively [34 –36]. The neuronal Nes enhancer resides in the 3′ portion of the second intron; it contains 2 region-specific enhancer elements, one for general expression in the central nervous system (CNS), and one for selective expression in the midbrain [37 –39]. TFs regulate the neuronal Nes enhancer notably through SOX- and POU-binding elements [40]. To what extent, however, epigenetic mechanisms, such as cytosine methylation in a cytosine-phosphate-guanine (CpG) methylation and posttranslational histone modifications, contribute to regulating the tissue-specific activity of the Nes enhancers remains unknown.
In this study, we show that upon a 2-step induction of human ASCs into the neurogenic pathway, NES is transiently upregulated, then repressed in connection with cell cycle arrest. NES activation is associated with transient site-specific CpG demethylation within the neuronal NES enhancer, while the muscle-specific enhancer is not markedly affected. CpG demethylation is associated with loss of transcriptionally repressive histone modifications in this region. Upon neurogenic induction per se, the neuronal NES enhancer is specifically remethylated in association with the re-emergence of repressive histone marks. NES induction is associated with a neurogenic transcriptional program. Our results support the potential to elicit an epigenetic program of neurogenic differentiation in human ASCs.
Materials and Methods
Reagents
All reagents were from Sigma-Aldrich (
Isolation and cultivation of adipose tissue stem cells
ASCs with a CD34+CD105+CD45−CD31− phenotype were isolated from the stromal vascular fraction of human lipoaspirates and cultured in the DMEM–F12 medium 10% fetal calf serum (FCS) (standard medium) as described [11,41] unless otherwise stated. Cells at passage 10 (corresponding to 30 population doubling) were used in this study. ASCs were cultured either under a standard medium as above, a condition referred to as “unstimulated” (UNSTIM). Alternatively, ASCs were mitogenically stimulated (“STIM” condition). STIM treatment consisted of culturing ASCs in the Knockout DMEM (containing 4.5 g/L glucose and glutamax) supplemented with 20 ng/mL epidermal growth factor (EGF), 20 ng/mL basic fibroblast growth factor (bFGF), and 1×B27 supplement (Gibco/Invitrogen;
Cell proliferation assay
ASC cultures from 2 donors, with 2 replicate flasks per donor, were seeded into 162-cm2 flasks at 0.5×106 cells per flask under STIM or UNSTIM conditions (see the Results section). After 7 days, cells were trypsinized, suspended in their respective treatment medium, and counted using an automated cell counter (Becton Dickinson;
Adipogenic, osteogenic, and chondrogenic differentiation
For adipogenic differentiation, ASCs (UNSTIM or STIM treated for 14 days) from 3 donors were plated at 2.5×104 cells per cm2 and allowed to settle for 4 h. After washing with phosphate buffered saline (PBS), cells were cultured for 3 weeks in adipogenic medium (DMEM–F12 with 10% FCS, 0.5 μM 1-methyl-3 isobutylxanthine, 1 μM dexamethasone, 10 μg/mL insulin, and 100 μM indomethacin) [11]. Cells were stained with Oil Red O to visualize lipid droplets [11]. For each of the 3 donors and for each condition, 10 images of the differentiated cells were acquired and thresholded using ImageJ [42]. The same threshold was used for all images. Percentages of total areas containing black pixels were counted for each image and the means±standard deviations (SDs) for each image series were compared using a Fisher's t-test. For osteogenic differentiation, ASCs seeded at 0.3×104 cells per cm2 were allowed to settle for 4 h, washed with PBS, and cultured for 3 weeks in the osteogenic medium (DMEM–F12 with 10% FCS, 100 nM dexamethasone, 10 mM β-glycerophosphate, and 0.05 mM
Neurogenic differentiation
ASCs cultured for 7 days under STIM conditions, or under UNSTIM conditions (where specified) were harvested and plated onto either 24-well plates (2×104 cells/well) with each well containing a sterile glass coverslip for immunostaining, or onto 10-cm dishes (0.5×106 cells/dish) for reverse transcription–polymerase chain reaction (RT-PCR) and chromatin immunoprecipitation (ChIP) analyses. At this stage, STIM ASCs were at 35–36 population doubling and UNSTIM cells were at 31–32 population doubling. Cells were allowed to settle for at least 6 h and washed twice with the serum-free Knockout DMEM. Neuronal differentiation was induced by culture in the Neurobasal medium (Invitrogen) containing 1% FCS, 1×B27 supplement, 0.5 mM 1-methyl-3 isobutylxanthine, 1 μM dexamethasone, 0.2 mM 8CPT-cAMP, 10 mM valproic acid, and 10 μM forskolin. The neuronal differentiation medium was changed weekly.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde in PBS for 30 min, rinsed 3×in PBS, and treated with 1 M ethanolamine-HCl in 0.1 M NaPi (pH 7.4). Cells were blocked for 1 h with 10% (v/v) normal goat serum, 3% (w/v) bovine serum albumin (BSA) in 0.1 M Tris-base (pH 7.4), 0.3 M sodium chloride, and 0.5% Triton X-100 (TBST) and incubated overnight at room temperature with primary antibodies (Supplementary Table S1; Supplementary Data are available online at
Proliferation assay
Proliferation rates were determined using the Clik-it™ 5-ethynyl-2′-deoxyuridine (EdU) Alexa fluor® imaging kit (Invitrogen) as previously reported [44]. Cells were incubated with 10 μM of EdU for 24 h. Cells were rinsed, fixed with 4% (w/v) paraformaldehyde in 0.1 M PBS (pH 7.4), washed twice with 3% BSA in PBS, and permeabilized with 0.5% Triton X-100 in PBS/3% BSA. Detection of EdU was performed according to kit instructions. DAPI+ and EdU+ nuclei were counted in digital fluorescent images (n=81 composite images, 5465 nuclei counted in total) using ImageJ [42] as previously described [44]. The proliferation rate was expressed as percent EdU+ nuclei relative to DAPI+ nuclei.
Reverse transcription–polymerase chain reaction
RT-PCR was done from 0.5 μg total RNA (Qiagen RNeasy;
Bisulfite genomic sequencing
DNA was purified using the DNeasy Blood and Tissue Kit (Qiagen;
Chromatin immunoprecipitation
ChIP was performed as described [45]. Cells were harvested, cross-linked with 1% formaldehyde, lysed, and sonicated using a Diagenode Bioruptor (
Flow cytometry
Flow cytometry analysis of cell surface marker expression was done as previously described [11] using antibodies listed in Supplementary Table S1.
Results
Characterization of ASCs cultured under active proliferation conditions
With the aim of eliciting neuronal differentiation of ASCs, cells were exposed to neurogenic induction conditions for 7 days (see the Materials and Methods section). This treatment elicited an apparent neuronal phenotype in a minor proportion (<5%) of the cells, in line with our earlier observations of the limited capacity of human ASCs to differentiate beyond mesodermal lineages (Boquest et al., 2005). During development, neurogenesis occurs through differentiation of neuroepithelial cells into radial glial cells, which in turn divide asymmetrically to give rise to either neurons or intermediate progenitor cells; these progenitors then proliferate before terminally differentiating into neurons [46]. Thus, we rationalized that active proliferation of ASCs before neurogenic induction might enhance their neurogenic potential.
To test this hypothesis, we promoted mitotic proliferation of ASCs for 7 days before neurogenic induction, by substituting the standard DMEM–F12/10% FCS culture medium with the Knockout DMEM/glucose/glutamax containing FCS, EGF, bFGF, and B27 supplement. Under this stimulation treatment (henceforth referred to as “STIM’), ASCs became smaller and more optically refractile than unstimulated ASCs cultured in the standard medium (“UNSTIM”; Supplementary Fig. S1A). STIM cells also became more sensitive to trypsinization (data not shown), a property likely linked to changes in the expression of cell adhesion molecules. Importantly, STIM treatment did not significantly alter the expression pattern of 22 cell surface antigens examined by flow cytometry, indicating that ASCs cultured under these conditions retain their surface immunophenotype (Supplementary Table S4).
STIM treatment elicited remarkable proliferation of ASCs, evidenced by the markedly enhanced weekly and cumulative increase in cell numbers in STIM relative to UNSTIM cultures (Supplementary Fig. S1B). The weekly fold increase of the STIM cell number declined over time, however, to reach by 8 weeks, a rate similar to that of UNSTIM cells (Supplementary Fig. S1B). STIM cells, as UNSTIM cells, eventually senesced, indicating that STIM treatment does not elicit immortalization.
In accordance with accelerated proliferation, gene expression analysis showed that STIM treatment upregulates cell cycle promoting genes (e.g., CCNA2, CCND1, and FGFR3B) and downregulates tumor suppressor genes (e.g., TP53, CDKN1A, and CDKN2A) (Supplementary Fig. S1C). Nonetheless, TERT expression (telomerase) remained unaltered, consistent with our finding that STIM cells show reduced proliferation capacity over time and that STIM treatment does not elicit unrestrictive proliferation. No significant changes were detected in expression of pluripotency-related genes (POU5F1, NANOG, KLF4, and SOX2), indicating that STIM treatment is not likely to promote the induction of a pluripotent state. In addition, STIM treatment downregulated transcripts for E-cadherins (CDH1 and CDH2), a hallmark of the epithelial-to-mesenchymal transition (EMT). As EMT occurs primarily during early development, wound healing, and tissue remodeling in the adult [47], this suggests that ASCs cultured under enhanced proliferative condition are likely to retain, or may expand, their differentiation repertoire.
Active proliferation of ASCs maintains their mesodermal differentiation capacity
STIM treatment of ASCs enhanced their subsequent ability to undergo adipogenic differentiation, as judged by qualitative and quantitative Oil Red O staining using pixel intensity measurements (Supplementary Fig. S2A, B). There was no obvious difference in the size of lipid-filled droplets between treatments; rather, more cells underwent adipogenesis in STIM pretreated cultures. In line with these findings, genes involved in adipogenesis showed a greater upregulation in adipogenic-differentiated STIM versus UNSTIM cells (Supplementary Fig. S2C). STIM treatment also enhanced osteogenic differentiation. Calcified extracellular matrices, visualized with Alizarin red, were more widespread with larger bone nodules in STIM versus UNSTIM pretreated cells after 3 weeks of differentiation (Supplementary Fig. S2D, E). Quantification of Alizarin red staining using pixel density analysis showed a 6-fold enhancement in bone mineralization conferred by STIM pretreatment (Supplementary Fig. S2D). STIM pretreatment also enhanced the upregulation of osteogenic marker genes after osteogenic induction (Supplementary Fig. S2F). In contrast, we find that chondrogenic differentiation was not improved by STIM treatment, and remained relatively inefficient from these cells, based on Alcian Blue staining and RT-PCR analysis of chondrogenic marker expression (Supplementary Fig. S3A, B). Thus, mitogenic stimulation of ASCs before differentiation positively affects their subsequent differentiation capacity into adipogenic and osteogenic lineages.
Transcriptional changes elicited by neurogenic induction of ASCs
To assess the neurogenic potential of ASCs, and with the aim of taking advantage of the enhanced mesodermal differentiation capacity of ASCs cultured under STIM conditions, we induced neurogenic differentiation of cells cultured for 7 days under STIM and UNSTIM conditions. Differentiation was induced in the neurobasal medium containing 1% FCS, B27, 1-methyl-3 isobutylxanthine, dexamethasone, 8CPT-cAMP, forskolin, and valproic acid. We noted that ∼50% of the cells died within 24 h of induction of differentiation; however, cell numbers remained constant thereafter (data not shown). STIM pretreatment greatly enhanced the formation of cells with neuron-like morphology compared with UNSTIM pretreatment (Fig. 1A). Approximately 50% of STIM-treated cells acquired a neuron-like morphology by 7 days (Fig. 1B), including interconnecting multipolar cells with phase-bright somata, multiply branched processes, and immunoreactivity to the neurofilament protein NEFH (neurofilament high molecular weight; Fig. 1C).

Neurogenic differentiation of ASCs.
Neurogenic differentiation was further evaluated by RT-PCR analysis of the expression of markers of progenitor cells, neurogenic cells (markers of immature neurons and regulators of neurogenesis), neurons (mature neuronal markers), glial cells (astrocytic and oligodendrocytic), and neuroendocrine cells (Fig. 2). The progenitor cell marker endoglin (ENG/CD105) was expressed in ASCs as expected, and appeared slightly downregulated upon neurogenic induction. CDK5, encoding cyclin-dependent kinase 5 and expressed in ASCs, was upregulated upon mitogenic stimulation, coincidently with its substrate nestin (NES). Expression of NES was detected on day 1 of neurogenic induction, but decreased markedly thereafter. Upregulation of NES upon STIM treatment is consistent with nestin being a marker of proliferating progenitor cells [33]. Transcripts of NEUROG1/2/3 (a designation reflecting the detection of a common transcript of the NEUROG1, NEUROG2, and NEUROG3 genes) was detected in undifferentiated ASCs and persisted during stimulation and day 1 of neurogenic differentiation, after which it was downregulated. Further transcript analysis using specific RT-PCR primers showed expression of NEUROG1 and NEUROG2, but not NEUROG3 transcripts at all stages examined (data not shown), consistent with a neurogenic induction process. Although not detected in ASCs, NEUROD1 expression was also induced by neurogenic differentiation, coincident with the peak expression of NEUROG1/2/3. This is consistent with the role of NEUROD1 as a downstream mediator of neurogenin [48] that provides functionality to the upregulated NEUROG transcripts. OLIG2 was also strongly upregulated by day 7 of neurogenic induction, coincident with NEUROG1/2/3 downregulation. This is consistent with the mutual antagonism of OLIG2 and NEUROG2 in modulating neural differentiation and specification [49]. Taken together, these results suggest a sequential activation and repression of neurogenic genes leading to a potentially neuralizing signaling cascade.
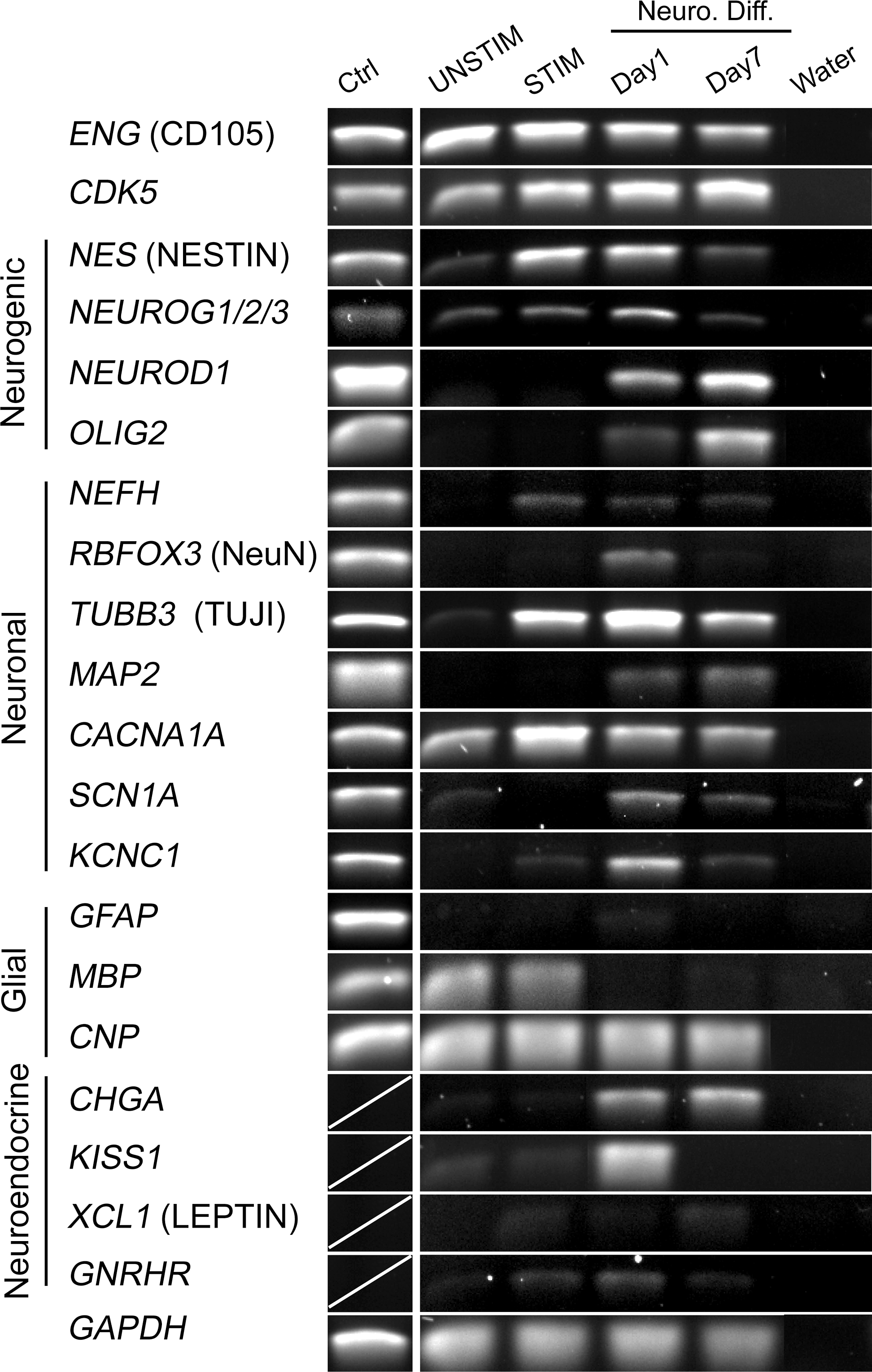
Induction of transcription of neurogenic, neuronal, glial, and neuroendocrine genes in ASCs. Reverse transcription–polymerase chain reaction analysis of expression of indicated genes in ASCs cultured under UNSTIM or STIM conditions, and after neurogenic differentiation of STIM-treated cells for 1 day or 7 days. Positive control (Ctrl) samples were RNA from human retina or white matter (the latter for OLIG2 only).
We next determined changes in expression of neuronal marker genes (Fig. 2). With the exception of CACNA1A (voltage gated calcium channel, P/Q type, alpha 1A subunit), and to some extent TUBB3 (beta-3-tubulin) and SCN1A (voltage-gated sodium channel, voltage-gated, type I, and alpha subunit), all other genes examined, namely, NEFH, RBFOX3 (NeuN), and MAP2 (microtubule assembly-promoting protein), and KCNC1 (voltage-gated potassium channel, Shaw-related subfamily, member 1) were upregulated under STIM or differentiation conditions, but not detected in UNSTIM conditions or in undifferentiated ASCs (Fig. 2). In particular, NEFH, though weakly expressed, was induced by STIM treatment and remained expressed throughout the differentiation period. RBFOX3, a marker associated with the initiation of terminal neuronal differentiation [29], was detectable only transiently on day 1 of neurogenic differentiation conditions. TUBB3 was sharply induced by STIM treatment and remained expressed throughout neurogenic induction. MAP2 was also expressed, although at a low level, upon differentiation. In particular, CACNA1A, SCN1A, and KCNC1 representatives of the three main ion channel types important for the electrophysiological properties that characterize neurons, were expressed. Collectively, these gene expression data support the view that ASCs have the potential to differentiate along the neuronal pathway in vitro.
Next, we investigated the expression of glial genes (Fig. 2). The astroglia-specific marker GFAP (glial fibrillar acid protein) was not detected before or after induction of neurogenic differentiation. Interestingly, the oligodendrocytic markers CNP (CNpase) and MBP (myelin basic protein) were expressed in both UNSTIM- and STIM-treated ASCs, though only CNP persisted during neurogenic differentiation. This is consistent with neurogenin being an inhibitor of glial differentiation [50] and suggests that the oligodendrocytic and astrocytic lineages become repressed in neurogenic-induced ASCs.
The last set of genes we examined included neuroendocrine genes (Fig. 2). These exhibited substantial variation in expression. CHGA (chromogranin A) and KISS1 (kisspeptin 1, a G-protein coupled receptor ligand for GPR54) were upregulated upon neurogenic differentiation, but KISS1 expression did not persist. XCL1 (leptin) and GNRHR (GnRH receptor) expression was barely detectable under STIM conditions and during neurogenic differentiation. Overall, our data suggest that neurogenic-induced ASCs differentiate predominantly toward the neuronal lineage.
Phenotypic markers elicited upon neurogenic induction
Using immunofluorescence, we further characterized the phenotype of ASCs induced toward the neurogenic pathway. In agreement with RT-PCR data, we found immunoreactivity for neurogenin1, neurofilament H, beta-3-tubulin, and MAP2 (Fig. 3A–D). The subcellular distribution of these markers was essentially as expected for neurons, with accumulations of the latter 3 proteins in the putative neurites. In a minor population of cells, we also detected antigens related to the synaptic vesicle release machinery, namely, MUNC13 (Fig. 3C), synapsin 1–2 (Fig. 3C), and synaptotagmin (Fig. 3D). However, these proteins were not observed in synaptic terminal-like structures, suggesting a lack of neuronal maturation. Moreover, immunolabeling for gephyrin, a scaffold protein usually associated with clusters of postsynaptic GABAergic and glycinergic receptors in the pericaryon and dendrites, was also detected (Fig. 3D), but only in the pericaryon and not in the putative dendrites, again suggesting a lack of neuronal maturation. These data argue that ASCs can be steered into a neurogenic differentiation program through the upregulation of TFs involved in neurogenesis, resulting in the expression of gene sets expressed uniquely in mature neurons.
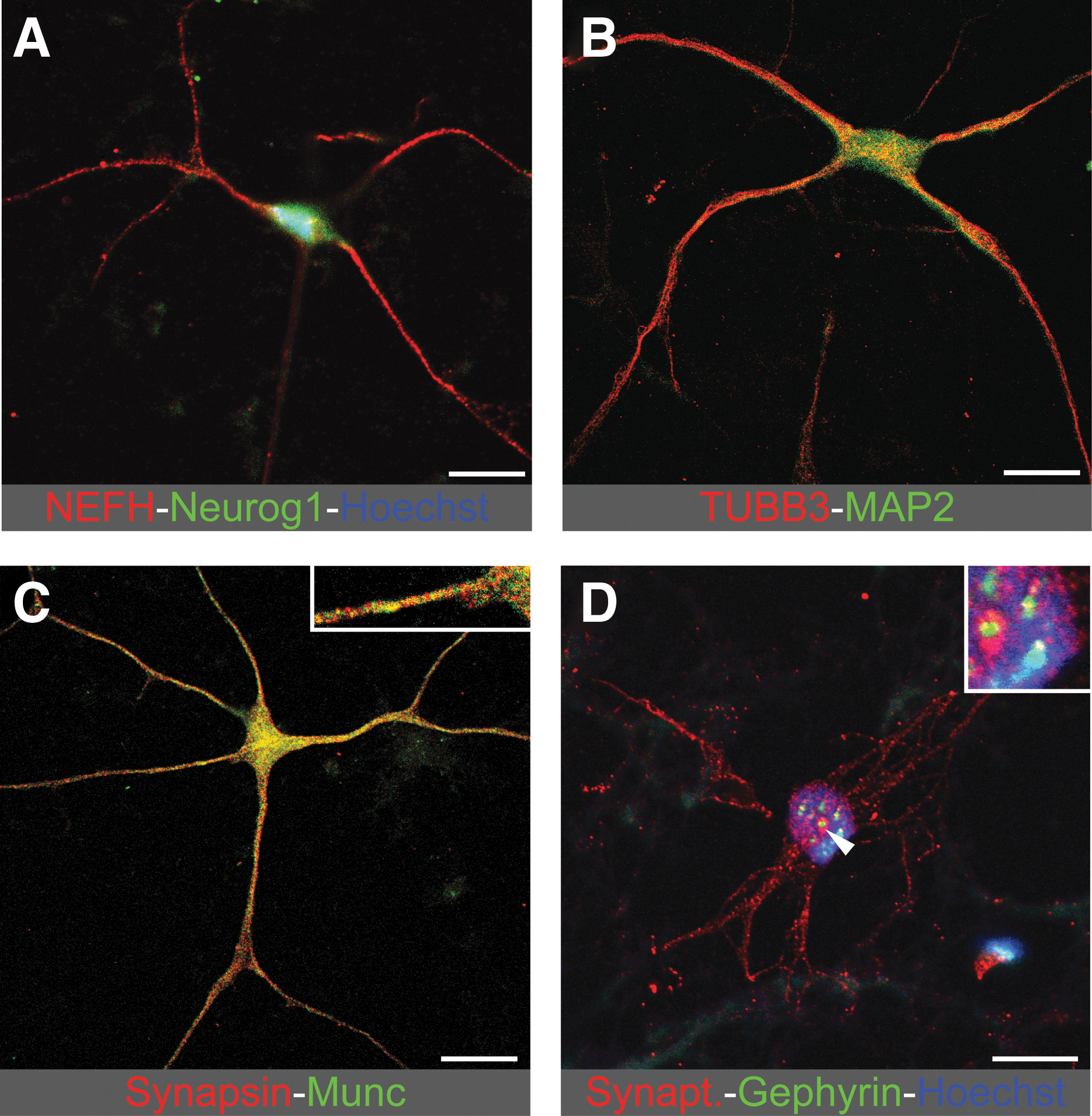
Induced expression of neurogenic and neuronal proteins in ASCs. Confocal microscope images of immunostaining for indicated proteins.
Induction of the postmitotic state
Downregulation of nestin has been correlated with cells leaving the proliferative state and becoming postmitotic [31]. Using immunofluorescence, we investigated nestin expression before and after induction of neurogenic differentiation. In line with our RT-PCR data (Fig. 2), the nestin protein was barely detectable in UNSTIM-treated cells, but strongly expressed in the majority of STIM-treated cells (Fig. 4A). During neurogenic differentiation, a gradual decline in nestin immunoreactivity occurred (Fig. 4A), suggesting progression into a postmitotic state.
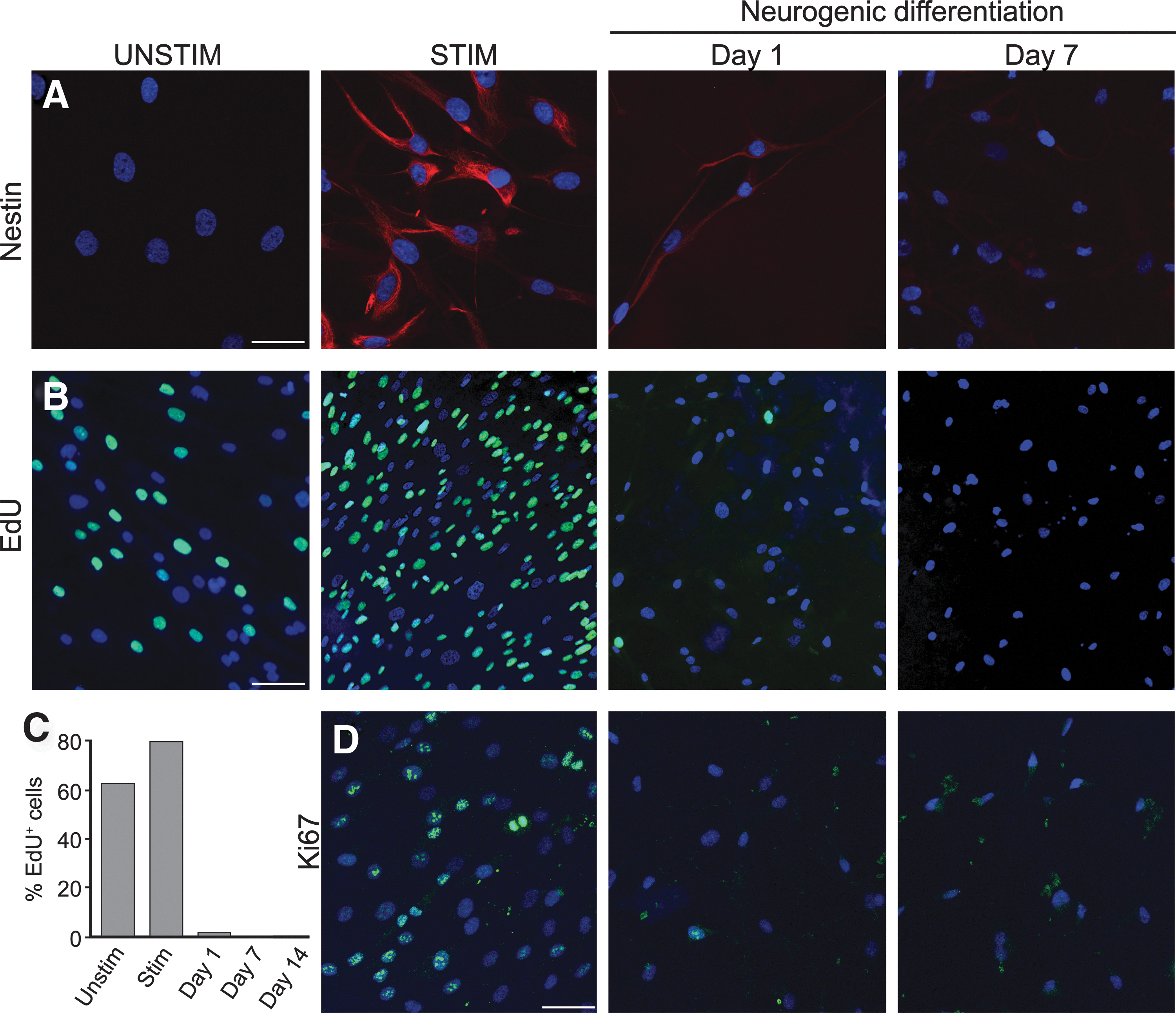
Neurogenic induction arrests the ASC cell cycle.
To further evaluate the effects of neurogenic induction on cell cycle arrest, we performed proliferation assays using a 24-h pulse-chase of EdU, a thymidine analog that incorporates into DNA during replication (Fig. 4B). We found a strong correlation between EdU labeling and nestin immunostaining. As for nestin, there was an increase in EdU labeling under STIM relative to UNSTIM conditions (Fig. 4B), and this was followed by an abrupt decrease on day 1 of neurogenic differentiation to a persistent low level (Fig. 4B, C). Finally, we stained cells for KI-67, a cell endogenous marker tightly associated with cell proliferation. Consistent with nestin expression and EdU incorporation, the majority of STIM-treated cells were positive for KI-67, but the proportion fell markedly during neurogenic differentiation (Fig. 4D). Overall, our results indicate that neurogenically induced ASCs become postmitotic, a characteristic of terminally differentiated cells.
Dynamic and site-specific demethylation and remethylation in the NES regulatory region
The dynamic changes in the expression of NES during STIM conditions and neurogenic differentiation strongly suggest changes in the epigenetic state of the NES gene. To investigate this possibility, we first determined the CpG methylation status within several regions of the NES gene by bisulfite sequencing in UNSTIM- and STIM-treated cells, and after neurogenic differentiation (Fig. 5A). The NES promoter regions examined (P1 and P2; Fig. 5A) remained completely unmethylated in undifferentiated and neurogenic-differentiated cells (Fig. 5B). This is consistent with its location in a CpG-rich region [51] and suggests that NES expression is not regulated by DNA methylation of the promoter.
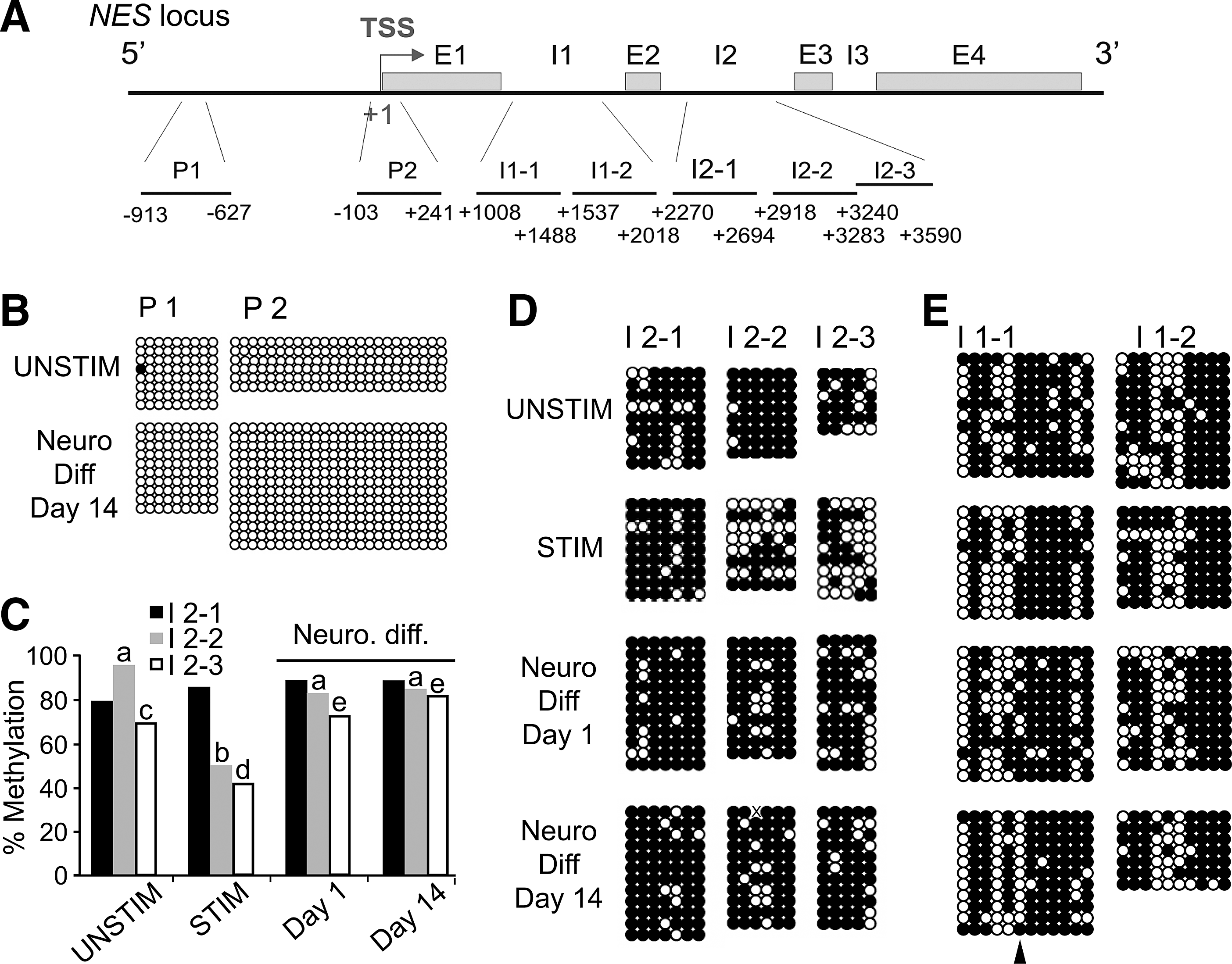
CpG methylation in the nestin gene (NES).
The second intron of NES (I2; Fig. 5A) contains neural-specific enhancer elements, notably the midbrain enhancer (in region I2-2) and the general CNS enhancer (in region I2-3). NES I2 as a whole showed marked DNA demethylation during STIM conditions (P≤0.02 relative to UNSTIM conditions, Fisher's test; Fig. 5C), and demethylation was remarkably restricted to those regions where the midbrain and CNS enhancers are believed to reside (Fig. 5C, D; STIM; I2-2, P<0.0001; I2-3, P=0.020; Supplementary Fig. S4A). In contrast, the 5′ region of I2 (I2-1) remained methylated regardless of STIM treatment or differentiation state (P>0.05; Fig. 5C, D). Thus, mitogenic stimulation of ASCs elicits demethylation of the neuronal and CNS enhancers of NES. During neurogenic differentiation, both of these enhancer regions were remethylated by day 1 (I2-2, P<0.001; I2-3, P=0.002) and remained methylated through day 14 (Fig. 5C, D). These methylation patterns are consistent with the transcriptional changes elicited by mitogenic stimulation and neurogenic induction of ASCs (see Fig. 2), in that CpG demethylation may create a permissive state for TF binding at these sites, enabling transcriptional upregulation.
We next assessed DNA methylation in the first intron of NES (I1; Fig. 5A), which is important for myoblast differentiation [34]. I1 is largely methylated in UNSTIM-treated ASCs and retains its global methylation level after STIM treatment and neurogenic differentiation (P>0.05; Fig. 5E; Supplementary Fig. S4B). We note, however, the demethylation of one specific CpG in the I-1 region (Fig. 5E, arrow; P<0.001) between UNSTIM-treated ASCs and day-14 neurogenic-induced ASCs. Although the muscle-specific enhancer of NES within I1 contains several E-boxes essential for enhancer activity [34], we could not identify any binding sites for known TFs overlapping this demethylated CpG in I1. Whether this region also plays a role in the regulation of NES expression during neurogenic differentiation of MSCs remains undetermined.
Changes in histone methylation in the promoter and the enhancer of NES
DNA methylation has been correlated with NES repression; however, DNA demethylation is not sufficient to mediate activation of NES transcription [52]. We therefore examined by ChIP-qPCR the enrichment of the NES promoter and I1 and I2 (see Supplementary Table S3 for the genomic position of the regions examined in the NES gene) in posttranslational modifications of histone H3 characteristic of transcriptionally permissive chromatin (H3K4me3) or repressive chromatin (H3K27me3 and H3K9me3).
UNSTIM ASCs were marked by H3K4me3, as well as H3K27me3 and H3K9me3 on the NES promoter, consistent with absent or very weak NES expression in these cells (Fig. 6A; see Fig. 2). The GAPDH promoter was in contrast marked by H3K4me3 only, in line with expression of this housekeeping gene (Fig. 6D; see Fig. 2). Whether codetection of H3K27me3 and H3K9me3 on the NES promoter reflects a dual repression mechanism in all cells or a differential means of transcriptional repression in distinct subpopulations of cells in the culture remains unknown at this stage. Regardless, the unmethylated state of the NES promoter in UNSTIM ASCs (Fig. 5B), suggests that NES is kept in a repressed state by H3K27me3 and/or H3K9me3. We also note occupancy of NES I1 and I2 by trimethylated H3K4, H3K9, and H3K27 in UNSTIM ASCs (Fig. 6A–C), indicating a spreading of these marks downstream of the transcription start site. STIM treatment promoted H3K27 and H3K9 demethylation of the NES promoter, I1 and I2 (Fig. 6A–C), consistent with induction of NES expression (Fig. 2). H3K27 and H3K9 demethylation of I1 (Fig. 6C), in the absence of CpG demethylation, suggests incomplete (or abortive) priming of NES for activation of the muscle-specific enhancer. In contrast, histone demethylation and CpG demethylation of I2 under STIM conditions suggest priming of the neural-specific enhancers for activation. After neurogenic differentiation, the NES promoter, I1 and I2 regained repressive H3K27me3 and H3K9me3 methylation (Fig. 6A–C), in line with inactivation of the gene (Fig. 2). Histone remethylation within I2 notably paralleled DNA remethylation in the midbrain and general CNS enhancers (Fig. 5D). These results are consistent with an epigenetic regulation of NES expression. Incidentally, we note occupancy of the GAPDH promoter by H3K27me3 and H3K9me3 after neurogenic differentiation (Fig. 6D). This is consistent with the ASCs becoming postmitotic (Fig. 4), and therefore less energy demanding than undifferentiated, proliferative ASCs.
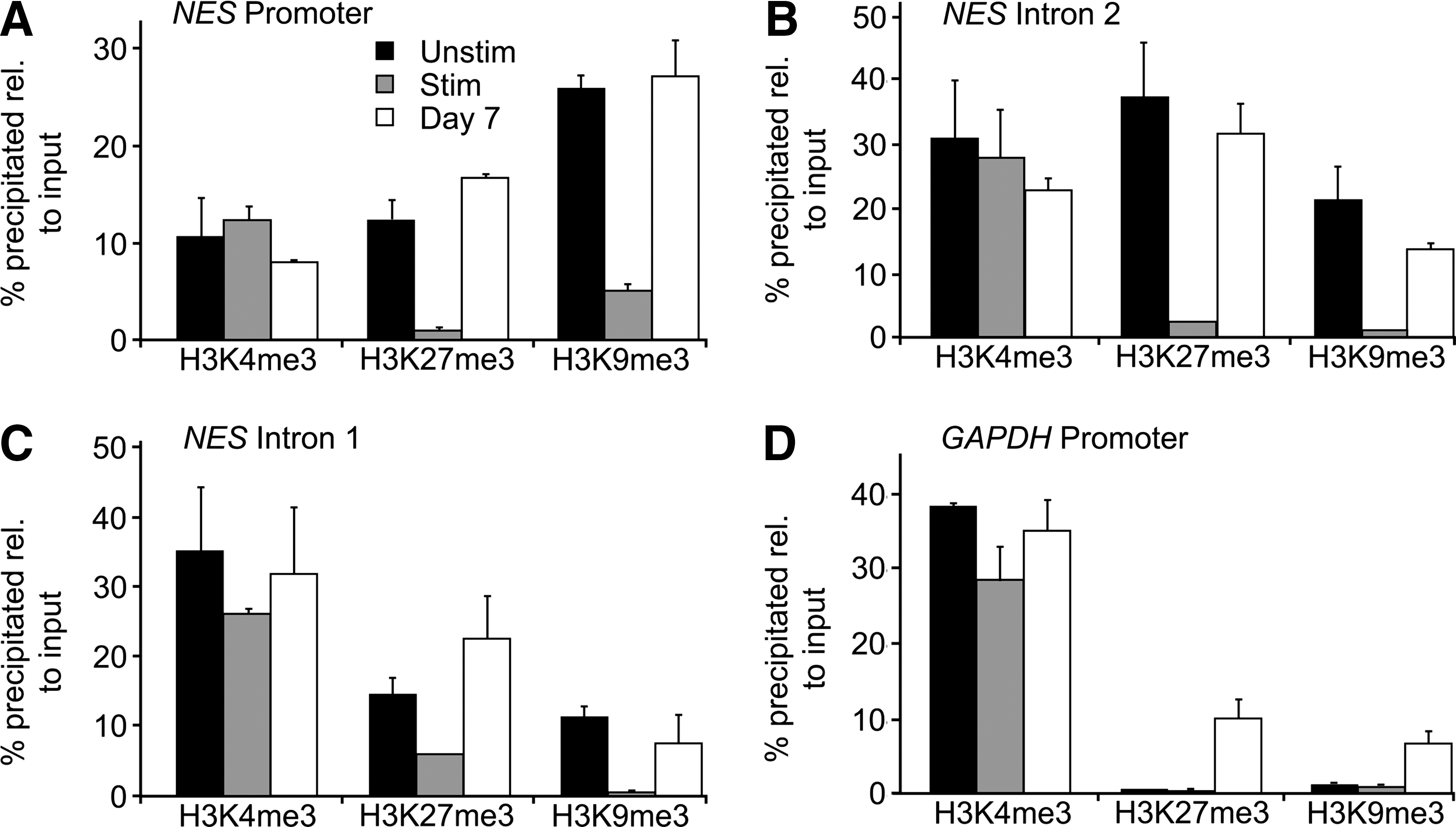
Histone modifications associated with the NES and GAPDH loci. ChIP-qPCR analysis of H3K4me3, H3K27me3, and H3K9me3 occupancy on the
Discussion
Proliferation and differentiation potential of ASCs following mitogenic stimulation
Our findings indicate that intense bFGF- and EGF-mediated mitogenic stimulation of ASCs confers 2 main benefits with respect to potential translational applications. First, it enables potent cell amplification over a relatively short time, with cell numbers ∼10-fold higher within a week compared to standard culture. Importantly, this rapid proliferation does not correlate with signs of neoplastic transformation, based on expression patterns of cell cycle-related genes, including tumor suppressors, or with the absence of proliferation of STIM cells in in vivo transplantation studies (J.L.B., M. Züchner, A.C.B., P.C., and J.C.G., unpublished data). Further, STIM ASCs induced to differentiate become postmitotic, supporting the view that cell cycle control is maintained. Thus, expansion of ASCs by STIM treatment is likely to be useful from a clinical perspective. Second, mitogenic stimulation leads to subsequent improved differentiation efficiency into adipogenic and osteogenic lineages, a property likely due to the proliferation requirement for differentiation into these lineages. Incidentally, chondrogenic (Supplementary Fig. S3) and endothelial (A.C.B. and P.C., unpublished data) differentiation potential is not improved, consistent with the absence of requirement for active proliferation before induction of these lineages. Additionally, mitogenic stimulation strongly primes ASCs for neurogenic differentiation, and therefore broadens the differentiation repertoire of ASCs to include an ectodermal lineage.
Upregulation of neurogenic and neuronal markers in ASCs
Gene expression and immunofluorescence data show substantial upregulation of nestin by STIM treatment. In agreement with reports showing that nestin is a marker of neurogenesis [32] and of proliferation of neural progenitors [30,31], we show that efficiency of neurogenic induction is greater in STIM-treated ASCs than in ASCs expanded in the standard medium. FGFs promote development of the nervous system [27] and both EGF and bFGF are critical for neurosphere growth [53 –55]. Neurogenic induction after STIM treatment elicits a neuron-like morphology in the majority of ASCs, accompanied by expression of neuronal markers, including neurofilament H, NeuroG1, beta-3-tubulin, and MAP2. A subset of cells expresses the synaptic markers synaptotagmin, gephyrin, synapsin, and MUNC13, supporting a neurogenic differentiation, although incomplete synaptic maturation during the time period studied. Note that we do not detect the astroglial marker GFAP, indicating that ASCs differentiated under these conditions are directed predominantly toward a neuronal as opposed to a glial fate. This may be explained by the presence of EGF in the STIM medium before neurogenic induction, as EGF tends to inhibit ASCs from entering astrocytic lineages [17].
DNA methylation changes associated with neurogenic induction of ASCs
Regulation of NES expression is conferred by 2 enhancer elements in the 3′ region of the NES intron 2, one specific for the developing midbrain and one targeting expression generally to the CNS [37]. These regions are methylated in UNSTIM ASCs, consistent with the low-level NES transcription in these cells. In mitogenically stimulated (STIM) ASCs, however, which strongly express nestin, we find that both enhancers become demethylated. This is likely to enable recruitment of TFs, such as SOX and POU family TFs and nuclear receptors, such as RARs, RXRs, and COUP TFs, that are involved in neurogenesis. Similarly, demethylation of the NES midbrain enhancer upon mitogenic stimulation may enable a permissive state for binding of Nurr1, an orphan nuclear receptor implicated in NES midbrain enhancer regulation [39]. Demethylation of the NES neuronal enhancer regions during mitogenic ASC stimulation may therefore prime ASCs for NES expression and neurogenic differentiation. The 5′ most region of NES intron 2, however, remains methylated. This region harbors an enhancer that confers suppression of neural enhancer activity in a neuronal stem cell line, yet activates NES expression in astrocytes [56]. This is consistent with the lack of ASC differentiation toward the astrocytic lineage under the differentiation conditions used here. In contrast to neuronal differentiation, myogenic differentiation of C2C12 myoblasts is regulated by a muscle-specific enhancer in the first intron of NES [34]. We show here a CpG-specific methylation change in NES intron 1 between unstimulated ASCs, stimulated ASCs, and neurogenic-induced ASCs (in contrast to intron 2, which displays more global changes). Whether intron 1 plays a role in NES expression during neurogenesis or whether ASCs also acquire a myogenic potential under the differentiation conditions used here is worthy of further investigation.
We have proposed earlier that promoter CpG methylation is not the primary determinant of the expression of lineage-specific genes in ASCs [57], nor is DNA demethylation sufficient to induce nestin expression [52]. We show here that in unstimulated, undifferentiated ASCs, the NES promoter and neural enhancer are marked by trimethylation of H3K4 and H3K27. This suggests a transcriptionally permissive and repressive bivalent chromatin state of NES in these cells [58], a condition that we identified on adipogenic promoters [59]. This potential dual marking also raises the possibility that ASCs are epigenetically primed for neurogenic differentiation. This view is reinforced by the acquired unmethylated state of the NES promoter upon mitogenic stimulation. This primed state is thought to be critical for determining the differentiation potential of neuronal precursors during development [60].
Our findings suggest that ASCs are epigenetically prepatterned to differentiate toward a neural cell fate. This prepatterning is enhanced by mitogenic stimulation preceding neurogenic induction, which promotes demethylation of critical NES enhancer elements. The neurogenic potential of adipose-derived progenitor cells is consistent with the view of a differentiation capacity of these cells toward the ectodermal lineage. Our findings provide molecular evidence that the differentiation repertoire of ASCs may extend beyond mesodermal lineages. Understanding the chromatin-linked principles governing lineage-specific differentiation of MSCs within and beyond their germ layer of origin may lead to novel avenues for manipulating the chromatin state and cell fate of ASCs in connection with therapeutic applications.
Footnotes
Acknowledgments
The authors are grateful to Kristin Vekterud and Amila Topalovic for assistance in the laboratory, and Jan E. Brinchmann and Aboulghassem Shahdadfar for expertise in flow cytometry. This work was funded by the Norwegian Center for Stem Cell Research, the University of Oslo, and grants from the Research Council of Norway to J.C.G. and P.C.
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.