
Abstract
Cancer stem cells (CSCs) constitute a subpopulation of cancer cells that have the potential for self-renewal, multipotent differentiation, and tumorigenicity. Studies on CSC biology and CSC-targeted therapies depend on CSC isolation and/or enrichment methodologies. Scientists have conducted extensive research in this field since John Dick's group successfully isolated CSCs based on the expression of the CD34 and CD38 surface markers. Progress in CSC research has been greatly facilitated by the enrichment and isolation of these cells. In this review, we summarize the current strategies used in our and other laboratories for CSC isolation and enrichment, including methods based on stem cell surface markers, intracellular enzyme activity, the concentration of reactive oxygen species, the mitochondrial membrane potential, promoter-driven fluorescent protein expression, autofluorescence, suspension/adherent culture, cell division, the identification of side population cells, resistance to cytotoxic compounds or hypoxia, invasiveness/adhesion, immunoselection, and physical property. Although many challenges remain to be overcome, it is reasonable to believe that more reliable, efficient, and convenient methods will be developed in the near future.
Introduction
A
Thus, strategies to identify CSCs and to efficiently and reliably isolate them from a heterogeneous tumor mass may have fundamental roles in CSC studies, the results of which will have profound implications both for tumor development and for therapeutic outcomes. In this review, we will briefly discuss the progress made in CSC isolation and enrichment during the past 10 years, particularly during the last 4–5 years.
It should be emphasized that putative CSC or CSC-enriched populations obtained using any of these strategies must be tested rigorously by serial xenotransplantation in immunocompromised mice, the gold standard for the identification of CSCs [6]. Self-renewal can be confirmed by this assay, in which prospectively re-isolated CSC populations are placed into secondary recipients. Multipotency is typically demonstrated by the ability of the cells to generate tumor xenografts that reflect the cellular heterogeneity of the original tumor [6,7].
Strategies for Isolating and Enriching CSCs
Surface markers
Cellular surface markers have been used for the isolation of CSCs. In 1994, Dick provided the first evidence of the existence of CSCs derived from acute myeloid leukemia using fluorescence activated cell sorting (FACS) based on CD34 and CD38 (CD34+CD38−) surface marker expression [8,9]. Since then, CSCs have been isolated from many types of solid tumors by FACS and magnetic cell sorting using the following specific surface markers: CD24, CD44, CD133, CD13, CD14, CD15, Stro-1, Cripto-1, CXC chemokine receptor type 4 (CXCR4), Lin, Thy1, stage-specific embryonic antigen-1 (SSEA-1), epithelial cell adhesion molecule (EpCAM), epithelial specific antigen, CD20, ATP-binding cassette (ABC) transporter B5, CD166, A2B5, leucine-rich-repeat-containing G-protein-coupled receptor 5 (LGR5), CD49f, CD90, CD117, stem cell antigen-1 (Sca-1), epidermal growth factor receptor (EGFR), CD271, and CD47 [8 –52]. This surface marker-based approach has become the most commonly used method to isolate CSCs from heterogeneous tumor cell populations and has significantly contributed to progress in CSC research. However, many of the surface markers used for sorting have been identified empirically and were identified on normal stem cells (SCs), such as embryonic stem cells (ESCs) and adult stem cells (ASCs). Questions have been raised regarding the specificity and reliability of these markers for the identification of CSCs. For example, CD133 is an important cell surface marker present on neural stem cells (NSCs) and has been the most extensively used marker for the isolation of CSCs from many types of cancers, such as glioblastomas, breast cancer, prostate cancer, colon cancer, pancreatic cancer, liver cancer, Wilms tumor, and neuroblastomas [53]. However, Beier et al. provided the first evidence that CD133+ CSCs maintain only a subset of primary glioblastomas. Kemper et al. found that glioma stem cells (GSCs) identified using antibodies against CD133 have inconsistent tumorigenic potential. The AC133 epitope, but not the CD133 protein, is lost upon CSC differentiation [54]. Some cell lines derived from primary glioblastomas grow adherently in vitro and are driven by CD133− tumor cells that exhibit apparent SC-like properties but distinct molecular profiles and growth characteristics in vitro and in vivo [55]. A subpopulation of A2B5+ glioma cells with the capacity to form tumors exists in some human gliomas that contain very few or no detectable CD133+ cells. These A2B5+ cells are phenotypically distinct from CD133+ cells [19]. In human lung cancer and rat glioma cell lines, both CD133+ and CD133− subpopulations possess clonogenic, self-renewal, and tumorigenic capacities [56,57]. A similar phenomenon has also been found in colon cancer. During the process of metastasis, CD133+ tumor cells may give rise to a more aggressive CD133− subset capable of tumor initiation in mice [58]. CD133 is expressed regardless of the differentiation state of colon cancer cells and may not be restricted to CSCs. The isolated CD133− cells still have the capacity to give rise to tumors in nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice [58]. The same result has also been recently demonstrated for head and neck squamous cell carcinoma [59]. Moreover, several studies have indicated that CD133 is expressed by differentiated epithelial cells in a variety of organs [60]. Park et al. performed immunohistochemical analyses of several stem cell-related markers in breast cancer cells with different stemness and differentiation characteristics. They found that the frequency of tumor cells positive for stem cell-like cell markers and markers of more differentiated cells varied according to the tumor subtype and histologic stage [61]. Furthermore, a recent study found that CD133 expression in colorectal cancer cells is modulated by the microenvironment [62]. In addition to colorectal cancer, there are other cancers in which the expression of CD133 is regulated, such as gliomas and prostate cancer. When GSCs were sorted into CD133+ and CD133− fractions, promoter methylation was observed in the CD133− fraction; this methylation was reversible using demethylation agents and could be reproduced by in vitro methylation. The researchers thought that CD133 might be a marker that is only co-regulated with other more relevant factors that help determine the stem cell characteristics, such as Sp1 and c-myc [63]. In both benign and malignant primary prostate tissues, the regulation of CD133 is independent of DNA methylation but is under the dynamic control of chromatin condensation [64].
Suspension culture
In 1992, Reynolds et al. established a serum-free culture method to obtain neurospheres from brain tissue [65]. This simple and convenient method for NSC enrichment has not only been widely used to study neurogenesis but has also been applied for CSC isolation from brain tumors and many other types of cancers with different combinations of supplemental factors. These tumor spheres express high levels of SC markers and exhibit a great degree of tumorigenicity. Using sphere assays for tumor cells, a number of groups have demonstrated that CSCs efficiently form tumor spheres in a clonogenic manner [66]. These tumor spheres are also chemoresistant and exhibit the upregulation of drug-resistance proteins [67]. However, the use of the sphere assay for CSC isolation has several problems. As shown by Pollard and others [68,69], the efficiency of successfully isolating CSCs from any given tumor is low (from 1% to 30%). The majority of cells in the sphere are differentiated and/or dying progeny [70]. During serial passaging, sphere cells spontaneously differentiate and/or undergo apoptosis [70]. Additionally, it is worth noting that sphere cells in some cancer cell lines are no more tumorigenic in vivo than cells grown on plastic and that cells that generate spheres in culture do not always give rise to tumors after more than 10 months following implantation [66]. Recently, Sudha Krishnamurthy designed an alternative strategy, termed the orosphere assay, for the propagation of head and neck CSCs. An orosphere is defined as a nonadherent colony of cells sorted from a tumor mass and cultured in three-dimensional soft agar or on ultralow attachment plates. This strategy is suitable for the propagation of head and neck cancer cells and can retain the stemness and self-renewal capacity of these cells [71].
Activity of intracellular enzymes
The aldehyde dehydrogenase (ALDH) family is composed of cytosolic isoenzymes that are responsible for oxidizing intracellular aldehydes [72]. Normal SCs, including ESCs and ASCs, have high ALDH activity [73 –78]. Increased activity of the Class 1 ALDH family (ALDH1) has been found in different types of CSCs [79 –84]. In these studies, researchers found that the ALDH1+ cancer cells isolated from tumor masses using the aldefluor assay and FACS analysis exhibited obvious CSC properties in vitro, including self-renewal, multipotency, and drug-resistance, and these isolated cells expressed SC markers. Additionally, these cells had CSC properties in vivo, including tumorigenicity and the generation of a heterogeneous cancer cell population. These data suggest that ALDH1 could be used as a CSC marker [79 –85]. Some researchers have also found that this ALDH1+ CSC pool is regulated by the expression of several growth factor receptors, including epidermal growth factor receptor type 2 (HER2, also known as ErbB), through the PI3-kinase/Akt pathway [86].
The 26S proteasome is a key regulator of many cellular functions, including cell cycle control, DNA repair, cell death, and survival. Cancer cells grown in sphere cultures enriched for CSCs exhibited decreased 26S proteasome activity relative to their respective monolayer cultures. Vlashi et al. engineered human glioma and breast cancer cells to express stably, ZsGreen fused to the carboxyl-terminal degron of ornithine decarboxylase, resulting in a fluorescent fusion protein that accumulates in cells in the absence of 26S proteasome activity. They found that the ZsGreen+ cells had increased sphere-forming capacity and higher expression levels of CSC markers than ZsGreen− cells in vitro. In vivo, ZsGreen+ cells were more tumorigenic than ZsGreen− cells [87]. Spheres from lung cancer cell lines have also been found to exhibit decreased 26S proteasome activity [88].
The Jumonji AT-rich interactive domain 1B (JARID1B), which belongs to the highly conserved family of Jumonji/ARID1 histone demethylases, is capable of removing three methyl groups from histone H3 lysine 4 (H3K4) [89]. Using the H3K4 demethylase JARID1B as a biomarker, Roesch et al. characterized a small subpopulation of slow proliferating melanoma cells and found that the JARID1B+ subpopulation is essential for continuous tumor growth. Compared with melanoma cells that do not express this enzyme [90], JARID1B+ cells cycled more slowly but also generated more progeny and were more tumorigenic [91]. The same result was found for uveal melanoma cells [92]. However, the presence of a subset of cells functionally enriched for tumorigenicity in melanoma is in contrast with the findings obtained by Morrison and coworkers. [93 –95].
Intracellular concentration of reactive oxygen species
Reactive oxygen species (ROS) have been implicated in many intracellular physiopathological processes, including cell cycle progression, proliferation, apoptosis, and differentiation. Normal SCs, such as ESCs, induced pluripotent stem cells, NSCs, and hematopoietic stem cells, have been shown to contain lower levels of ROS than their more mature progeny. This difference is essential for maintaining SC function [96 –103]. Interestingly, the results obtained by Diehn et al. indicated that as observed for normal SCs, subsets of CSCs in some tumors also contain lower ROS levels and enhanced ROS defenses compared with their differentiated descendants and non-CSCs [104,105]. Following the purification of CD44+CD24−/loLin− CSCs from breast tumors, 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) staining revealed that the CSC-enriched population contained a considerably lower DCF-DA intensity than the corresponding non-CSC population. A similar phenomenon has also been found in head and neck tumors. These mechanisms may be attributed to the upregulation of the ROS defense system via the overexpression of scavenging molecules and an enhanced capacity for glutathione synthesis and protection against ROS [106]. Although the detailed mechanisms remain unknown, it may be possible to establish a method to isolate CSCs based on the intensity of fluorescent stains for ROS, such as DCF-DA and dihydroethidium. In our laboratory, researchers are attempting to isolate CSCs using this method.
Mitochondrial membrane potential
The mitochondrial and energy/metabolism-related features of ASCs and ESCs have sparked the interest of an increasing number of researchers [107 –110]. It is thought that mitochondrial function and integrity may affect SC viability, the proliferative and differentiation potential of these cells, and their lifespan [111]. Heerdt et al. found that the mitochondrial membrane potential (Δψm) is associated with a cell's tumorigenicity. Two isogenic cell lines were subcloned and established from the SW620 colonic carcinoma cell line, and they exhibited significant and stable differences in the intrinsic Δψm [112]. These differences in Δψm are linked to important tumorigenic properties. Compared with cells with a lower Δψm, cells with an intrinsically higher Δψm exhibit significantly higher resistance to apoptotic inducers and hypoxia, increased invasive behavior, and enhanced initiation of angiogenesis [113,114]. Our recent results have indicated that side population (SP) cells and sphere-forming A549 lung cancer cells have a fairly high Δψm (tumor sphere, 90.92±18.2; monolayer subpopulation, 50.53±3.35; P<0.05; SP, 133.48±29.33; non-SP, 111.37±20.7; P<0.05) [105, 115]. In most of the A549 cells with Δψm values in the highest 5% (ΔψmH), CD133 expression was clearly detected. However, the expression of this SC marker was nearly absent in the subpopulation with Δψm values in the lowest 5% (ΔψmL) [115]. Michelakis et al. measured the Δψm in cells from freshly excised glioblastoma multiforme (GBM) tissue, primary cell lines, GBM-SCs isolated from tumors, and differentiated cells derived from GBM-SCs. The highest potential was found in the putative GBM-SCs. Both the primary and GBM-SC-derived secondary GBM cells (15-day differentiation) had Δψm values similar to those of the parent tumors. Simultaneous staining with a CD133 antibody and tetramethylrhodamine methyl ester (TMRM) demonstrated that CD133+ cells had a higher Δψm than the neighboring non-GBM-SCs in vivo [116]. These data suggest that differences in intrinsic Δψm of carcinoma cells are likely to be associated with subtle shifts in the biochemical pathways and/or cell phenotypes that play fundamental roles in determining the probability of tumorigenesis. Compared with their differentiated descendants, CSCs may have a different Δψm. Using approaches, such as flow cytometry, Δψm heterogeneity can be detected based on varying fluorescence intensities (Rhodamine-123 (Rh123), TMRM, and JC-1), and difference in the Δψm can used to isolate and/or enrich CSCs from a tumor population [115].
Promoter-driven fluorescent protein expression
ESCs and multiple types of CSCs share a genetic expression pattern [117]. Some core transcription factors associated with the stemness of ESCs, such as octamer-binding transcription factor 4 (Oct-4), sex determining region Y-box 2 (Sox2), Nanog, and c-kit, are also overexpressed in CSCs. In contrast, the activities of these factors are weak or even absent in the differentiated descendants [118,119]. Thus, one can isolate CSCs from a heterogeneous tumor mass based on the different activities of these stemness factors. Compared with surface marker-based strategies, this approach may provide a more target-specific tool with a wider range of applicability to isolate CSCs due to the critical roles of these transcription factors in controlling cell stemness. However, due to the intracellular expression of these proteins, one cannot isolate CSCs directly using these proteins but must use a reporter system in which a specific gene promoter drives the expression of a reporter protein.
In recent years, reporter systems employing a specific gene promoter driving green fluorescent protein (GFP)/red fluorescent protein (RFP) expression have been widely used to isolate CSCs. These genes include SH2 domain containing 5′-inositol phosphatase-1 (s-SHIP) [120], Oct4 [121], Nanog [122], nucleostemin [123], T-cell factor/lymphoid enhancer factor [124], and Sox2 [125]. Additionally, early transposon (ETn), which is highly transcribed during early mouse embryogenesis in ESCs and in embryonic carcinoma cell lines [126], could potentially be used to isolate CSCs [127]. In our laboratory, we used the Nanog promoter as a reporter system to successfully isolate a small subpopulation of Nanog-positive cells from hepatocellular carcinoma (HCC) tissue. These cells demonstrated an enhanced ability for self-renewal, clonogenicity, and tumor initiation. Moreover, these cells were also multipotent and chemoresistant [128]. Recently, we used the EOS system (based on a lentiviral vector with the ETn promoter and Oct4/Sox2 enhancer) developed by Hotta et al. to isolate CSCs from the glioma cell line U87, obtaining very exciting preliminary results. As shown in Fig. 1, GFP+ U87 cells can form tumor spheres in serum-free stem cell culture medium, and they express several stem cell markers, such as CD133, Sox2, Oct4, nestin, and SSEA-1. Because of the complex gene expression pattern, the selection of the appropriate vector and the number of promoter/enhancer copies should be carefully considered.
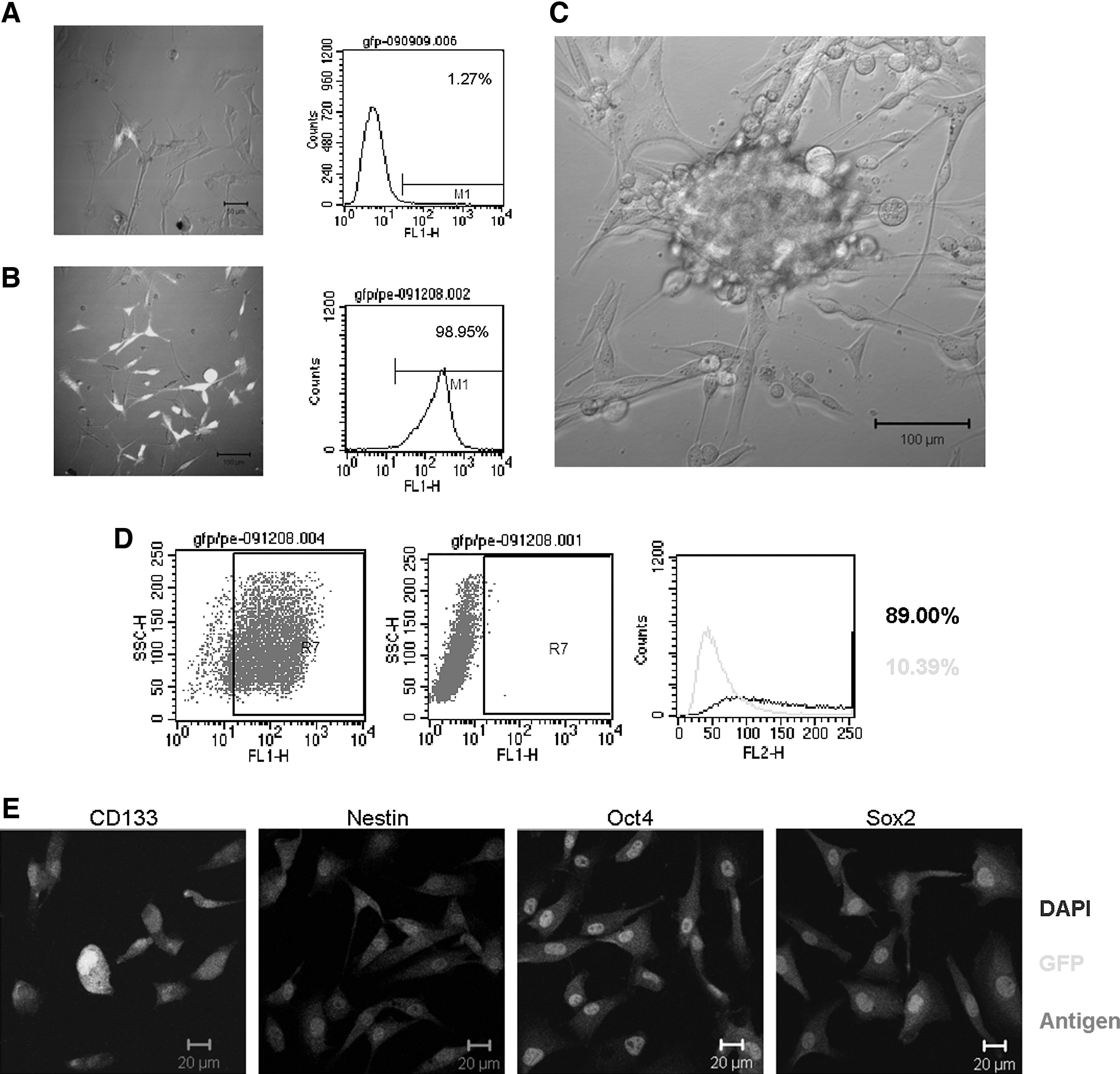
Isolation of cancer stem cells (CSCs) based on EOS system (based on a lentiviral vector with the ETn promoter and Oct4/Sox2 enhancer, plasmid purchased from Addgene).
Autofluorescence
It has been found that some types of stem cells exhibit intrinsic autofluorescence, which can be used to isolate and track stem cells [129 –131]. Radovanovic and coworkers utilized intrinsic autofluorescence and distinctive morphology to isolate a subpopulation of CSCs from human gliomas. Excitation at 488 nm causes these cells to autofluoresce, with emission at approximately 520 nm (in the FL1 channel). These cells, termed FL1+ cells, possess many SC-like characteristics, such as a capacity for self-renewal in vitro and tumorigenesis in vivo, and these cells preferentially express the SC genes Nanog, Oct4, Sox2, and Notch1. Although the underlying mechanism remains unknown, these data suggest a novel approach for isolating and enriching CSCs from a tumor mass [132].
Adherent culture
Morphologic heterogeneity is a typical feature of malignant cells and has been attributed to genetic instability and clonal evolution [133]. Studies have revealed a close relationship between the clone morphology and stemness of cancer cells. After being plated at low densities, epithelial-derived malignant cells exhibit a spectrum of colony morphologies, ranging from round colonies of small, closely packed cells to irregularly shaped colonies consisting of larger, loosely packed cells; these different clone types are referred to as holoclones, meroclones, and paraclones [134]. The holoclones, which have a regular and compact shape, are enriched in CSCs and have the highest proliferative capacity. A similar phenomenon has been observed in head and neck squamous cell carcinoma and prostate carcinoma cells [135,136]. Recent studies have demonstrated that adherent culture is better than suspension culture because of the lower rate of spontaneous apoptosis and the high tumor-initiating ability in vivo [137,138]. Our observations also support the possibility of predicting “stemness” based on colony morphology in nonepithelial tumors. Tight clones formed by glioma cells also possess many CSC features, including the expression of SC markers (e.g., nestin, CD133, Sox2, Oct4, and CD44s), self-renewal, and multipotency [139].
By applying methodologies previously used for NSC culture in vitro [140], Pollard et al. demonstrated that CSCs can be grown as adherent monolayers using a simple modification of the suspension sphere culture. They obtained a monomorphic population that had many characteristics possessed by CSCs, such as the expression of SC markers (e.g., nestin, Sox2, and oligodendrocyte transcription factor 2), multipotency, genetic abnormalities consistent with the parental tumor, and tumorigenicity. Most importantly, these adherent glioma cells appear to be capable of long-term serial passaging; they contain a high percentage of true CSCs, with significantly fewer spontaneously differentiating or apoptotic cells; and they can be derived from nearly 100% of primary malignant gliomas [68]. However, what is interesting about this model is that these cells can recapitulate the original tumor phenotype after orthotopic injection. This model augments the previous validation of the serum-free de novo cell culture model [141,142].
Side population (SP) cells
A small population of cells, termed SP cells, was first described by Goodell et al. based on these cells' distinct ability to pump out Hoechst 33342. These cells were defined as a small subpopulation of cells with enriched SC activity, including clonogenic capacity, tumorigenicity, multipotency, long-term repopulating properties, and chemoresistance [143,144]. In recent years, some studies have indicated that the SP cells may be useful for the FACS-based identification and isolation of CSCs from human cancers [145 –151]. However, it has been demonstrated that both sorted SP and non-SP cells from several cancer cell lines have a similar capacity for clone formation, tumorigenicity, and multipotency. Additionally, the phenotypes of these two cell fractions are interconvertible, and one cell type can give rise to the other [152]. Thus, one hypothesis is that the SP and non-SP populations, albeit phenotypically distinct, do not differ with respect to the number of SC-like cells or behavior. Similarly, although SP cells from the adrenocortical NCI-h295R cell line have a less differentiated phenotype, the proliferation rates of SP and non-SP cells are the same. Furthermore, both cell types have the capacity to give rise to the original SP-containing and non-SP-containing cell populations. SP cells exhibit no survival enhancement after exposure to cytotoxic agents commonly used to treat adrenocortical carcinomas. In addition, the growth kinetics of SP cells does not differ from those of the non-SP cells [153]. SP cells from the gastric cancer BGC-823 cell line also do not behave as CSCs do [154]. However, some studies have yielded conflicting results. Wu et al. isolated SP cells from the same cell line and demonstrated that these cells had stem-like properties [155]. In short, these results suggest that the concept of the SP phenotype as a universal CSC marker does not apply to some cancers. Additionally, the potential cytotoxicity of Hoechst 33342 is controversial, and some researchers have proposed that the differences in clonogenicity and tumorigenicity between SP and non-SP cancer cells might be the result of the dye itself [57]. However, other researchers have reported that this dye is not harmful to some cancer cells, such as small cell lung cancer cells [156].
Cancer cell division
SCs are defined by their ability to generate more SCs (“self-renewal”) and produce differentiating cells. These two tasks can be accomplished through a single mode of self-renewing mitotic division (“asymmetric self-renewing division”), in which one progeny cell retains the SC identity, and the other (progenitor) undergoes multiple rounds of division before entering a fully differentiated state. The two cells generated by the asymmetric division differ markedly in their proliferative potential; the SC remains quiescent or proliferates slowly, whereas the progenitor cell actively divides. This process ensures the production of a large number of differentiated progeny cells while maintaining a relatively small pool of long-lived SCs [157]. Although CSCs possess a greater ability to divide through symmetric self-renewing division than normal SCs, the cell division time is still shorter compared with their differentiated descendants.
PKH-26 is a fluorescent dye that binds to cell membranes and segregates in daughter cells after each cell division. Therefore, the intensity of staining correlates inversely with the rate of cell division [158]. The division rates of different subpopulations in a tumor mass vary during the same time span. Cicalese et al. established a novel strategy for isolating CSCs from breast cancer based on the PKH-26 staining intensity [13,159]. The PKH-26hi cells are highly enriched in CSCs (∼1:1), and represent the only cell subset capable of reconstituting a mammary tumor; thus, suggesting that breast CSCs undergo a limited number of divisions. Other researchers have also used this strategy to isolate CSCs from different types of human tumors, such as gastrointestinal cancers, glioblastomas, and nasopharyngeal carcinomas [160 –163].
Carboxy fluorescein succinimidyl ester (CFSE) is a fluorescent dye that has been used to track the cell division frequency in several types of solid tumors. Recent studies have found that CFSE labeling can be used to identify and isolate a slow proliferating population of cells from glioblastomas. These dye-retaining brain tumor cell populations are enriched in CSCs, and the progeny that differentiate from these label-retaining cells exhibit all the pathological features of the primary disease [164,165]. Kamohara et al. used Hoechst 33342 and Pyronin Y to sort Huh7 cells into G0, G1, and G2/M fractions by FACS. They found that the G0 fraction was located within the neck of the SP fraction. These cells were capable of sphere formation and marked tumorigenesis. G0 cells, which do not express Ki67, were weakly positive for albumin expression and positive for keratin 19 expression. In contrast, the G1 cells were positive for Ki67 and albumin expression but negative for keratin 19. These findings suggest that the G0 Huh7 cell are promising CSC candidates [166]. Thus, most CSCs, if not all, possess slow proliferating characteristics. Furthermore, label-retaining techniques are unique methods for functionally identifying and isolating CSCs [167].
Cytotoxic and hypoxic resistance
It has been experimentally demonstrated that CSCs can resist apoptosis induced by cytotoxic drugs and radiation through multiple complicated mechanisms [4,5,168 –178]. Enhanced chemoresistance is associated with a SC-like phenotype in tumor cells. These undifferentiated CSCs express high levels of specific drug resistance-associated proteins, such as ABC, that preferentially activate DNA damage checkpoint genes in response to radiation or cytotoxic drugs and can repair DNA damage more effectively than the progenitors and committed cells [5,177,178]. Additionally, CSCs have been shown to overexpress antiapoptotic genes [173,179] and downregulate some tumor suppressor genes [180]. This characteristic of functional CSCs has been confirmed by many studies in recent years and has been proposed to be the root cause of chemoresistance [181]. Moreover, the fraction of CSCs increases significantly following chemotherapy and radiation [5,149]. Furthermore, the proportion of CD133-positive cells in U251 glioma cells increases after the administration of rotenone and ethidium bromide [182]. Recent research has revealed that melanoma cells are enriched in ABCB5-expressing cells after temozolomide treatment and that these ABCB5-expressing cells exhibit enhanced tumorigenicity, with SC-like properties [183]. Ionizing radiation can activate stemness pathways in HCC cells and can result in the enrichment of a CSC subpopulation with higher resistance to radiotherapy [184]. Furthermore, methotrexate (MTX)-resistant U2OS/MTX300 osteosarcoma cells, termed osteosarcoma SCs, are enriched by chemotherapy. These cells have a greater ability to generate sarcospheres, can form the original tumor, and express SC surface markers, including CD117 and Stro-1 [185]. These data indicate that resistance to cytotoxic compounds could be used to enrich CSCs from a tumor mass [186 –189]. However, Pajic et al. did not observe CSC enrichment in breast cancer 1 (BRCA1)- and p53 -deficient breast cancers after cisplatin treatment [190].
The link between hypoxia and CSCs derived from solid tumors is fairly strong; hypoxia maintains the undifferentiated status and promotes the self-renewal capability of the SC population and a more stem-like phenotype in the nonstem population [180,191,192]. Several core stemness-related transcription factors, including Oct4, Nanog, c-myc, and Notch, are the direct or indirect targets of hypoxia-inducible factors [180,192]. More importantly, solid tumor CSCs are predominantly localized in hypoxic zones in vivo [180]. Regarding the observation that the proportion of the CD133-positive cells is increased under hypoxic conditions, another possibility may be that CSCs possess greater resistance to hypoxic conditions and can therefore, be enriched under these conditions. One study revealed that intratumoral hypoxia can enrich the CSC population in breast cancer and can limit the effect of antiangiogenic agents [193]. A similar result was found in another study, which identified a stem-like cell population in a metastatic breast cancer cell line subjected to repeated cycles of hypoxia and reoxygenation [194].
Invasiveness and adhesion
There is increasing evidence indicating that CSCs may possess a distinct potential for adherence, migration, and invasion. In glioma specimens, many nestin+/Sox2+ glioma cells, which can infiltrate into the surrounding normal tissue, are detected at the tumor-brain interface [141]. Huang et al. demonstrated that CSCs are more aggressive and invasive than core glioma cells [195]. Additionally, colorectal carcinoma cells with an enhanced migration and invasion capacity at the tumor's invading edge express SC markers [196]. Several studies have demonstrated that a CSC subset with the capacity for migration in the CD133+ CSC compartment express high CXCR4 or matrix metalloproteinase-2 (MMP-2) levels, and this high expression was correlated with metastatic potential [197,198]. A greater proportion of holoclone colonies with an indefinite proliferation capacity and enriched in CSCs is formed by early adherent cells [134]. Moreover, distinct CSCs were identified as having a CD133+ CXCR4+ phenotype at the invasive front of pancreatic tumors. The depletion of this subpopulation almost completely abrogated the metastatic phenotype of pancreatic tumors [28]. Many invasion-associated molecules are overexpressed in CSCs [4,168]. Extracellular signal-regulated kinase (ERK), N-cadherin, and integrinα6 have been found to play an important role during glioma stem cell invasion [199]. CD44+ prostate CSC-like cells possess a higher capacity for matrigel invasion, which is in contrast with the noninvasive nature of CD44− cells. More importantly, the genotype of the invasive cells closely resembles that of the CD44+CD24− prostate CSCs. These invasive cells are more tumorigenic than noninvasive cells [200]. In addition, transforming growth factor-β (TGF-β), snail and forkhead box protein C2 (FOXC2) -mediated EMT has been suggested to induce a CSCs phenotype accompanied with high metastatic potential [201 –203]. Although many studies have demonstrated a relationship between CSCs in the primary tumor and metastasis in the secondary tumor, we still lack evidence for a causal relationship between the primary tumor and distant metastases. However, current studies and recent data strongly suggest that basement membrane invasion can be used as a tool to isolate and enrich CSCs from a tumor mass. In our previous study, we proposed a novel protocol to isolate and enrich CSCs from a tumor mass based on the heterogeneity of tumor cells with respect to invasiveness and adherence. Using this method, we successfully isolated SC-like subpopulations from glioma cell lines and resected samples [204].
Notably, based on the prediction that CSCs are readily detachable from tissue-culture plastic, Walia et al. established another interesting method to isolate CSCs. After dividing immortalized or transformed mammary epithelial cells into trypsin-sensitive and trypsin-resistant populations, these researchers demonstrated that the trypsin-sensitive cells were mesenchymal in morphology and expression profile, had enriched SC properties, and displayed mammosphere-forming properties, drug resistance, and CD44 expression. After several rounds of differential trypsinization, the trypsin-sensitive pool had an 80-fold higher mammosphere-forming ability than the trypsin-resistant population and a 20-fold higher ability than the starting population. Trypsin-sensitive cells are regarded as breast CSCs, and this method is faster, more affordable, and more efficient than other enrichment methods. Thus, differential adhesion may serve as the basis for an enrichment strategy to increase the size of the SC pool for subsequent manipulations for relatively differentiated epithelial cell types [205].
CSC immunoselection with natural killer cells
The responses of different tumor cell subpopulations to immune cells are heterogeneous. Reim et al. observed that sphere-forming MCF7 cells, which display a CD44hiCD24lo ‘‘CSC–like’’ phenotype, preferentially survived following treatment with natural killer (NK) cells. These surviving tumor cells displayed increased clonogenicity and tumorigenicity [206]. Similar results have been observed for glioma CSCs, suggesting that human leukocyte antigen-E (HLA-E) plays an important role in inhibiting NK cell-mediated lysis. HLA-E gene silencing in glioma CSCs can enhance the susceptibility to NK-cell mediated lysis [207]. Similarly, the CD44hiCD24lo population has also been found to be more immunoresistant in other breast cancer cell lines, such as SK-BR3, MDA-MB-231, and BT474 cells. Recently, CXCR4+ metastatic CSCs were shown to be induced by interferon-γ (IFN-γ) from NK resistant CSCs harbored in the highly tumorigenic CD44hiCD24lo subset [208]. Thus, a novel strategy based on immunoselection with NK cells could be used to isolate and enrich CSCs from cancer cell populations [209].
However, other studies have demonstrated that CSCs and non-CSCs derived from a melanoma cell line are equally susceptible to NK cell-mediated lysis [209]. Tseng et al. demonstrated that CSCs were more sensitive than their differentiated descendants. When NK cells were coincubated with primary oral squamous CSCs, increased cytotoxicity and augmented IFN-γ secretion were observed compared with differentiated cells [210]. Similar results for primary oral squamous carcinoma demonstrated that CSCs were significantly more susceptible to NK cell-mediated cytotoxicity than their differentiated counterparts or the parental cells [211]. Thus, it is questionable whether the immunoselection of tumor populations with NK cells can be used as a strategy for CSC isolation and enrichment.
Density gradient centrifugation
Recently, an interesting strategy has been developed to isolate CSCs from a primary rat HCC model using the physical properties of CSCs. Hepatic tumor cells were first isolated from the diethylnitrosamine-induced F344 rat HCC model by Percoll discontinuous gradient centrifugation (PDGC), followed by purification using differential trypsinization and differential attachment (DTDA). Among the four cell fractions (FI–FIV) obtained using this strategy, the third fraction (FIII) was enriched in cancer stem-like cells. This fraction had a higher nuclear-to-cytoplasmic ratio and expressed higher levels of SC markers, including alpha fetoprotein, EpCAM, and CD133. Furthermore, FIII exhibited a greater self-renewal capacity, multipotency, membrane invasiveness, and chemoresistance. The cells in this fraction were also found to form tumors in both the subcutaneous tissue and livers of nude mice in vivo. Taken together, the results of that study provide the basis for a novel strategy to isolate and enrich CSCs using the physical properties of these cells [212].
Conclusions and Perspectives
As mentioned above, there are many strategies for the isolation and/or enrichment of different types of CSCs (the suitable cancer types, CSC frequencies in the enriched populations, and references are summarized in Table 1). However, the scope, advantages, and limitations of these different strategies remain to be elucidated. In addition, the level of phenotypic similarity among cells obtained from the same starting material using different techniques remains to be determined. Thus, comparisons of different methods in the same lab using single tumors as the starting material are needed in the future. Kuch et al. performed an extensive cross-comparison of sphere formation capability, cell division, and putative CSC marker expression with the tumor-initiating properties of different melanoma and mammary tumor cells. Their observations suggest that it cannot be assumed that there is always a direct relationship between the sphere-forming ability, the expression of CSC markers, label retention, and the number of TICs in vivo. Indeed, there can be discrepancies in the identification of CSC-containing subpopulations when using different methods [213].
In addition, some studies have indicated that CSCs in the same type of human cancer are heterogeneous [214]. This heterogeneity is not limited to the differential expression of surface markers but also involves various functional subsets of CSCs, which may be the result of genetic mutations and epigenetic modifications. For example, in pancreatic cancer, both CD133+CXCR4−and CD133+CXCR4+ populations were found to exhibit similar levels of tumorigenicity. However, the metastatic phenotype of the individual tumor was determined by the distinct subpopulation of CD133+CXCR4+ CSCs, and the metastatic phenotype, but not the tumorigenic potential, of pancreatic tumors was nearly abrogated after the depletion of this subset of cells [28]. In breast cancer, both ALDH1+ and CD44+CD24–/lo cells are thought to be CSCs. Interestingly, the overlapping subset of these two populations, ALDH1+CD44+CD24–/lo cells, exhibits greater tumorigenicity than the cells expressing either marker alone [73]. These results suggest that different phenotypes may exist in the overlapping tumorigenic population; thus, combination strategies rather than single strategies are needed to enrich or isolate CSCs.
In summary, research into CSCs is still in its infancy. CSC isolation is viewed as the foundation and starting point for better understanding the characteristics of this specific subpopulation. An ideal method for isolating CSCs should have the following characteristics. (i) Specificity: The isolated cells should not contain any nontumorigenic cells, progenitor cells, or differentiated cells and should be able to form the original tumor in mice using as few as one cell. (ii) Sensitivity: All CSC subgroups that can initiate tumor formation should be included in the isolated cells. (iii) Versatility: The method should be applicable for the isolation of CSCs from different types of tumors. (iv) Convenience: The isolation procedures should be simple, use limited resources (such as time, effort, and energy), and be user friendly. The obvious limitations include surface marker sorting, SP isolation, and sphere culture; however, many researchers have expended great effort to establish a simpler, universal method for isolating CSCs. It is reasonable to believe that additional reliable, efficient, and convenient methods for isolating CSCs from tumor masses will be developed in the near future.
Finally, it should be emphasized again that the CSC hypothesis is based on the functional identification of TICs in vivo using the serial xenotransplantation assay, which serves as the gold standard [6].
Footnotes
Acknowledgments
We would like to thank Prof. Peng Zhang, Dr. Zhi-hua Zhou, and Dr. Lang Yang for their constructive suggestions. This study was supported by grants from the National Natural Science Foundation of China (no. 81172071, 81272598), the National Basic Research Program of China (973 Program, no. 2010CB529400), and the National S&T Major Special Project on New Drug Innovation of China (no. 2011ZX09102-010-02).
Author Disclosure Statement
No competing financial interests exist.