
Abstract
Human embryonic stem cell (hESC)-derived natural killer (NK) cells are a promising source of antitumor lymphocytes for immunotherapeutics. They also provide a genetically tractable platform well suited for the study of antitumor immunotherapies in preclinical models. We have previously demonstrated the potency of hESC-derived NK cells in vivo. Here we use both bioluminescent and fluorescent imaging to demonstrate trafficking of hESC-derived NK cells to tumors in vivo. Our dual-imaging approach allowed us to more specifically define the kinetics of NK cell trafficking to tumor sites. NK cell persistence and trafficking were further evaluated by flow cytometry and immunohistochemistry. This integrated approach provides a unique system to apply the use of human pluripotent stem cells to study the kinetics and biodistribution of adoptively transferred lymphocytes, advances broadly applicable to the field of immunotherapy.
Introduction
N
To address the issue of how the NK cells interact with tumor cells (directly or indirectly), our studies have focused on using hESC-derived NK cells as a system to test lymphocyte engraftment and trafficking in vivo. Here we used a combined bioluminescent and fluorescent imaging system using luciferase-expressing hESC-derived NK cells. Immunodeficient NOD/SCID/γC−/− (NSG) mice received intraperitoneal (IP) or intravenous (IV) injections of hESC-derived NK cells and were followed for biodistribution and trafficking. Interestingly, we found persistence of the cells for more than 25 days in vivo by bioluminescent imaging and flow cytometric analysis. We next engineered the leukemia cell line K562 to express a membrane-bound Gaussia luciferase (mbGluc) construct, which is distinct from the Firefly luciferase (FLuc) constitutively expressed in our hESC-derived NK cells. These two luciferase reporters have been used in combination before to demonstrate trafficking and serve as a genetic reporter in the same cell type [8]. However, to our knowledge, this is the first study to utilize both in a cancer immunotherapy model. This permitted parallel monitoring of both the tumors and NK cells in vivo, noninvasively over time. Although this approach allows dual-bioluminescense imaging, it is technically difficult and limiting because of the need to deliver the mbGluc substrate intravenously to each mouse. To overcome this, we subsequently modified K562 tumor cells with the fluorescent protein turboFP650. TurboFP650 is a far-right shifted fluorescent protein that allows optimal in vivo imaging because of its reduced background compared to other fluorescent reporters in vivo. By doing this, we were able to successfully recapitulate our findings from the dual-luciferase model by showing NK cell trafficking with similar kinetics. As a third measure of NK cell trafficking, we used immunohistochemistry to evaluate the invasion of hESC-derived NK cells into tumor sites.
These results confirm the ability of hESC-derived NK cells to persist and traffic to the site of tumor in a xenograft model. The data also validate hESC-derived blood cells as a system to study in vivo trafficking and are broadly applicable across a variety of models.
Materials and Methods
hESC maintenance and hematopoietic differentiation
hESCs were maintained on low-density (90,000 cells/well of a six-well plate) mouse embryonic fibroblasts (MEFs). Generation of hematopoietic progenitor cells from hESCs was accomplished using an established method [9]. To generate spin embryoid bodies (EBs) amenable to aggregation, hESCs and iPSCs were passaged in TrypLE Select (Invitrogen) on low-density MEFs (90,000 cells/well). To follow the hESC-derived NK cells in vivo, we used an H9 line modified with a GFP/firefly luciferease construct [10]. TrypLE adapted hESCs around 60%–70% confluency were dissociated and filtered through a 70-micron sterile filter. Cells were then counted and placed at a concentration of 3000 cells per well (100 μL volume) of a round-bottom 96-well plate in the BPEL medium containing the stem cell factor (SCF, 40 ng/mL), vascular endothelial growth factor (VEGF, 20 ng/mL), and bone morphogenic protein 4 (BMP4, 20 ng/mL) [9]. The outer wells of the plate were filled with sterile water to prevent any evaporation of the media. Plates were then spin aggregated at 1,500 RPMs for 5 min at room temperature and placed undisturbed in a 37°C incubator with 5% CO2.
NK cell differentiation from spin EBs
At day 11 differentiation, six wells of a 96-well plate were directly transferred to one well of a 24-well plate in NK cell initiating cytokines (IL-3, IL-7, IL-15, SCF, fms-like tyrosine kinase receptor-3 ligand (FLT3L), all from Peprotech) [5]. NK cell cultures were refreshed with 0.5 mL of cytokine containing media every 4–5 days. Mature NK cells were measured at 28–35 days of culture. Following 4 weeks of NK cell culture, cells were further expanded using artificial antigen presenting cells (aAPCs) [11]. aAPCs were kindly provided by Dr. Dean A. Lee (MD Anderson Cancer Center).
Cell lines
K562 cells were obtained from American Type Culture Collection (ATCC). K562 cells expressing mbGLuc were generated as follows. First, membrane-bound Gaussia luciferase (mbGluc) was PCR amplified using the following primers: 5′-CATACA
In vivo fluorescent and bioluminescent imaging to follow trafficking of hESC-derived NK cells
At 24 h before tumor inoculation, 6- to 8-week-old nonobese diabetic/severe combined immunodeficiency with gamma-chain knockout (NOD/SCID/γC−/−) mice (Jackson Labs) were given a sublethal dose of irradiation (225–250 cGy). A total of 1×106 mbGluc+ or mbGluc+/turboFP650+ K562 cells were resuspended in 200 μL of the Iscoves modified Dulbecco's medium (IMDM) (HyClone Laboratories) supplemented with 20% FBS (Life Technologies). Cells were then injected subcutaneously into the upper left thorax of the mice. The tumors were allowed to engraft for 4 (mbGluc+) or 7 days (turboFP650+). Mice were then given an IP injection of 10×106 hESC-NK cells resuspended in 300 μL of the IMDM supplemented with 20% FBS. For all experiments, mice receiving no NK cell infusion were included as a negative control and tumor-only mice were included as a positive control for tumor engraftment. All mice received IP injections of IL-2 (1×104 U/mouse) and IL-15 (10 ng/mouse) every day for the first 7 days after NK cell injection followed by IL-2 only every 2 to 3 days until mice were sacrificed. All mice were housed, treated, and handled in accordance with the guidelines set forth by the University of Minnesota Institutional Animal Care and Use Committee and the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
To follow tumor progression and NK cell trafficking simultaneously, we utilized two dual-imaging schemes. To track the mbGluc+ K562 cells and Fluc+ hESC-NK cells, bioluminescent imaging was performed using a Xenogen IVIS Spectrum imaging system (Caliper Life Science). Before imaging, mice were anesthetized with isoflurane. A bioluminescent image was acquired using a 1-min exposure 10–15 s following IV injection of coelenterazine (320 μg; Nanolight Technology) or 10 min after IP injection of D-luciferin (150 mg/kg; GOLD Bio Technology). Mice were imaged individually following injection of coelenterazine and allowed to recover before injection of D-luciferin. Optical images were analyzed with Living Image software version 4.2 (Caliper Life Science).
To track turboFP650+ K562 cells and Fluc+ hESC-NK cells, fluorescent and bioluminescent imaging was performed using a Xenogen IVIS Spectrum imaging system. Before imaging, mice were anesthetized as described. A bioluminescent image was acquired for a total 1-min exposure 10 min after IP injection of D-luciferin. Immediately following, a fluorescent imaging sequence was acquired by performing an emission scan for turboFP650 (Excitation: 605, Emission: 660–720) and background signal (Excitation: 570, Emission: 640–720) using autoexposure settings. To separate the tumor and background signal, fluorescent imaging sequences were spectrally unmixed and set to a standard scale using Living Image software version 4.2 (Caliper Life Science).
Immunohistochemistry
Tumor tissue collected at the time of sacrifice was fixed in 10% formalin for 24–36 h and embedded in paraffin. Four micron sections were cut using a microtome, mounted onto uncharged slides, and rehydrated according to standard protocols. Slides were pretreated with a citrate buffer, 6.0 pH, in a Oster steamer for 30 min, and allowed to cool for 15 min. Primary antibodies were used at the following concentrations: Human NKp46 (R&D Systems; AF1850, 1:100), IgG1 kappa isotype control (eBioscience; Cat # 14-4714-82, 1:50). Antibody detection was by horseradish peroxidase-labeled streptavidin and DAB chromagen (Covance). Tissue sections were counterstained in hematoxylin. In every experiment, human tonsil tissue was stained as a positive control and tumor tissue from mice receiving no NK cell injection as a negative control. Images were taken at 10×, 40×, and 63× objective magnifications.
Results
Generation of NK cells from luciferase-expressing hESCs for in vivo tracking
To study lymphocyte trafficking in a mouse xenograft model, we used a well-characterized differentiation protocol to derive NK cells from hESCs [2,3,5]. We have previously demonstrated the derivation and function of NK from both hESCs and iPSCs [3,5]. hESC-derived NK cells have a potent antitumor activity both in vitro and in vivo. However, our previous studies did not directly implicate tumor clearance as a direct result of trafficking NK cells. Using hESCs that we have modified to express firefly luciferase and GFP [10], we first demonstrated their ability to differentiate into hematopoietic progenitor cells and subsequently NK cells (Fig. 1). To further explore the function of hESC-derived NK cells in vivo, we developed a model to monitor NK cell persistence and trafficking, as well as tumor burden. We compared the survival of hESC-derived NK cells upon transfer into immunodeficient mice via the IV or IP route. Typically, effector cells for adoptive immunotherapy have been administered IV. For the treatment of leukemia with adoptively transferred NK cells, it is important that these effectors can traffic to sites of tumor involvement, including the spleen and bone marrow. However, this may not be an optimal delivery system for all malignancies, such as ovarian cancer. Additionally, injection of NK cells into a nonhematopoietic or nonlymphoid compartment using an IP approach can provide a more rigorous test of trafficking. Mice also received injections of IL-2 (10,000 units/mouse) and IL-15 (10 ng/mouse) for the first 7 days followed by injections of IL-2 (10,000 units/mouse) every other day until the end of the study. Mice receiving IP delivery of NK cells had prolonged persistence compared to those injected IV (Fig. 2). IV delivery of NK cells first trafficked to the lungs of the mice, but were absent by day 4, whereas IP delivery of NK cells lead to persistence for greater than 4 weeks. At day 19, we sacrificed mice and looked for engraftment within the peripheral blood, spleen, bone marrow, and peritoneum. Both routes of NK cell delivery had low levels of engraftment within the peripheral blood, bone marrow, and spleen as measured by GFP+ and CD56+CD45+ cell surface antigens. However, there was a high level of NK cells in the peritoneum of IP injected mice compared to IV injected mice or controls (Fig. 2). This corresponds with the bioluminescent imaging data and led us to conclude that IP delivery of NK cells allows enhanced persistence of NK cells in vivo and would be optimal for our trafficking studies.
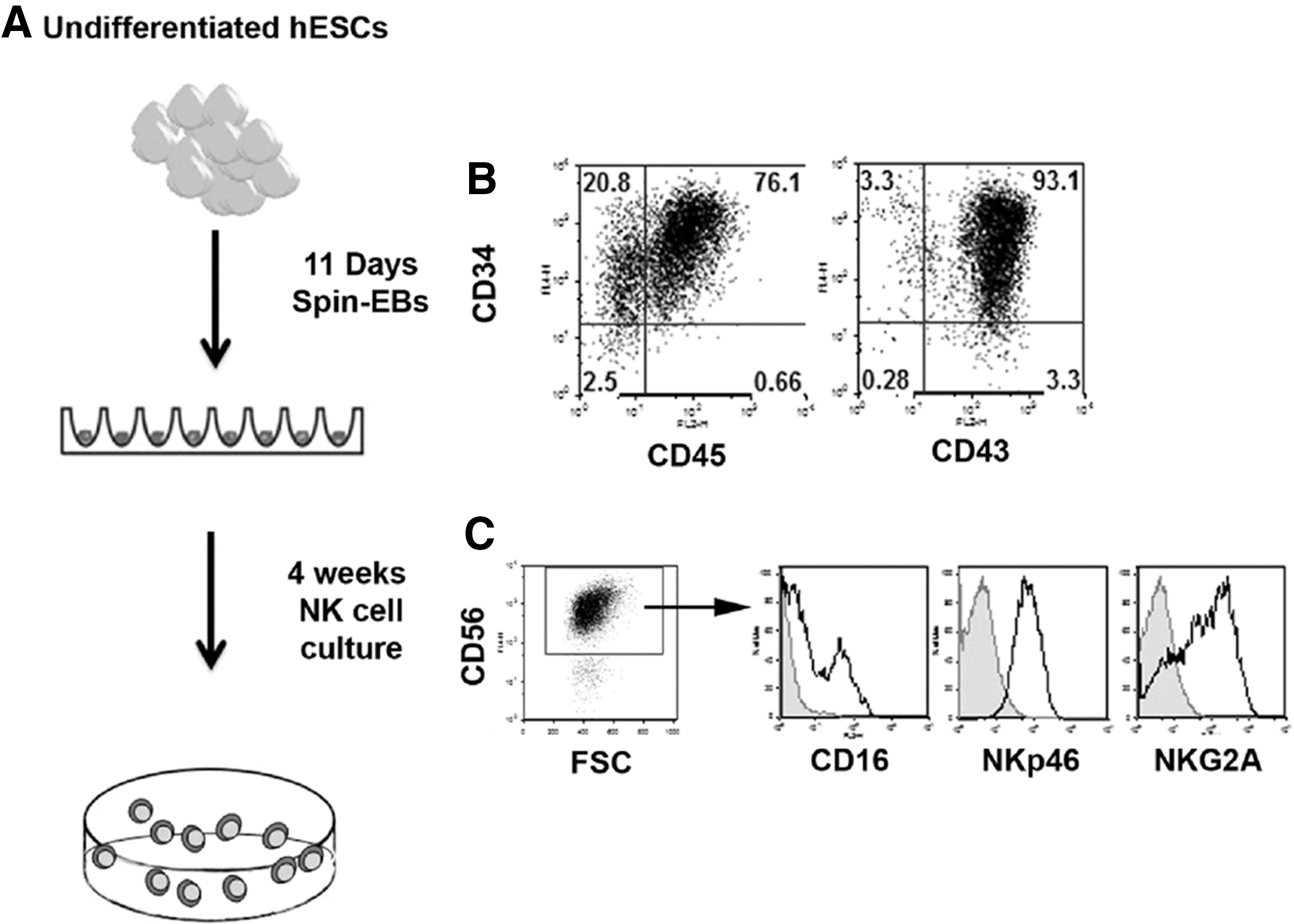
Derivation of natural killer (NK) cells from human embryonic stem cells (hESCs) expressing firefly luciferase.
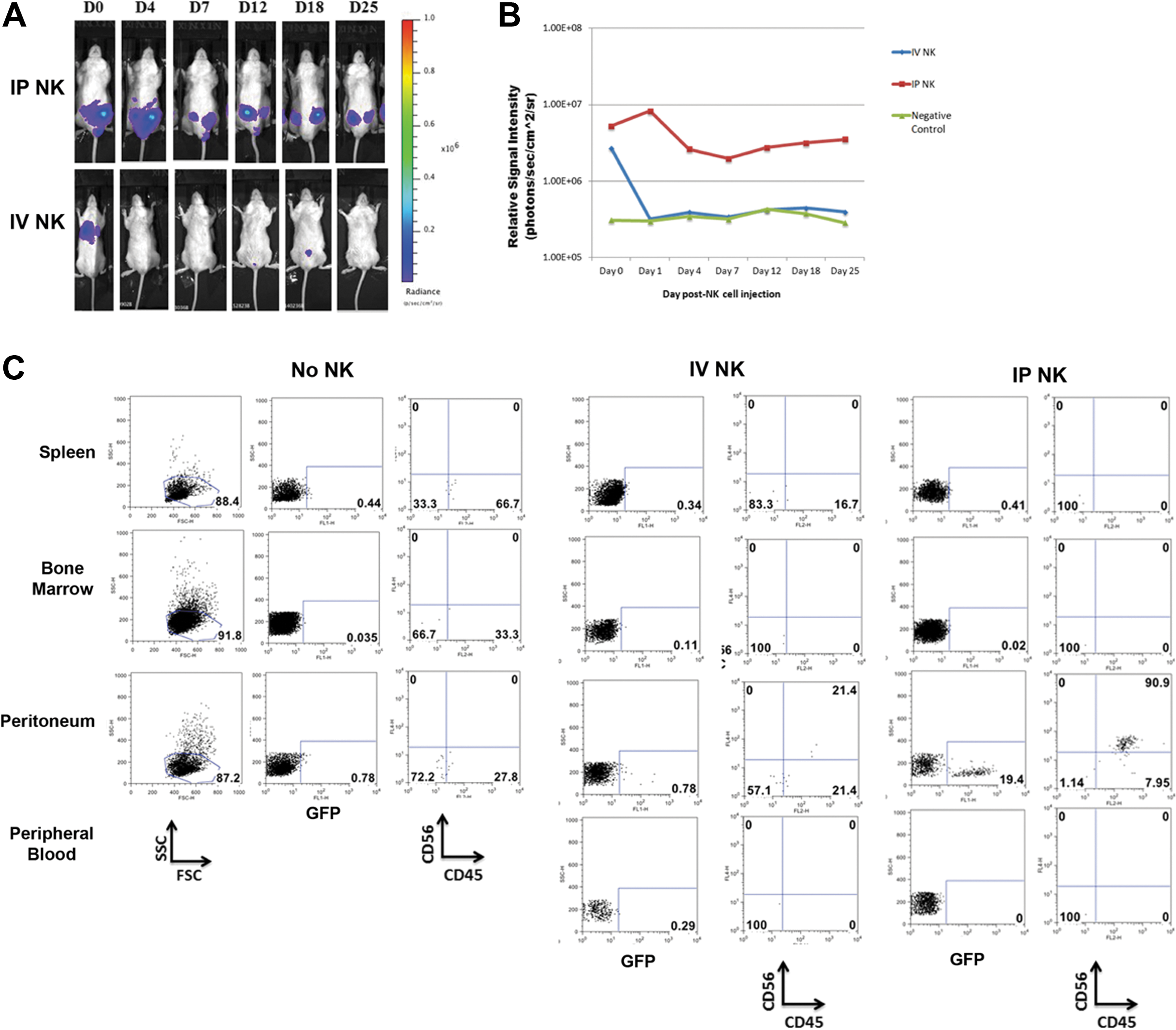
Persistence of hESC-derived NK cells.
NK cells persist and colocalize with tumors in vivo
Next, we took advantage of firefly luciferase stably expressed by our hESC-derived NK cells, as a well-characterized reporter of lymphocyte persistence in vivo [8,12]. Our previous studies demonstrating the powerful antitumor activity of hESC-derived NK cells in vivo used luciferase-positive tumor cells; however, we were unable to follow NK cells concurrently. To image both NK cells and tumors in the same mouse, we adopted another luciferase reporter of the Gaussia luciferase family. Using a recombinant form of the Gaussia protein that has been modified to be tethered to the membrane (membrane-bound Gaussia lucifearse, mbGluc) [13], we were able to utilize two different substrates to image both tumors and NK cells in the same mouse. We initially subcloned the mbGluc gene into a Sleeping Beauty backbone driven by an mCAGS promoter (Fig. 3). We were able to stably transduce K562 tumor cells and select for cells with the luciferase activity in response to the substrate coelenterazine (activated by Gaussia luciferase), but not luciferin (activated by Firefly luciferase).

Generation of K562 tumor cells expressing membrane-bound Gaussia luciferase.
To use both reporters in vivo, we took advantage of the fact that coelenterazine, the substrate for mbGLuc, is rapidly degraded in vivo [13]. Firefly luciferase (expressed in the hESC-derived NK cells) is reciprocally stable in vivo [13,14] and was delivered second. Using these two reporters, we were able to initially image mbGluc+ tumor cells, and then image the firefly luc-expressing hESC-derived NK cells in the same mouse. We replicated our initial model by allowing K562 tumor cells to engraft in sublethally irradiated mice for 4 days before NK cell injection [5]. Because the aim of this study was to study NK cell trafficking, we increased the tumor dose to 1 million cells to better allow tumor growth. Our previous studies, which demonstrated a complete tumor clearance, used a dose of 200,000 cells per mouse. At day 0, NK cells were given IP and mice were treated with cytokines. Mice were evaluated for both tumor size and NK cell trafficking at days 0, 4, 7, 9 to 12. Here NK cells were capable of trafficking to tumor sites (Fig. 4). This typically occurred between day 9 and 12, but was variable among mice. Additionally, not every mouse demonstrated trafficking by bioluminescence. (4 out of 6 mice showed trafficking). We conclude from this that hESC-derived NK cells can be followed for trafficking in this dual-bioluminscent system. Although not every mouse demonstrated trafficking, this could be due to the absolute number of luciferase+ cells needed to demonstrate the bioluminescent signal and the negative mice could be below the limit of detection. Additionally, we found that increasing the tumor burden of the mice (1×106 cells vs. 200,000 cells) was necessary to allow enough NK cells to accumulate and give a bioluminescent signal over background.

Dual-bioluminescent imaging to monitor hESC-derived NK cell trafficking in vivo. Monitoring of a single mouse over a period of 9 days for the presence of both tumor cells (mbGluc+, top row) and NK cells (firefly luciferase+, Fluc, bottom row). NK cell trafficking to the tumor site can be seen on day 9 in this particular mouse. Color images available online at
Improved dual reporter imaging with firefly luciferase and the fluorescent protein turboFP650
The use of mbGluc in conjunction with firefly luciferase provided a reliable, yet technically challenging model to study NK cell trafficking. This is primarily because the substrate for Gaussia luciferase, coelenterazine, needs to be delivered intravenously. This was difficult for several reasons. First, due to the decay kinetics of the substrate, a limited number of mice could be imaged simultaneously [13]. Also, repeated injection of the coelenterazine substrate leads to injury of the tail vein over time. To overcome this, we took advantage of a more recently described fluorescent reporter that can be imaged in vivo [15,16]. TurboFP650 is a red-shifted fluorescent reporter (excitation 592 nm, emission 650 nm) with tissue penetrance for optimal in vivo imaging. It also does not require delivery of a second substrate. We used a similar cloning approach to express the turboFP650 protein in K562 cells using Sleeping Beauty (see Materials and Methods). Stable expression of TurboFP650 was determined by flow cytometry. Following confirmation of stable transduction for more than 1 week, cells were sorted using the same parameters as above. Sorted cells maintained expression of the TurboFP650 protein and were used for further in vivo studies (Fig. 5).
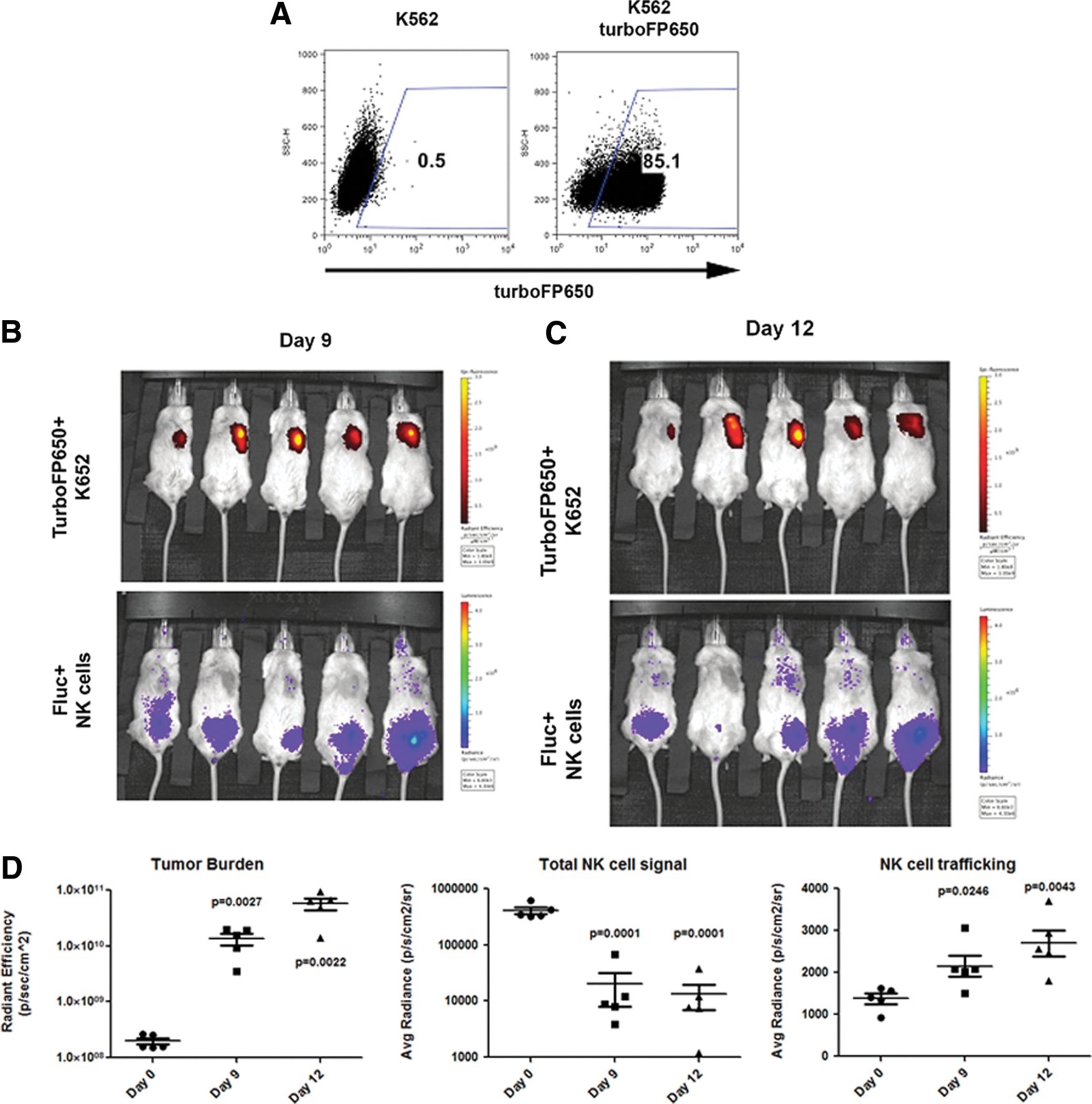
Enhanced dual-reporter imaging utilizing TurboFP650-expressing K652 cells.
Using the same in vivo model as above, mice were engrafted with 1 million TurboFP650+ K562 tumor cells and allowed to engraft for 7 days. At day 0, mice were then given firefly luciferase expressing NK cells intraperitoneally and followed for trafficking. Similar to our dual-luciferase studies, we found that NK cells were able to track to turboFP650+ tumor cells in four of the five mice, which occurred within 9–12 days post-NK cell injection (Fig. 5). Importantly, the tumor burden and quantity of NK cells can be determined simultaneously. First, turboFP650 provides a reliable in vivo marker of tumor progression. Here we saw a significant increase in tumor burden in all mice at both days 9 (P=0.0027) and 12 (P=0.0022). We next measured the level of total NK cell signal in each of the mice, and as before, saw that it decreases significantly from day 0 to day 9 (0.0001) or day 12 (P=0.0001). Reciprocally, we saw increased NK cell signal at the site of tumor at day 9 (P=0.0246) and day 12 (P=0.0043), indicating successful trafficking of hESC-derived NK cells to the tumor site. These data support the use of turboFP650 as a reporter compared to mbGluc. By overcoming the technical limitations of using coelenterazine-based reporters, these studies provide an enhanced in vivo system to monitor two cell populations over time.
Trafficking of hESC-derived NK cells to tumor confirmed by immunohistochemistry
To definitively show NK cells at the tumor site, we more closely examined tumors taken from mice. To confirm the presence of human NK cells at the tumor site, we extracted the tumors, paraffin embedded them, and used immunohistochemistry as a qualitative, confirmatory measure of NK cell trafficking. Compared to the tumor-only (no NK cell injection) group and isotype controls, mice with demonstrated NK cell trafficking by bioluminescent imaging had human NK cells present as demonstrated by NKp46 staining on IHC (Fig. 6). Human tonsil tissue was used as a positive control. NKp46 is a more specific marker of human NK cells, as CD56 can also mark other tissue types. The NK cells positive by IHC staining were uniformly dispersed throughout each tumor tissue section stained, with 15–20 hESC-derived NK cells per section. These data further support the trafficking of NK cells to the tumor site and that bioluminescent imaging using firefly luciferase is an effective model to study lymphocyte trafficking in vivo.
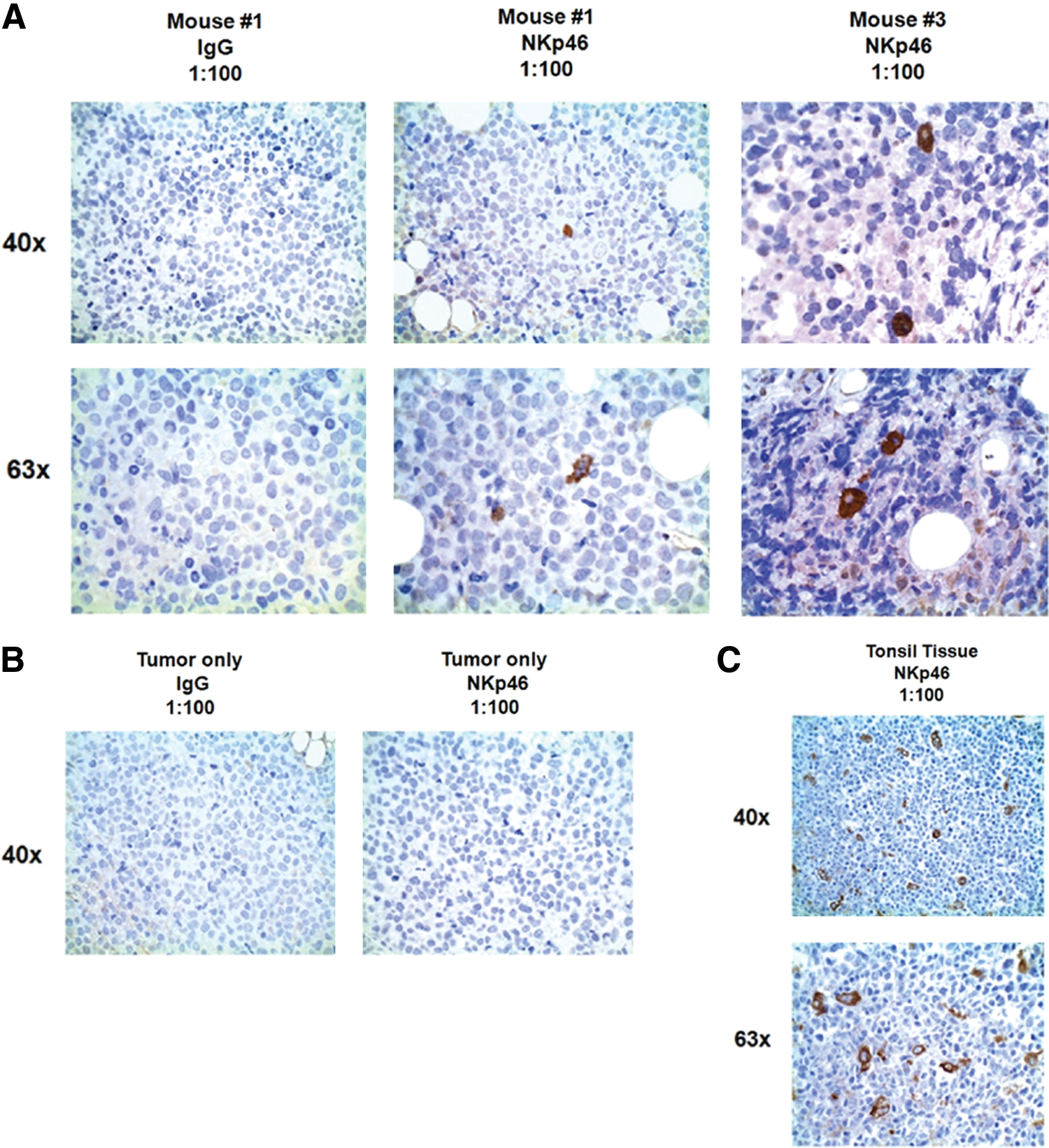
Immunohistochemical (IHC) confirmation of in vivo NK cell trafficking.
Discussion
The use of hESCs and iPSCs to study blood and lymphocyte development has several advantages. First, each line provides an unlimited number of cells and serves as an individual donor allowing a homogenous starting source. Our most recent studies have improved on the method in generating NK cells from hESCs and iPSCs, allowing the generation of enough cells to treat a single patient from 250,000 starting cells [17]. Second, hESCs and iPSCs are genetically amenable platforms allowing stable transgene expression. Here we have utilized hESCs that constitutively express firefly luciferase, although there are other reporters that can be used. Additionally, having a genetically tractable system allows modification of the input cells. Some recent studies have used transfer of certain chemokine receptors into effector cells to get enhanced homing to the tumor site [18]. This could be tailored on a patient, disease-specific basis if a bank of hESC-derived effector cells was used. Finally, there are numerous well-defined protocols leading to the development of almost all hematopoietic lineages, not only NK cells [19 –23]. Studies have aimed at genetic correction of erythroid progenitor cells to potentially provide a novel source of red blood cells to patients with hemoglobinopathies [19 –23]. The major challenge facing the translation of hESCs and iPSCs is the continued inability to generate and isolate functional HSCs capable of long-term, multilineage engraftment [24,25]. Until a protocol to generate definitive HSCs is found, delivery of more differentiated blood types (such as lymphocytes or erythroid cells) is the best available option of hESC- and iPSC-based hematopoietic cell therapies.
By using hESC-derived NK cells to monitor trafficking, we found that delivery of cells intraperitoneally leads to enhanced persistence of NK cells, not surprisingly within the peritoneum. IV injected NK cells are most likely diluted in the periphery, whereas the IP injected NK cells persist longer. We did not aim to study the optimal conditions for expansion of IV delivered NK cells; however, this will be important for further preclinical trials. Higher doses of IL-2 or the use of additional cytokines (such as IL-15) would likely lead to the expansion of IV delivered NK cells. Of note, these studies will also be important to determine whether or not successful engraftment and expansion of IV delivered NK cells is required for clearance of spleen and bone marrow disease. Enhanced persistence of IP delivered NK cells could be due to several reasons. First, although firefly luciferase is a sensitive in vivo reporter, the signal is most prevalent when there are collections of cells. IV delivery simply has a wider distribution as the cells traffic through the circulation, thereby minimizing the luciferase signal. Alternatively, IP delivered NK cells persist for longer periods of time because they have more direct access to injected cytokines, which are also delivered IP.
Importantly, these studies were able to demonstrate trafficking of hESC-derived NK cells to the site of tumor and strongly support the hypothesis that hESC/iPSC-derived NK cells directly kill tumor cells in vivo. Without the utility of a dual-bioluminescent imaging model, trafficking would have been much more difficult to discern. Additionally, use of bioluminescent imaging allowed easier and more rapid quantification than IHC. Not all mice grossly demonstrated trafficking by bioluminescent imaging and signs of trafficking ranged over 9–12 days. However, bioluminescent quantification indicates a significant difference in NK cell signal accumulation in the tumor region (Fig. 5). It is possible that some NK cells traffic and are below our limit of detection. Or, NK cells can indeed traffic to tumor, but do not receive the correct signals to stay within the tumor environment. In this case, one could modify the NK cells with tumor-specific receptors to enhance intratumoral persistence and activity [18]. This has been recently accomplished using chimeric antigen receptors in human T cells [26,27]. hESCs and iPSCs provide an optimal platform for such a modification. The studies also confirm our initial findings that unmodified hESC-derived NK cells traffic to tumor sites to clear disease.
Immunohistochemistry was performed to confirm the above evidence and validate the ability of this dual reporter system. As NSG mice are deficient of all lymphocytes, and the NKp46 marker is specific to human NK cells, we can definitively show trafficking of NK cells to the tumor site. By having effector cells labeled with firefly luciferase, we were able to monitor whole body trafficking, whereas other methods are limited to longitudinal analysis of engraftment in the peripheral blood. Although we did not see high levels of engraftment in other organs such as the spleen or bone marrow by bioluminescent imaging, the NSG mouse may not be the optimal host to support the hESC-derived NK cell persistence at these sites. Alternatively, the hESC-derived blood cells may lack the surface molecules necessary to establish engraftment at these sites.
Together, these data provide a model system to follow two different populations of human cells in mice and should not be limited to antitumor therapies. The use of two diverse reporter systems combined with hESC/iPSC-derived cells has broad applicability to a number of biological systems. For therapeutic purposes, the ability to derive almost any blood cell from hESCs could provide banks of cells for new therapeutics [19]. Alternatively, given the relative ease of manufacturing iPSCs and technological advances in this process, the thought of using patient's autologous cells for therapy is also reasonable [28]. hPSCs also provide a platform to take gain- and loss-of-function approaches in studying lymphocyte development and trafficking. Constitutive or conditional knock down of particular molecules affecting these processes would be beneficial and a major advantage over using cells isolated from primary sources (HSCs or peripheral blood), which are intrinsically resistant to genetic modification.
Footnotes
Acknowledgments
The authors would like to thank Laura E. Bendzick, Zhenya Ni, and Michael Lepley for their technical assistance with this work. The authors would also like to thank Colleen Forster for her expertise and invaluable assistance with IHC, Brad Taylor at Caliper Life Sciences for his expert technical advice, and Dr. Peter Southern for kindly providing tonsil tissue for use in IHC.
This work was supported by the NIH/NHLBI R01-HL77923 (D.S.K.) and by NIH MSTP grant T32 GM008244 (D.A.K.), Stem Cell Biology Training Grant T32 (D.A.K.) (T32HD060536), Undergraduate Research Opportunities Program (UROP) Grant, the University of Minnesota (A.M.B), the Leukemia Research Fund of the University of Minnesota Cancer Center, and the William L and Blanche Hughes Foundation.
Author Disclosure Statement
The authors declare no competing financial interests.