
Abstract
Several strategies have been developed to facilitate the prospective isolation of bone marrow-derived mesenchymal stem/stromal cells (BM-MSCs) based on the selective expression or absence of surface markers. Recently, we described the monoclonal antibodies W3D5 and W5C5, which selectively react with BM-MSCs, but not with hematopoietic cells. Both antibodies showed an identical reactivity pattern, indicating that they may recognize the same molecule. To identify the cognate antigen, cultured MSCs were sorted for cells expressing either very high levels of W5C5/W3D5 antigen or for cells which were negative for this antigen. Further processing of these cells for microarray analysis revealed a 20-fold enrichment of the type 1 integral membrane protein Sushi domain containing 2 (SUSD2) in the in W5C5+ subset. To confirm the identity of the W5C5/W3D5 antigen to SUSD2, HEK293 cells were transfected with the full-length coding sequence of human SUSD2 followed by reactivity analysis of W5C5 and W3D5 antibodies with the transfected line. Flow cytometric analysis showed that both antibodies selectively recognized HEK293/huSUSD2 cells, but not the parental cell line. In line with this, SUSD2 siRNA treatment of SUSD2+ WERI-RB-1 retinoblastoma cells reduced the expression levels of W3D5 and W5C5 antigens to ∼39% and 37%, respectively. Finally, FACSorting and colony assays revealed that only SUSD2+, but not SUSD2− BM cells give rise to colony-forming units-fibroblasts and are able to differentiate into osteoblasts, adipocytes, and chondrocytes. In conclusion, we identified SUSD2 as a novel and specific marker for the prospective isolation of BM-MSCs.
Introduction
M
In conventional protocols, unfractionated bone marrow (BM)-derived cells are used as the starting population for the culture of MSCs. This isolation process is hampered by the unpredictable influence of cocultured hematopoietic or other unrelated cells and by the potential removal of late adhering MSCs after media replacement. The remaining adherent cells are not completely defined and give rise not only to heterogeneous MSC populations, but also to osteoblasts and/or osteoprogenitor cells, fat cells, reticular cells, macrophages, and endothelial cells [16,17]. To circumvent these limitations, many techniques for the isolation of human BM-MSCs have become available. In particular, immunomagnetic isolation and fluorescence-activated cell sorting (FACS) using MSC-specific markers have been employed. These markers include STRO-1, CD73, GD2, and CD271 for the positive selection and CD45 and CD235 for the negative selection [18 –24].
Previously, we have introduced a panel of novel surface molecules expressed on primary human MSCs or MSC subsets, including CD56, CD140b, HER-2/erbB2 (CD340), frizzled-9 (CD349), TNAP, and SSEA-3, as well as cell-surface antigens defined by the antibodies W1C3, W3D5, W4A5, W5C4, W5C5, W7C6, 9A3, 58B1, F9-3C2F1, and HEK-3D6. Of these, CD56, TNAP, frizzled-9, SSEA-3, and CD140b have been proved to be suitable for the prospective isolation of MSCs [3,18,19,25 –28].
Sushi domain containing 2 (SUSD2) is a recently identified type I transmembrane protein of 820 amino acids consisting of a large extracellular region containing a Somatomedin B (SMB), an adhesion-associated domain in MUC4 and other proteins (AMOP), a von Willebrand factor (vWF), and a Sushi domain (Fig. 1). SMB is a small cysteine-rich serum factor of unknown function, proteolytically cleaved from the N terminus of the cell–substrate adhesion protein vitronectin [29]. Cysteine-rich SMB-like domains are found in a number of extracellular proteins, including the membrane glycoprotein PC1, ENPP-1, ENPP-3, and placental protein 11 [29,30]. AMOP domains are considered to be involved in cell adhesion and represent an important component of MUC4, the most related protein to SUSD2 [31 –33]. Similar to SMB domains, AMOP domains contain a high number of cysteine residues. It is supposed that these residues play a role in homodimerization by forming disulfide bridges between adjacent molecules. The vWF domain is found in a variety of plasma proteins, integrins, and collagens [34,35]. Proteins containing vWF domains participate in numerous biological events, including cell adhesion, migration, homing, and signal transduction, and are involved in interactions with a large array of ligands [34]. Sushi domains, also known as the Complement Control Proteins (CCP) module or Short Consensus Repeats, are components of a variety of complement and adhesion proteins [36]. The function of the short cytoplasmic region of SUSD2 is not known. In a recent report, SUSD2 was described to be differentially expressed in skeletal muscle during the development of obesity [37] and to inhibit cellular growth and to reverse tumorigenic phenotypes of cancer cells in vitro [38,39]. The biological role and molecular mechanism of action of SUSD2 in tumor cells was investigated by analyzing the effect of SUSD2 overexpression in human HT1080 fibrosarcoma and in HeLa cervical carcinoma cells, which resulted in reduced or abrogated tumorigenic features such as the anchorage-independent growth, migratory and invasive activity of the cells [39].

Domain architecture of the SUSD2 protein. SUSD2 consists of a type I single spanning TM region separating a short cytoplasmic tail with unknown function from a large extracellular region containing SMB, vWF, AMOP, and Sushi domains. SUSD2, Sushi domain containing 2; SMB, Somatomedin B; AMOP, adhesion associated domain in MUC4 and other proteins; vWF, von Willebrand factor; TM, transmembrane.
Very recently, the MSC marker W5C5 was introduced as a marker for the prospective isolation of highly clonogenic endometrial MSCs, which were able to reconstitute endometrial stromal tissues in vivo [40,41]. In this report, we identify SUSD2 as the cognate antigen of antibodies W5C5 as well as of W3D5 and introduce this molecule as a novel stand alone marker to isolate human BM-MSCs.
Methods
Isolation of cells from peripheral blood and bone marrow
Bone marrow was harvested from the femoral shafts of patients undergoing total hip replacement at the BGU Center for Traumatology, Tübingen, Germany. The age of patients (three female, three male) ranged from 45–73 years. Cells were collected in 5,000 U heparin (Sigma-Aldrich) after informed consent and approval of the Ethics Committee of the University of Tübingen. Mononuclear cells (MNCs) were isolated by Ficoll Histopac (Biochrom AG) density gradient fractionation and remaining erythrocytes were lysed in an ammonium chloride solution. Peripheral blood (PB) samples from healthy volunteers were drawn in Vacutainer tubes at the University clinic of Tübingen after informed consent and approval of the Ethics Committee of the University of Tübingen. Before analysis, erythrocytes were lysed in the ammonium chloride solution.
Culture of BM-MNC
2×107 Ficoll-separated or 1×104 FACS-enriched bone marrow cells were cultured in 0.1% gelatin-coated T-75 or T-25 culture flasks in MSC media containing knockout Dulbecco's modified Eagle medium (DMEM) supplemented with 20% knockout serum replacement (Invitrogen), 1 mM
CFU-F assays
CFU-F assays were performed by plating either 1×105 unselected or 500–5,000 FACS selected BM-MNCs in 0.1% gelatin-coated T-25 flasks in the presence of the MSC medium. After 10–14 days of culture, adherent cells were washed twice with phosphate-buffered saline (PBS), fixed with methanol (Sigma-Aldrich) for 5 min at room temperature, air-dried, and stained with the Giemsa solution (Merck). CFU-F colonies were macroscopically enumerated. Colonies consisted of at least 50 cells.
Osteoblast and adipocyte differentiation
Passage 2 MSCs derived from sorted or unfractionated BM cells were cultured in the NH OsteoDiff or NH AdipoDiff medium (Miltenyi Biotec), respectively. In brief, 2×104 (osteogenesis) or 4×104 (adipogenesis) MSCs were cultured in 24-well Falcon plates (Becton Dickinson). For negative controls, MSCs were grown in the MSC medium in place of the differentiation medium. After 10 days of culture in the NH OsteoDiff medium, an alkaline phosphatase (AP) activity in osteoblasts (methanol-fixed, 5 min, −20°C) was determined using the FAST BCIP/NBT substrate (Sigma-Aldrich). Calcium deposition in fixed cells (4% paraformaldehyde, 15 min) was analyzed by staining with 2% Alizarin Red S (Merck) for 10 min at room temperature. The formation of adipocytes was evaluated after 25 days of culture in the NH AdipoDiff medium and staining of methanol-fixed cells with Oil Red O dye (Sigma-Aldrich) for 45 min at room temperature. Pictures were taken using an Observer.Z1 AX10 microscope (Carl Zeiss).
Chondrocyte differentiation
MSCs (4×105 cells) derived from sorted or unfractionated BM cells were cultured as a pellet for 4 h in a U-bottom 96-well plate at 37°C in 20 μL of the chondrogenic induction medium containing DMEM-high glucose (PAA Laboratories) supplemented with 1% ITS, 175 μM
Generation of MSC-reactive mAbs W3D5 and W5C5
The mAbs W3D5 (IgG2a) and W5C5 (IgG1) were raised by immunization of a 6–8-week-old female Balb/c mouse (Charles River WIGA) with the retinoblastoma cell line WERI-RB-1, as previously described [18]. The isotypes of mAbs were determined by enzyme-linked immunosorbent assay (ELISA) (Boehringer Mannheim) [18].
Immunofluorescence staining for flow cytometry
Phycoerythrin (PE)-conjugated antibodies against CD34, CD45 CD73, and CD133, fluorescein isothiocyanate-conjugated antibodies against CD2, CD3, CD4, CD14, CD20, CD34, CD45, CD71, and CD235a, and allophycoyanin-conjugated antibodies against CD271 and CD90 were purchased from Becton Dickinson. PE-conjugated antibodies against CD140b, CD164, CD166, and CD200 were purchased from BD PharMingen. PE-conjugated antibodies against CD29, CD105, W3D5, and the Brilliant violet 421 (BV)-conjugated antibodies against CD45 and CD235a were purchased from BioLegend. The PE-conjugated W5C5 antibody was obtained from Miltenyi Biotec. Unconjugated W5C5 and W3D5 antibodies were generated in-house as described [18].
BM-MNCs were washed twice with PBS (Lonza) containing 1% fetal bovine serum (FBS) and 0.01% NaN3 (FACS buffer) and incubated with polyglobin for 15 min on ice to block the nonspecific binding. In the next step, the cells were stained with direct antibody conjugates or with unlabeled proprietary antibodies for 15 min on ice. Cells labeled with proprietary antibodies were stained with F(ab)2 fragments of a R-PE-conjugated goat anti-mouse antibody (Dako Cytomations) for 15 min, washed twice, and analyzed on a FACSCanto flow cytometer (Becton Dickinson) and FCS express software (De Novo Software) or FlowJo software (Tree Star, Inc.).
Immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde in PBS for 15 min at 4°C, permeabilized with 0.2% Triton X-100 in PBS for 5 min, and blocked in 5% bovine serum albumin (BSA) for 1 h at room temperature. After blocking, cells were incubated with 10–20 μg/mL of the W5C5 antibody for 2h at room temperature or 4°C overnight. In the next step, cells were incubated with goat anti-mouse IgG-conjugated Alexa Fluor 488 (Invitrogen) for 1h at room temperature. Cells were mounted with 4′6′- diamidino-2 phenylindole (DAPI) containing the Vectashield mounting medium (Vector Labs) and photographed with the Zeiss Observer.Z1 AX10 microscope with ApoTome and AxioVision 4.8 imaging software 488 (Carl Zeiss).
Isolation of W5C5+ cells and W5C5− cells by magnetic activated cell sorting and FACS
In selected experiments, the cells were pre-enriched by magnetic activated cell sorting (MACS) using the W5C5-PE microbead kit (Miltenyi Biotec), according to the instructions provided by the manufacturer. In brief, at least 108 Ficoll-isolated BM cells were incubated with 100 μL of the W5C5-PE antibody together with the Fc receptor blocking reagent for 30 min. After washing, the cells were incubated with 200 μL of anti-PE microbeads for 15 min and loaded onto a LS MACS column. After separation, unbound cells were collected and used as the negative fraction. W5C5+ cells retained on the column were eluted with 3 mL of PBS containing 0.5 M EDTA and 0.5% bovine serum albumin (BSA; Sigma-Aldrich). In the next step, MACS-selected W5C5+ and W5C5− cells were further purified on a FACSAria cell sorter (Becton Dickinson). FACS-sorted W5C5+ cells were exclusively derived from MACS-selected W5C5+ cells and FACS-sorted W5C5− cells from MACS-selected W5C5− cells.
Gene chip analysis of W5C5+/− MSC
Passage 1 MSCs derived from unseparated BM-MNCs were stained with the W5C5-PE antibody and fractionated by FACS gating on W5C5bright and W5C5− cells, respectively. About 2.5–5×105 cells from each sample were used for commercial gene chip analysis (Miltenyi Biotec) to perform human whole genome oligo microarray (Agilent Technologies). RNA was isolated using standard RNA extraction protocols (RNeasy, Qiagen, Hilden, Germany). In brief, 25 ng RNA from each sample was amplified and labeled with Cy3 using the Agilent Low Input Quick Amp Labeling Kit (Agilent Technologies). The samples were hybridized for 17 h at 65°C on Agilent's Whole Human Genome Oligo microarray using the Agilent Gene Expression Hybridization Kit. Gene chip scanning and data analysis was carried out using the Microarray Scanner System and The Agilent Feature Extraction Software (Agilent Technologies).
Real-time RT-PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen) and treated with DNase I (Invitrogen) according to the manufacturer's instructions. One microgram of RNA was reverse transcribed using the ImProm-II Reverse Transcription system (Promega) as described by the manufacturer's protocol. Real-time quantitative PCR was performed using the SYBR Green reagent (Eurogentec) on a Light Cycler480 Real-time PCR instrument (Roche, Mannheim, Germany). Primer sequences were used as listed in Table 1. All primers were used at annealing temperature of 58°C.
Generation of HEK293/huSUSD2 transfectant cell line
Transfection-ready ORF clone of Homo sapiens SUSD2 (accession No.: NM_019601.3) was obtained from OriGene. HEK293 cells were selected for the transfection because of their lack of reactivity with the antibody W5C5. About 3×105 cells were transfected according to the manufacturer's protocol in the presence of Xtreme Gene 9 DNA transfection Reagent (Roche) in the RPMI 1640 medium containing 10% FBS (PAA Laboratories) for 24 h. The transfected cells and mock transfected cells were grown for additional 2 days in the RPMI 1640 medium containing 10% FBS, 2 mM
siRNA treatment
Trilencer-27 siRNAs (includes three unique 27mer siRNA duplexes; Cat. No. SR311132) for human SUSD2 were purchased from OriGene. Transfection was performed on WERI-RB-1 cells using Lipofectamine RNAiMAX reagent 22 (Invitrogen) at siRNA concentrations ranging from 20 to 100 nM. After 24 h of transfection, knockdown efficiency was tested by qPCR analysis. Scrambled universal negative control RNA duplex (OriGene) was used as a negative control.
Cross-blocking analysis of SUSD2-specific mAbs
For epitope mapping studies, 2×10 6 cells/mL WERI-RB-1 cells were incubated with W3D5 or with W5C5 antibody or with isotype-matched negative control antibodies at concentrations of 1–30 μg/mL for 30 min on ice. After washing, cells were stained either with W5C5-PE or with W3D5-PE. After washing, cells were analyzed on a FACSCanto II flow cytometer. The percent blocking was calculated as follows:
100 − [(MFI of cells incubated with blocking mAb and stained with test mAb÷MFI of cells incubated with negative control mAb and stained with test mAb)×100].
Blocking of more than 50% was considered to be significant.
Results
Antibodies W3D5 and W5C5 recognize the extracellular region of SUSD2
In an initial screening effort, the reactivity analysis of W3D5 and W5C5 antibodies with several cell lines revealed that both antibodies showed identical reactivity patterns, indicating that they recognize the same antigen (Supplementary Fig. S1; Supplementary Data are available online at
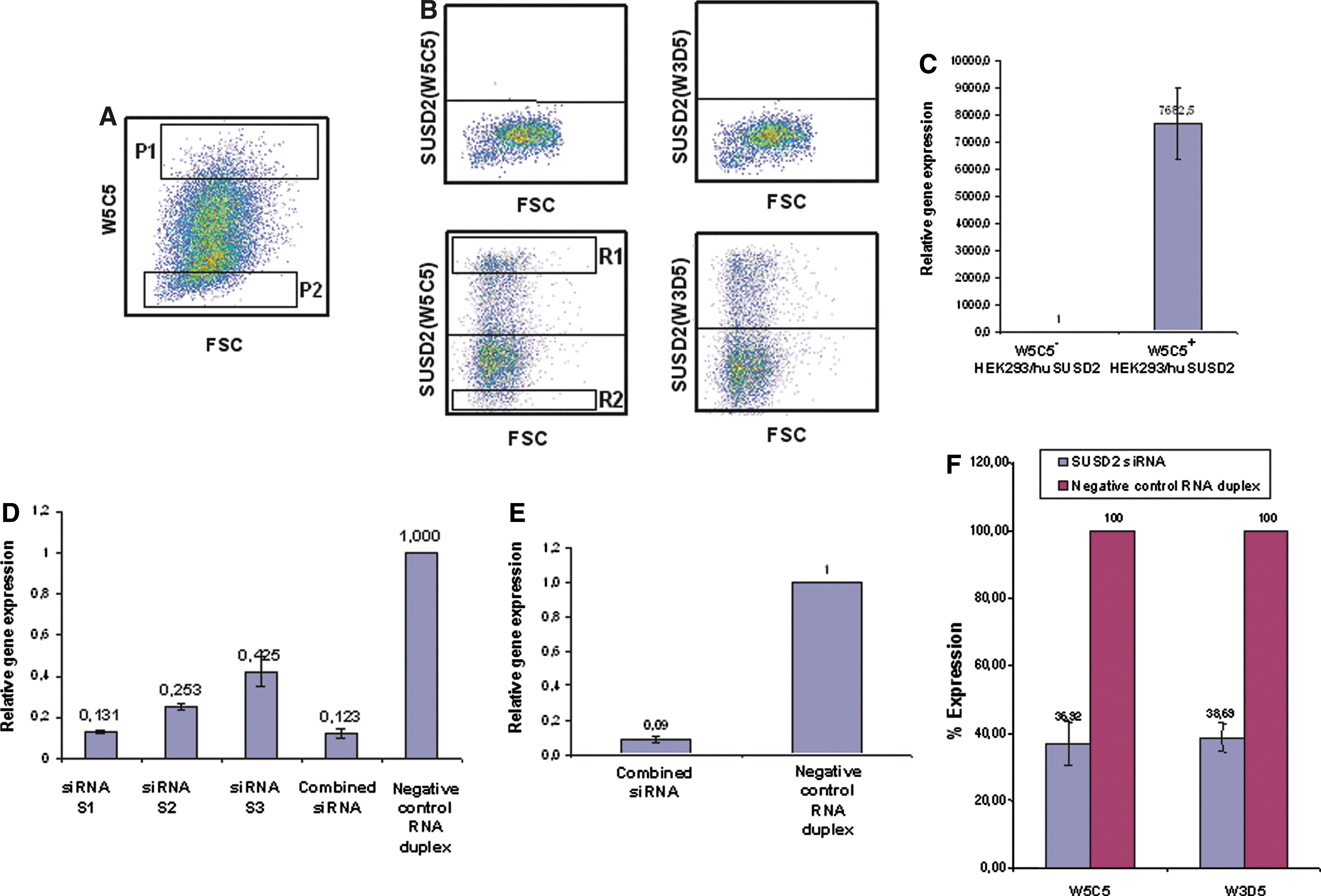
Antibodies W3D5 and W5C5 recognize SUSD2.
To further confirm the specificity of antibodies W3D5 and W5C5 for SUSD2, loss of function experiments were performed by treating cells with SUSD2 siRNA. Negative control RNA duplex or three siRNA sequences S1, S2, and S3 against human SUSD2 were transfected into HEK293/huSUSD2 cells, either individually or in combination. Figure 2D shows that compared to negative control RNA duplex, transfection of HEK293/huSUSD2 cells with S1, S2, and S3 and the combined siRNAs (S1, S2, S3) reduced the steady-state levels of mRNA for SUSD2 to ∼87%, 75%, 58%, and 88%, respectively. As the combined siRNAs were slightly more effective than S1 in reducing SUSD2 mRNA levels, the combined siRNAs were used for further studies. In the next set of experiments, WERI-RB-1 cells were transfected with combined siRNAs. About 24 h after transfection, the steady-state levels of SUSD2 mRNA were analyzed and compared with the negative control RNA duplex treated WERI-RB-1 cells. Figure 2E shows ∼90% reduction of SUSD2 expression in combined siRNA-treated cells compared to control RNA-treated cells. To determine whether SUSD2 cell surface expression is affected by siRNA treatment, the reactivity of W3D5 and W5C5 antibodies was compared on siRNA and negative control RNA duplex treated WERI-RB-1 cells using flow cytometric analysis. As shown in Fig. 2F, the percentage SUSD2 expression of cells stained with W3D5 and W5C5 antibodies was reduced after siRNA treatment to 39% and 37%, respectively, when compared to negative control RNA duplex treated cells. Collectively, these data demonstrate that the target of W3D5 and W5C5 antibodies is SUSD2.
Analysis of SUSD2 expression on PB and bone marrow cells
To analyze SUSD2 expression on hematopoietic cells, PB and BM cells were stained either with W5C5 alone or in combination with lineage-specific antibodies. Flow cytometric analysis revealed that SUSD2 was negative on all hematopoietic cells from both PB and BM, including hematopoietic progenitors, T cells, B cells, leukocytes, macrophages/monocytes, or nucleated erythroid cells (Table 2 and Supplementary Fig. S2). In contrast, SUSD2 expression was detected on CD271bright CD45− nonhematopoietic cells in bone marrow cells (Fig. 3A, B), indicating a selective expression on MSCs.
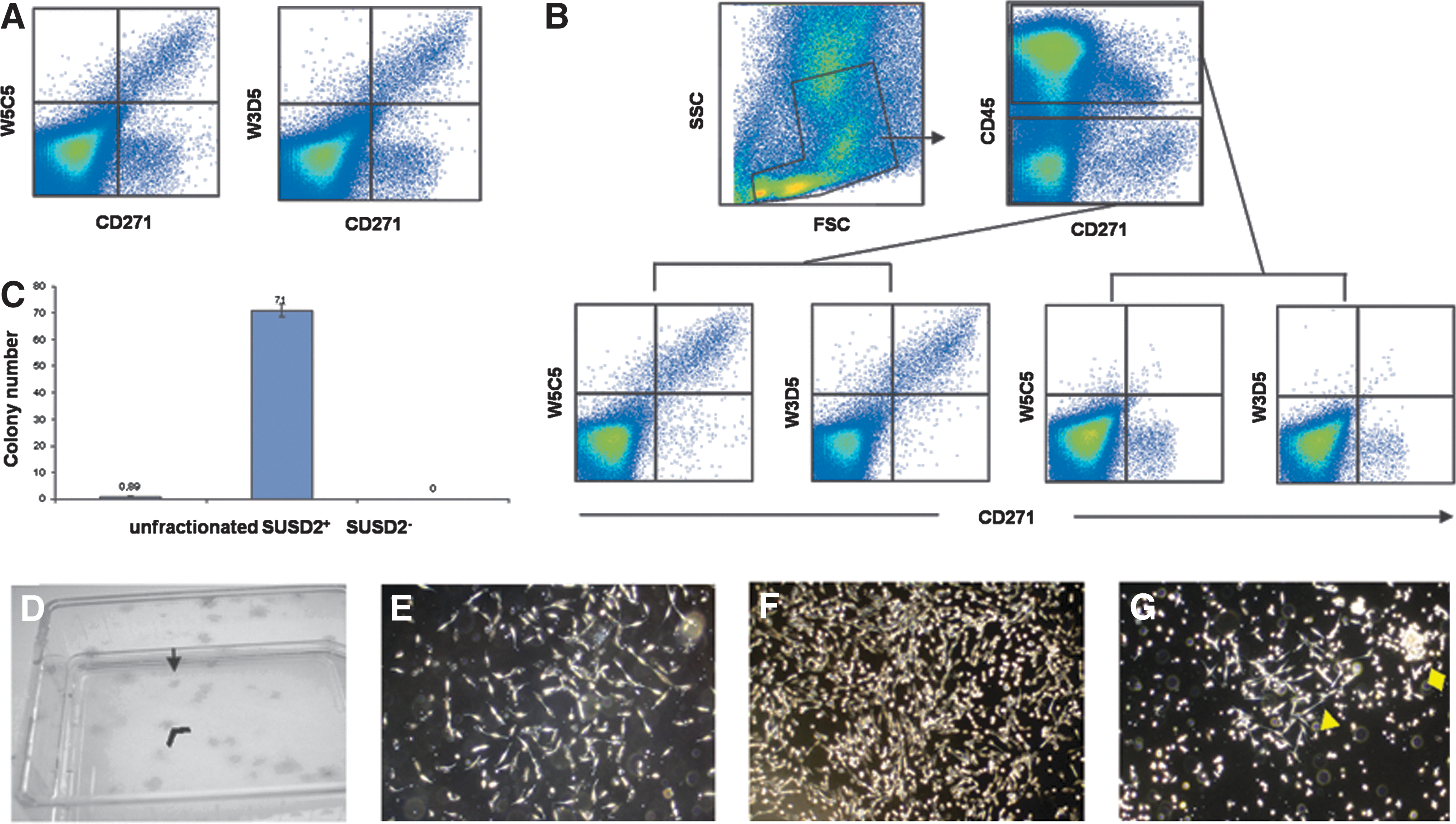
BM, bone marrow; MSC, mesenchymal stem/stromal cells; PB, peripheral blood.
SUSD2+ cells, but not SUSD2− cells give rise to CFU-F
To determine the clonogenic potential of SUSD2+ cells, CFU-F assays of fractionated and unfractionated BM cells were performed. Staining of BM cells with mAb W5C5 revealed that W5C5 exclusively reacted with CD271bright CD45− cells (Fig. 3A, B). After pre-enrichment of SUSD2+ cells by MACS with the W5C5 antibody, cells were further fractionated by FACS sorting into SUSD2+ and SUSD2− cells and plated into culture flasks at defined cell numbers. Enumeration of the resulting CFU-F after 12 days of culture showed that only W5C5+ cells, but not W5C5− cells gave rise to CFU-F. Compared to unfractionated cells, CFU-F were ∼71-fold enriched in the positive fraction (Fig. 3C). Both, small and large colonies were observed in the SUSD2+ fraction (Fig.3D–F) in the approximate ratio of 10:8 (small versus large). Small colonies were more loosely packed with fewer cells compared to the large colonies. SUSD2+ cells gave rise only to fibroblast-like colonies (Fig. 3E, F), while unfractionated cells gave rise to both fibroblast- and macrophage-like colonies (Fig. 3G).
Phenotype of cultured SUSD2+-derived MSCs
To confirm that sorted SUSD2+ BM cells give rise to MSCs fulfilling the standard phenotypic criteria, the selected cells were cultured until passage 2. Adherent cells were trypsinized and stained with markers indicated in Fig. 4. Flow cytometric analysis revealed that MSCs derived from SUSD2+ primary BM cells expressed characteristic MSC markers, including CD29, CD73, CD90, CD105, CD106, CD140b, and CD166, but lacked CD11b, CD34, CD45, and CD133. However, a small subpopulation of MSCs (<1%) was positive for CD31.
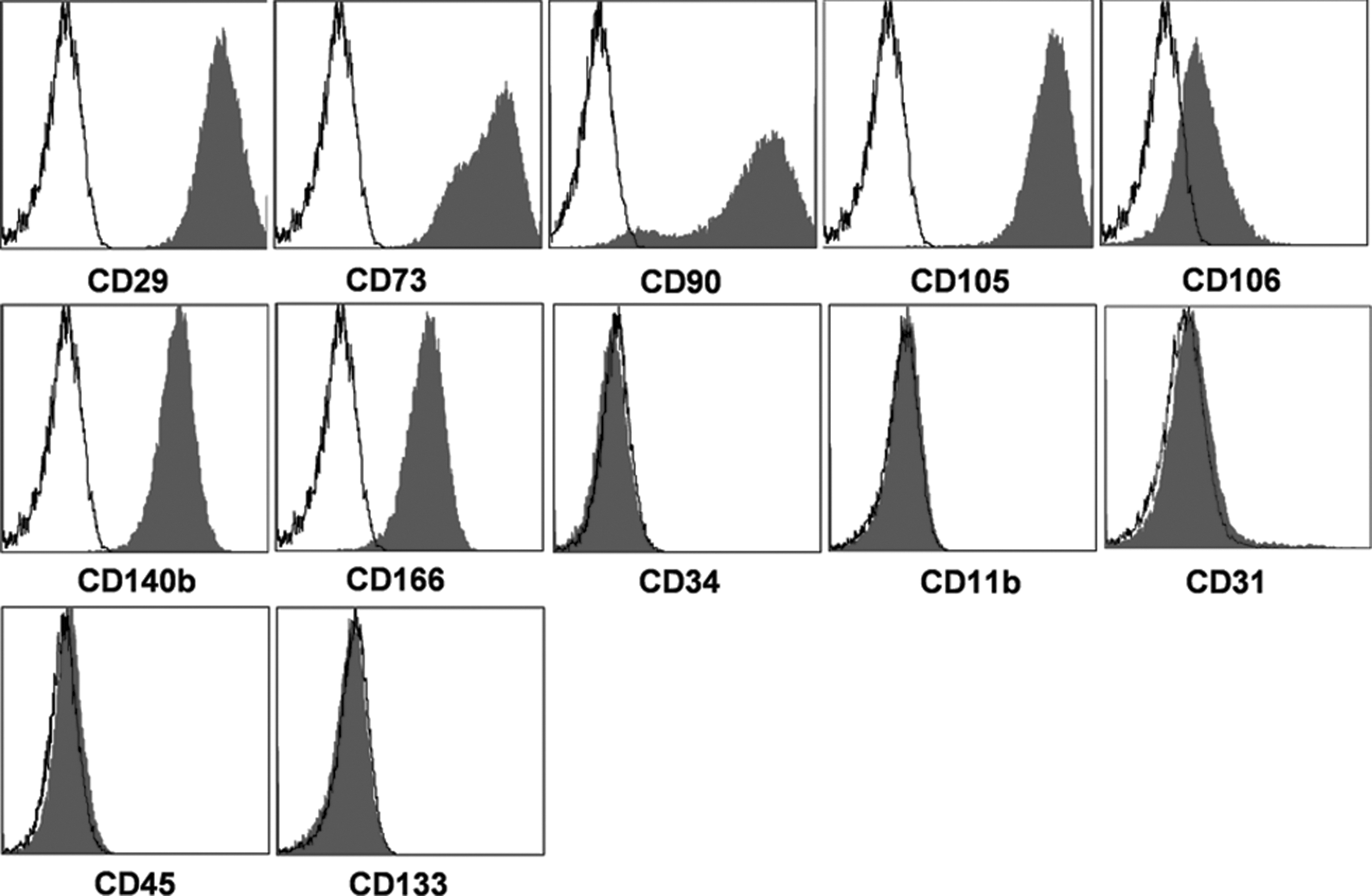
Immunophenotype of cultured MSCs derived from SUSD2+ BM cells. Flow cytometric analysis of passage 2 MSCs demonstrates that cultured MSCs express standard MSC markers CD29, CD73, CD90, CD105, CD106, CD140b, and CD166 and are negative for CD11b, CD34, CD45, and CD133. <1% of MSCs expressed low levels of CD31.
Cultured MSCs derived from SUSD2+ BM cells are able to differentiate into osteoblasts, adipocytes, and chondrocytes
To examine the osteogenic, adipogenic, and chondrogenic differentiation potential of SUSD2+ BM cells, FACS sorted SUSD2+ cells were expanded in the MSC medium for about 3 weeks until passage 2, and the resulting MSCs were induced to differentiate into osteoblasts, adipocytes, and chondrocytes. As determined by Alizarin Red Staining and AP activity, osteoblasts were generated in MSCs derived from SUSD2+ cells after 10 days of culture in the osteogenic differentiation medium, but not in the MSC medium (negative control) (Fig. 5). After induction in the adipogenic and chondrogenic differentiation medium, adipocytes and chondrocytes appeared after 25 and 30 days of culture, as determined by Oil Red O and Alcian Blue staining, respectively. Adipocytes were not detected when cultured in the MSC medium. Chondrocyte pellet formation did not occur when TGF-β3 was omitted in the differentiation medium.
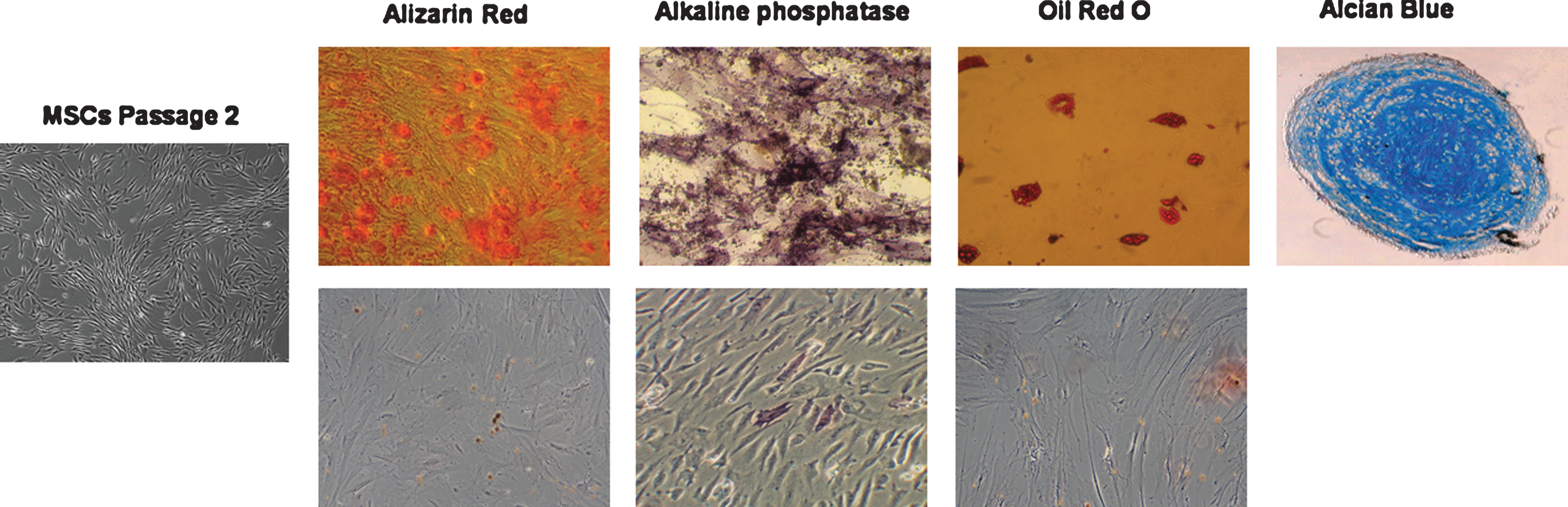
Differentiation potential of MSCs derived from SUSD2+ BM cells. Expanded MSCs were induced to differentiate into osteogenic, adipogenic, or chondrogenic lineages and stained as described in the Methods. Photographs were taken on a Zeiss Observer.Z1 AX10 microscope. Note that MSCs (Passage 2) derived from SUSD2+ BM cells were able to differentiate into osteoblasts (Alizarin Red, alkaline phosphatase), adipocytes (Oil Red O), and chondrocytes (Alcian Blue). The lower panel shows negative controls for differentiation as detected by Alizarin Red or alkaline phosphatase (osteoblasts) or Oil Red O (adipocytes) staining.
Cellular localization of SUSD2 in MSCs, WERI-RB-1, and HEK293/huSUSD2 cells
To analyze the cellular localization of SUSD2, immunofluorescence analysis on a fluorescent microscope was performed. In WERI-RB-1 and HEK293/huSUSD2 cells, SUSD2 accumulated in a honeycomb-like pattern, predominantly at the cell–cell contact sites of the plasma membrane. In contrast, only a weak SUSD2 signal was observed in other regions of the plasma membrane, which were not in contact with other cells (Fig. 6A, B). In cultured SUSD2+ MSCs, SUSD2 accumulated in zipper-like structures similar to filopodia that extend toward the extracellular matrix or interlocked with similar structures from the neighboring cells (Fig. 6C). Weak staining of SUSD2 was detected in the cytoplasm of all the cell types studied.
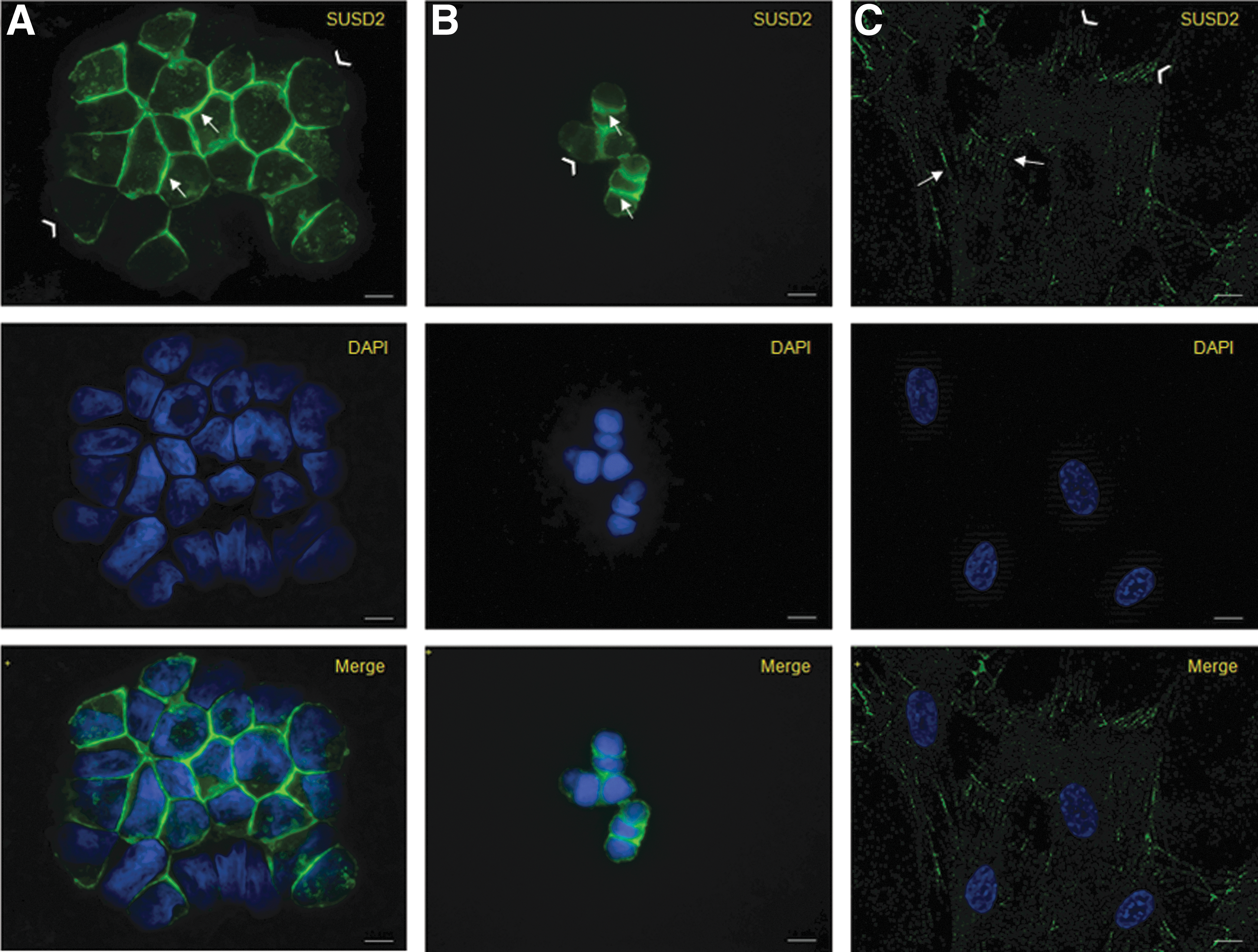
Cellular localization of SUSD2. After 48h of culture, cells were fixed, permeabilized, labeled with the W5C5 antibody followed by staining with Alexa Fluor 488-conjugated secondary antibody and mounted in DAPI-containing media as described in the Methods.
mAbs W3D5 and W5C5 recognize related epitopes of SUSD2
To determine whether W3D5 and W5C5 mAbs recognize identical or distinct epitopes of SUSD2, cross-blocking experiments were performed. As shown in Table 3, incubation of WERI-RB-1 cells with mAb W5C5 significantly blocked the binding of W3D5 and vice versa. Hence, mAbs W3D5 and W5C5 recognize identical or highly related epitopes of SUSD2.
Discussion
Aim of the current study was to identify the target of the MSC-reactive antibodies W5C5 and W3D5 and to verify their specific recognition of MSCs in human bone marrow. By cell sorting of antigen-positive and -negative MSCs and whole genome microarray analysis of the sorted fractions, we observed a 20-fold increased expression of SUSD2 mRNA in the positive fraction. In subsequent experiments, we could demonstrate that both antibodies selectively recognize HEK293/huSUSD2 transfectant cells, but not their parental counterpart. The specificity for SUSD2 was further supported by the fact that SUSD2 mRNA was found to be selectively expressed in SUSD2+-transfected cells and that SUSD2 siRNA treatment of WERI-RB-1 cells significantly reduced SUSD2 cell surface expression. Collectively, we demonstrate that the MSC-reactive antibodies W3D5 and W5C5 recognize a cell surface epitope of SUSD2.
SUSD2 is a type I transmembrane protein of 820 amino acids that localizes in the plasma membrane [38]. Although the SUSD2 protein harbors several functional domains inherent to adhesion molecules, the biological role of SUSD2 remains to be clarified. In our studies, analysis of the cellular localization of SUSD2 with fluorescent microscopy revealed a characteristic pattern of distribution in the cell-to-cell contact region. Our observation is in accordance with the findings of Sugahara et al. who showed an accumulation of SUSD2 in the cell-to-cell contact region of the plasma membrane of HeLa/SUSD2-transfected cells [38,39]. A similar pattern of accumulation and biological activity of membrane proteins at cell-to-cell contact sites has also been reported in the case of other adhesion molecules, including E-cadherin, TSLC-1, and nectins [42 –45]. In line with this finding, large-scale mapping of human protein–protein interactions by mass spectrometry revealed that SUSD2 may be involved in polysaccharide binding through interactions with UGGT1 (UDP-glucose ceramide glucosyltransferase-like 1) or LGALS1 (galactoside-binding, soluble, 1; galectin-1) [46].
A recent report has shown that overexpression of SUSD2 in HeLa cells induced cell aggregation and suppressed migration and invasion of cells [39]. Although the function of SUSD2 appears to be similar to that of a number of studied tumor suppressor molecules [38,39], the final assignment of this molecule as a tumor suppressor requires additional studies focusing on genetic/epigenetic alterations and abnormal expression patterns of SUSD2 in tumors. To get more insight into the biological functions of SUSD2 and to identify a potential extracellular ligand, cell-binding assays are in progress. For this purpose, we generated a soluble fusion protein consisting of the extracellular region of SUSD2 to determine its binding capacity to a large panel of cell lines.
In our report, we could demonstrate that SUSD2 is expressed with high selectivity on human bone marrow-derived MSCs. We have shown that only W5C5+, but not W5C5− cells gave rise to CFU-F and were able to differentiate into osteoblasts, adipocytes, and chondrocytes. This is in line with our previous report, in which we showed that W5C5 and W3D5 selectively recognize CD271bright and no other cells in bone marrow and that MSCs reside in the CD271bright, but not CD271dim population [18]. Although several additional surface markers have been identified for the prospective isolation of BM-MSCs, SUSD2 appears to be the most suitable target for MSC isolation because of its superior selectivity for these cell types. Previously published markers, including CD73, CD105, GD2, TNAP, STRO-1, and several others are not stringently expressed only on CD271bright cells, but are also found on other cell types [47 –52]. Although GD2 was introduced as a suitable marker for the prospective isolation of MSCs [22], we were unable to detect this molecule on primary BM-MSCs, which is in contrast to cultured MSCs, on the surface of which we could confirm a heterogeneous GD2 expression. CD73 and CD105 are not selective for MSCs because these molecules are also expressed on lymphocyte subsets and on erythroblasts, respectively [47,49,51,52], as well as on vascular endothelial cells in the marrow. TNAP (tissue nonspecific AP) is highly expressed on the majority of MSCs, but only weakly positive or negative on bone-lining MSC subsets [3,28,53]. Another negative aspect to choose TNAP as a selection marker is its additional expression on neutrophils [48]. Although STRO-1 is a well-established marker for the isolation of MSCs [24], only STRO-1bright cells in BM consist of MSCs. However, many other cell types in BM express this molecule at lower levels [24,50]. Taken together, SUSD2 appears to be a superior stand alone marker to prospectively isolate primary BM-MSCs at high purity.
Masuda et al. described W5C5 as a novel stand alone marker to purify endometrial MSCs, which can self-renew, differentiate into adipogenic, osteogenic, chondrogenic, and myogenic cell lineages and which are able to reconstitute mesodermal tissue in vivo [40]. In their study, the W5C5 antibody was described to be superior to the previously published markers CD146 and CD140b. Only the combined use of CD146 and CD140b was equally effective for the prospective isolation of endometrial MSCs as W5C5 alone. Immunostaining of endometrial tissue revealed that the W5C5 antigen (SUSD2) is located in perivascular regions of not only the basalis layer, but also of the functionalis layer, which is shed during menstruation and regenerates in the following menstrual cycle. However, the functional role of SUSD2 in this process remains open.
Bone marrow-derived MSCs are the best characterized postnatal stem cells. Due to their niche-forming, immunomodulatory, and regenerative properties, BM-MSCs are not only discussed as a potent niche source for HSCs, but also as attractive cells for applications in the field of regenerative medicine and for cell-based clinical therapies. A major concern about the use of cultured MSCs derived from unselected cells for clinical purposes is the fact that no information about the initiating cell population exists. To overcome these limitations and to avoid the coculture and the growth factor-induced influence of unrelated cells, SUSD2 may serve as a selection marker of choice to isolate MSCs, which may be used for the treatment of an increasing number of diseases, in particular, for the reconstruction of bone, cartilage, muscle, and blood vessels. In conclusion, we introduce SUSD2 as a novel marker for MSCs and show that mAbs W5C5 or W3D5 are useful tools for their selection.
Footnotes
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG), Sonderforschungsbereich SFB-685 (Immunotherapy: Molecular Basis and Clinical Applications) project C10: Development of therapeutic antibodies for the elimination of tumor stem cells, by the DFG project BU 516/2-1: Identification and functional analysis of MSC-specific molecules, and by a research collaboration with TETEC GmbH, Reutlingen, Germany entitled: Isolation and identification of stem cells with adipogenic, chondrogenic, and pancreatic differentiation potential. We thank Sabrina Grimm and Flavianna Cerabona for excellent technical assistance in cell sorting, cell culture, and antigen identification experiments.
Note Added in Proof
After submission of this manuscript, Watson AP et al. published in an online report that human SUSD2 interacts with galectin-1 (Molecular Cancer Research. DOI: 10.1158/1541-7786.MCR-12-0501-T)
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.