
Abstract
α2-macroglobulin (α2M) is an acute-phase protein released upon challenges like cardiac hypertrophy and infarction. α2M signals via the low density lipoprotein receptor-related protein (LRP-1) and may induce stem cell activation. In the present study, the effects of α2M on vasculogenesis/angiogenesis and underlying signaling cascades were investigated in mouse embryonic stem (ES) cells. LRP-1 was expressed in ES cells and upregulated during differentiation. α2M dose dependently increased CD31-positive vascular structures in ES cell-derived embryoid bodies, the early cardiovascular markers isl-1, Nkx-2.5, and flk-1 as well as numbers of VE-cadherin and flk-1-positive cells, but downregulated α-smooth muscle actin. Enhancement of vasculogenesis/angiogenesis by α2M was abolished by the LRP-1 antagonist receptor-associated protein (RAP) and LRP-1 blocking antibody. Notably, α2M stimulated vascular growth in the chicken chorioallantois membrane assay, but not in a human umbilical vein endothelial cell spheroid model. α2M increased fibroblast growth factor-2 (FGF-2) protein expression, which was abolished by RAP, induced nitric oxide (NO) generation as determined by 4,5-diaminofluorescein diacetate microfluorometry, and activated nitric oxide synthase-3 (NOS-3) as well as extracellular-regulated kinase 1,2 (ERK1/2) and phosphatidyl inositol 3-kinase (PI3K). NO generation, the increase in FGF-2 expression, and the stimulation of vasculogenesis/angiogenesis by α2M were blunted by the NO synthase inhibitor L-NAME, the ERK1/2 inhibitor PD98059, and the PI3K inhibitor LY294002. Furthermore, vasculogenesis/angiogenesis by α2M was inhibited in the presence of the FGF receptor 1 antagonist SU5402. In conclusion, α2M stimulates endothelial and early cardiac, but not smooth muscle differentiation of ES cells through generation of NO, activation of ERK1/2 as well as PI3K, and induction of FGF-2 expression.
Introduction
T
The earliest organ system developed during embryonic development is the cardiovascular system. Blood vessel formation occurs by a process referred to as vasculogenesis, whereas angiogenesis is referred to as vascular growth from already existing blood vessels. In mammals, extraembryonic vasculogenesis precedes intraembryonic vascular development and is initiated shortly after gastrulation. Endothelial precursor cells emerge mainly from mesodermal precursors [13]. During vasculogenesis, major embryonic vessels form by fusion of endothelial progenitor cells or angioblasts that arise de novo from extraembryonic and embryonic lateral mesoderm. These progenitors form vesicles and cords of attached vascular endothelial cells (ECs) that undergo further morphogenesis to form epithelial tubes [14].
The present study was undertaken to investigate whether the acute phase protein α2M may stimulate vasculogenesis/angiogenesis of stem cells via LRP-1 signaling. Vasculogenesis/angiogenesis in embryoid bodies derived from embryonic stem (ES) cells is well described and has been shown to mimic the development of the embryonic yolk sac vasculature [15,16]. Indeed our data demonstrate that α2M enhances vasculogenesis/angiogenesis of ES cells via a signaling pathway that involves an NO-mediated increase of the proangiogenic growth factor FGF-2, and exerts a proangiogenic effect in the in vivo chicken chorioallantois membrane (CAM) assay.
Materials and Methods
Materials
α2M isolated from human plasma was obtained from Sigma (Deisenhofen, Germany). PD98059, LY294002, L-NAME, and SU5402 were obtained from Calbiochem (Bad Soden, Germany). The receptor-associated protein (RAP) was purchased from Research Diagnostics (Concord, MA).
Spinner-culture technique for cultivation of embryoid bodies
In the present study, the ES cell line CCE isolated in 1986 by Robertson et al. from a 129/Sv mouse strain [17] was used. To obtain embryoid bodies, ES cells were grown on mitotically inactivated feeder layers of primary murine embryonic fibroblasts in the Iscove's medium (Gibco; Live Technologies, Helgerman Court, MD) supplemented with 18% heat-inactivated (56°C, 30 min) fetal calf serum (FCS) (Sigma), 2 mM glutamine, (PAA, Cölbe, Germany), 100 μM β-mercaptoethanol (Sigma), a 1% (v/v) NEA nonessential amino acids stock solution (100×) (Biochrom, Berlin, Germany), 0.8% (v/v) minimal essential medium amino acids (50×) (Biochrom), 1 mM Na+-pyruvate (Biochrom), 0.25% (v/v) penicillin/streptomycin (200×) (Biochrom), and 1000 U/mL leukemia inhibitory factor (Chemicon, Hampshire, UK) in a humidified environment containing 5% CO2 at 37°C, and passaged every 2–3 days. At day 0 of differentiation, adherent cells were enzymatically dissociated using 0.05% trypsin-EDTA in phosphate-buffered saline (PBS) (Gibco), and seeded at a density of 3·106 cells mL−1 in 250-mL siliconized spinner flasks (Integra Biosciences, Fernwald, Germany) containing 100 mL of Iscove's medium supplemented with the same additives as described above. Following 24 h, 150 mL of the medium was added to give a final volume of 250 mL. The spinner flask medium was stirred at 20 r.p.m. using a stirrer system (Integra Biosciences), and 150 mL of the cell culture medium was exchanged every day. For incubation with α2M, 3-day-old embryoid bodies were removed from spinner flasks, transferred to 6-cm bacteriological Petri dishes (Greiner-Bio One, Frickenhausen, Germany) ref. 628102, lot F090801C, filled with 5 mL of the serum-replacement medium (Gibco; Life Technologies, Darmstadt, Germany), and treated with α2M in concentrations as indicated from day 3 to day 8. The cell culture medium was replaced every day.
Immunohistochemistry
As primary antibodies, a rat monoclonal anti-CD31 antibody (Chemicon International, Hampshire, UK) (dilution 1: 100), a polyclonal goat anti-rabbit phospho-NOS 3 (Chemicon, Temecula, CA), and a monoclonal mouse anti-FGF-2 antibody (Millipore, Schwalbach, Germany) were used. Whole mount embryoid bodies in suspension culture were fixed in 4% paraformaldehyde for 15 min, subsequently in ice cold methanol for 20 min at −20°C, and washed 3 times with PBS containing 1% Triton X-100 (PBST) (Sigma). Blocking against unspecific binding was performed for 60 min with 10% FCS dissolved in 0.01% PBST. The cells were subsequently incubated for 60 min at room temperature with primary antibodies dissolved in PBS supplemented with 10% FCS in 0.01% PBST. The cells were thereafter washed 3 times with PBST (0.01% Triton) and reincubated either with a Cy5-conjugated goat anti-rabbit IgG (H+L) (Dianova, Hamburg, Germany) or a Cy5-conjugated goat anti-rat IgG (H+L) (Dianova) at a concentration of 3.8 μg/mL in PBS containing 10% FCS in 0.01% PBST. After washing 3 times in PBST (0.01% Triton), the cells were stored in PBS until inspection. Fluorescence recordings were performed by means of a confocal laser scanning setup (Leica TCS SP2, Bensheim, Germany). The confocal setup was equipped with a 5 mW helium/neon laser single excitation 633 nm (excitation of Cy5). Emission was recorded at >665 nm.
Flow cytometry
Embryoid bodies were treated with 4 μg/mL α2M in the presence/absence of RAP (10 nM) from day 3 to day 9 of differentiation. Subsequently, embryoid bodies were washed twice with prewarmed PBS and incubated for 10 min at 37°C in 2 mg/mL (PBS) collagenase B (Roche, Grenzach-Wyhlen, Germany). They were then dissociated by gentle pipetting and passed through a 40-mm cell strainer. Single-cell suspensions were centrifuged at 400g for 5 min at 4°C, resuspended at 1×106 cells/mL in precooled PBS containing 2% fetal calf serum [flow cytometry (FCM) buffer], and stained for 20 min at 4°C with the PE-conjugated flk-1 antibody (BD Biosciences, Heidelberg, Germany), rat anti-CD31 antibody (Chemicon-Millipore Billerica, MA), and rabbit anti-VE-cadherin antibody (Abcam, Cambridge, UK). As secondary antibodies, Alexa 488 donkey anti-rat and Alexa 488 donkey anti-rabbit (both from Life Technologies, Darmstadt, Germany) were applied. In each experiment, IgG-matched isotype controls were used. For analysis, cells were resuspended in 300 μL of the FCM buffer and data were acquired and analyzed by a fluorescence-activated cell sorter Calibur instrument (BD) and BD CellQuest Pro software.
Quantification of vasculogenesis/angiogenesis in embryoid bodies
For the quantification of vasculogenesis/angiogenesis within embryoid bodies, an optical sectioning routine based on confocal laser scanning microscopy (Leica TCS SP2) was used. Images (512×512 pixels) were acquired from CD31-labeled embryoid bodies using the extended depth of focus algorithm of the confocal setup. In brief, 5 full-frame images separated by a distance of 20 μm in the z-direction were recorded that included the information of the vascular spatial organization in a tissue slice 100 μm thick. From the acquired images, an overlay image giving a 3-dimensional projection of the vascular structures in the scanned tissue slice was generated. By use of the image analysis facilities of the confocal setup, the branching points of vascular structures within the 3-dimensional projection of vascular structures were identified and counted in relation to the size of the respective embryoid body.
Spheroid/embryoid body sprouting assay
Spheroids were generated from human umbilical vein endothelial cells (HUVECs) (3 • 103 cells/spheroid) and embryoid bodies were cultured from ES cells (1 • 103 cells/embryoid body) using the hanging drop method in the Iscove's cell culture medium supplemented with 20% methylcellulose (Sigma). They were incubated for 24 h, during which spherical cell aggregates formed. HUVEC spheroids or embryoid bodies were separately embedded into a 3D fibrinogen matrix (Calbiochem, Bad Soden, Germany) and stimulated with either α2M (4 μg/mL) or FGF-2 (10 ng/mL) in the Iscove's cell culture medium and cultured for further 24 h before fixing in 4% para formaldehyde. The cumulative sprout length was determined as a measure of vascular growth.
CAM assay
The CAM assay was performed as described previously [18]. Briefly, fertilized white chicken eggs were incubated at 37°C and 60% humidity in an egg incubator for 48 h. After albumen removal, a square window was cut into the shell with the aid of small dissecting scissors. On day 8 of incubation a sterilized gelatine sponge (1 mm3) (Gelfoam, Pfizer, NY) was implanted onto the CAM. A volume of 3 μL of α2M (0.5–2 mg/mL) was pipetted once onto the sponge. Eggs with sponges loaded with vehicle (PBS) were used as appropriate controls. At day 12, photographs were taken from the CAM and a vascular index was determined by enumerating all the vessels that converge toward the implant and are contained inside a 1-mm diameter ring superposed on the CAM.
Western blot analysis
The western blot assays were carried out after washing the embryoid bodies in PBS and lysing in the lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulphate (SDS), 1% Nonidet P-40, 2.5) supplemented with protease inhibitor cocktail (Biovision, Hannover, Germany), and phosphathase inhibitor cocktail (Sigma) for 20 min on ice. Samples were centrifuged at 13,000g for 10 min to pellet the debris. After determination of the protein concentration using a Lowry protein assay (Bio-Rad, Munich, Germany), 30 μg of protein samples was boiled, separated in 10% SDS polyacrylamide gels, and transferred to polyvinylidene fluoride membranes by the iBlot dry blotting system (Invitrogen, Karlsruhe, Germany) at 20 V for 7 min. Membranes were blocked with 5% (wt/vol) dry fat-free milk powder in Tris-buffered saline with 0.1% Tween (TBST) for 60 min at room temperature. Incubation with a primary antibody was performed at 4°C overnight. The primary antibodies used were a monoclonal rabbit anti-FGF-2 antibody (Cell Signaling Technology, Frankfurt, Germany), a monoclonal rabbit anti-mouse LRP-1 antibody (Abcam, Cambridge, UK), a polyclonal rabbit anti-phospho-phosphatidyl inositol 3-kinase (PI3K) p85 (Tyr458)/p55 (Tyr199) antibody (New England Biolabs, Frankfurt, Germany), a polyclonal rabbit anti-mouse α-smooth muscle actin antibody (Abcam), a polyclonal rabbit anti-Nkx-2.5 antibody (Abcam), a polyclonal rabbit anti-isl-1 antibody (Chemicon, Temecula, CA), a polyclonal rabbit anti-flk-1 antibody (Cell Signaling Technology), and a polyclonal goat anti-rabbit NOS3 antibody (Chemicon). After washing with 0.1% TBST, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology) for 60 min at room temperature. The blot was developed using the electrochemiluminescence detection kit (Perkin Elmer, Freiburg, Germany) to produce a chemiluminescence signal.
Measurement of NO generation
NO generation was evaluated by the use of the cell permeable-specific fluorescent NO indicator 4,5-diaminofluorescein diacetate (DAF-2DA). After incorporation into cells, DAF-2 reacts rapidly with NO in the presence of oxygen to yield the highly fluorescent compound triazolofluorescein (DAF-2T). Cells grown on coverslips to confluency were incubated for 30 min with 10 μM DAF-2DA dissolved in the E1 buffer, containing (in mM) NaCl 135, KCl 5.4, CaCl2 1.8, MgCl2 1, glucose 10, and HEPES 10 (pH 7.4 at 23°C). Subsequently, coverslips were transferred to an incubation chamber mounted to the inspection table of the confocal setup, and DAF-2T fluorescence was recorded in single cells using the 488 nm of the argon-ion laser of the confocal setup. Emission was recorded at >515 nm.
Statistical analysis
Data are given as mean values±SD, with n denoting the number of experiments unless otherwise indicated. One-way ANOVA for unpaired data and the Student's t-test were applied as appropriate. A value of P<0.05 was considered significant.
Results
Stimulation of vasculogenesis/angiogenesis by α2M and expression of LRP-1 during differentiation of ES cells
ES cells grown in the 3-dimensional tissue of embryoid bodies spontaneously differentiate toward endothelial cells, which line the lumen of capillary-like blood vessels (see Supplementary Fig. 1; Supplementary Data are available online at
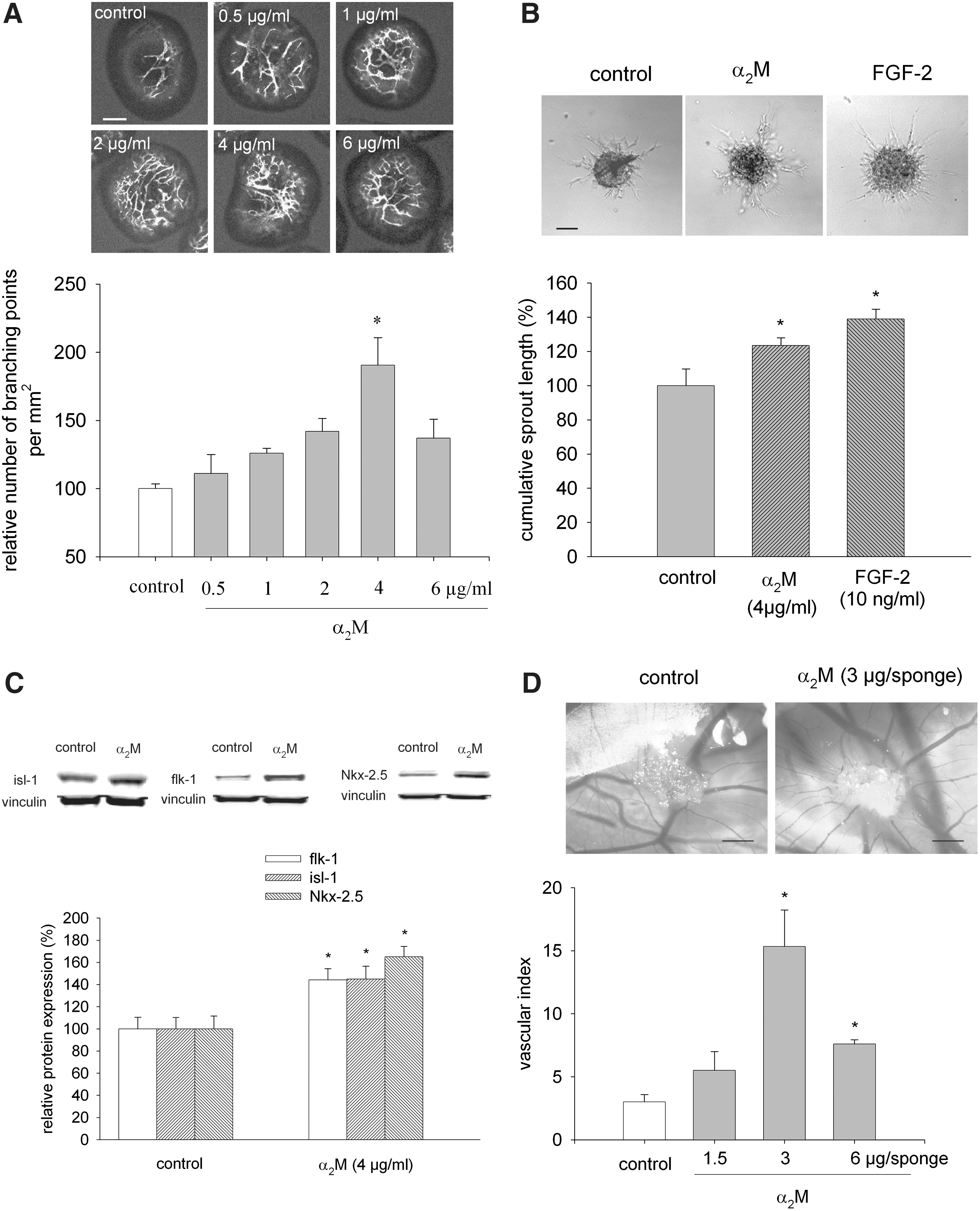
Stimulation of vasculogenesis/angiogenesis of embryonic stem (ES) cells by α2-macroglobulin (α2M).
To test the proangiogenic effects of α2M in an in vivo model, the CAM assay was applied. After 3 days of treatment with α2M (0.5–2 mg/mL, i.e., 1.5 μg, 3 μg, and 6 μg α2M, respectively, per sponge), vascular sprouting toward the gelatin sponge loaded with α2M was observed, which was maximum with an amount of 3 μg α2M/sponge (Fig. 1D, n=4 per experimental condition). Interestingly, α2M failed to significantly induce vascular growth in a HUVEC sprout assay, indicating that this substance was acting only on vascular progenitor cells (data not shown).
Since α2M may act via LRP-1, its expression was monitored in undifferentiated ES cells and during the differentiation process (Fig. 2A, n=3). LRP-1 was already prominently expressed in undifferentiated ES cells. At day 2 of differentiation, LRP-1 was downregulated and re-expressed during the time where vasculogenesis/angiogenesis of ES cells occurs. When embryoid bodies were coincubated with either an antibody directed against the α2M receptor LRP-1 (0.2 μg/mL) (Fig. 2B, n=6) or RAP (10 nM) (Fig. 2C, n=5), which inhibits binding of all known ligands to LRP-1, the increase in vasculogenesis/angiogenesis by α2M was significantly inhibited, suggesting that α2M was acting via the LRP-1 receptor. To corroborate our results based on the computer-assisted image analysis of CD31-positive confocal images, we performed the FCM to analyze changes in cell numbers positive for VE-cadherin, which is expressed in terminally differentiated endothelial cells and flk-1, which is a marker of cardiovascular progenitor cells. Indeed, treatment with α2M (4 μg/mL) significantly increased the relative numbers of VE-cadherin as well as flk-1-positive cells, which was completely abrogated in the presence of RAP (10 nM) (Fig. 2D, E, n=3). Comparable data were achieved when the number of CD31-positive cells was assessed in the absence or presence of RAP (data not shown). To investigate whether α2M was acting on proliferation of mature endothelial cells rather than differentiation of stem cells, HUVECs were treated with α2M and the number of cell nuclei that were positive for the proliferation marker Ki-67 assessed. However, no stimulation of endothelial cell proliferation was observed under these experimental conditions (data not shown).

Expression of low density lipoprotein receptor-related protein (LRP-1) in ES cells and during ES cell differentiation
Activation of extracellular regulated kinase 1,2 and PI3K in embryoid bodies upon treatment with α2M
Binding of α2M to LRP-1 may activate distinct signaling cascades known to be involved in vasculogenesis/angiogenesis of ES cells [19,20]. We have previously demonstrated that α2M activates extracellular regulated kinase 1,2 (ERK1/2) and PI3K in mouse ventricular cells and stimulates hypertrophic cell growth [21]. To investigate whether comparable signaling pathways existed in ES cells, 3-day-old embryoid bodies were incubated with 4 μg/mL α2M and phosphorylation of either ERK1/2 or PI3K was assessed by western blot. It was apparent that α2M activated ERK1/2 (Fig. 3B, n=4) as well as PI3K (Fig. 3A, n=3) with maximum activation achieved at 15–30 min.

Activation of phosphatidyl inositol 3-kinase (PI3K) and extracellular regulated kinase 1,2 (ERK1/2) upon treatment of embryoid bodies with α2M. Four-day-old embryoid bodies were treated with 4 μg/mL α2M, lysed at times as indicated, and the phosphorylation of either PI3K
Generation of NO and activation of NOS3 by α2M
NO has been previously shown by us to be a prominent stimulator of cardiovascular differentiation of ES cells [22]. We therefore hypothesized that α2M may increase NO generation by activation of NOS3. To corroborate this assumption, 5-day-old embryoid bodies were stained with the NO indicator 3-amino, 4-aminoethyl-2′,7′difluorofluorescence diacetate and the DAF fluorescence increase was monitored over 50 min. Upon incubation with α2M, an increase in DAF fluorescence was observed, which reached significant values after ∼30 min (Fig. 4A, n=3). Furthermore, activation of NOS3 was assessed by semiquantitative immunohistochemistry (data not shown) and western blot analysis. Phosphorylation of NOS3 (Fig. 4B, C, n=4) was observed after α2M treatment and remained elevated for at least 240 min, whereas the expression of native NOS3 remained unchanged. To investigate the signaling cascade involved in the generation of NO, embryoid bodies were treated with α2M either in the presence or absence of the ERK1/2 inhibitor PD98059 (40 μM), the PI3K inhibitor LY294002 (20 μM), or the NOS inhibitor L-NAME (100 μM) (Fig. 4C, n=3). Under these experimental conditions, the increase in NO generation was totally abolished, suggesting an involvement of the PI3K, ERK1/2 signaling cascade in NOS3 phosphorylation and NO generation upon treatment with α2M.

Generation of NO upon treatment of embryoid bodies with α2M.
Increase of FGF-2 expression upon α2M treatment
FGF-2 is an important proangiogenic growth factor and has been previously shown to stimulate vasculogenesis/angiogenesis of ES cells [15]. Angioblasts (endothelial cell precursors) appear to be induced by FGF-2 [23]. To investigate the mechanism of the proangiogenic pathway elicited by α2M, 4-day-old embryoid bodies were treated for 24 h with increasing concentrations of α2M ranging from 0.5–6 μg/mL α2M, and FGF-2 expression was monitored by western blot. Our data demonstrated that FGF-2 expression was significantly and dose dependently increased upon treatment of embryoid bodies with α2M, which may suggest that α2M was activating the FGF-2 signaling pathway (Fig. 5A, n=4). To investigate whether α2M increased FGF-2 expression via activation of LRP-1, embryoid bodies were treated with α2M either in the presence or absence of RAP. Under these experimental conditions, the stimulation of FGF-2 expression by α2M was totally abolished (Fig. 5B, n=6). This clearly demonstrates that α2M acted via the LRP-1 receptor, which may then downstream elicit a signaling cascade resulting in the stimulation of vasculogenesis/angiogenesis. To directly correlate the α2M-mediated increase in FGF-2 expression with the stimulation of vasculogenesis/angiogenesis of ES cells, 4-day-old embryoid bodies were coincubated with the SU5402 (20 μM), which binds to the tyrosine kinase domain of the FGF-receptor 1, thereby inhibiting the downstream signaling pathway in either the presence or absence of α2M. Under these experimental conditions, the stimulation of vasculogenesis/angiogenesis by α2M was totally abolished (Fig. 5C, n=3). Interestingly, SU5402 strongly inhibited vasculogenesis/angiogenesis already in the absence of α2M, suggesting an important role of intrinsic FGF-2 for vasculogenic differentiation of ES cells. Notably, exogenous addition of FGF-2 (10 ng/mL) strongly stimulated vasculogenesis/angiogenesis of ES cells (data not shown).
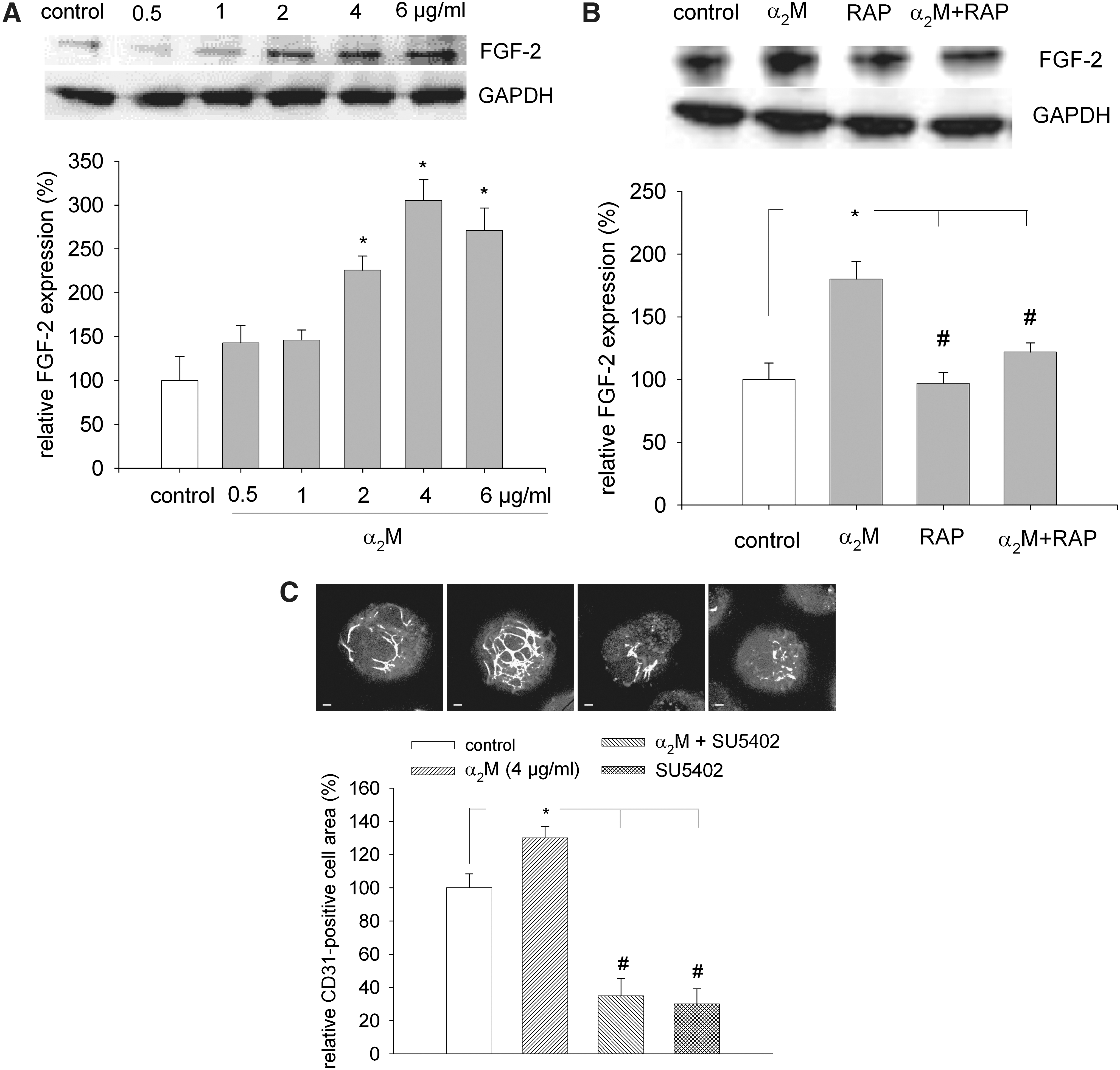
Increase of FGF-2 expression upon treatment of embryoid bodies with α2M and effects of RAP and FGF-2 receptor antagonist.
Since our data demonstrated that α2M activated NOS3, PI3K, and ERK1/2, we finally investigated whether pharmacological inhibition of the respective effectors would abolish the increase in FGF-2 protein expression by α2M. Indeed, preincubation with PD98059 (40 μM), LY294002 (20 μM), or L-NAME (100 μM) (Fig. 6, n=7) totally abolished FGF-2 upregulation by α2M as assessed by semiquantitative immunohistochemistry, indicating that α2M upregulated FGF-2 by NO generation and activation of ERK1/2 and PI3K (see Fig. 7). Furthermore, coadministration of PD98059, LY294002, and L-NAME with α2M abolished the proangiogenic activity of the compound (data not shown).
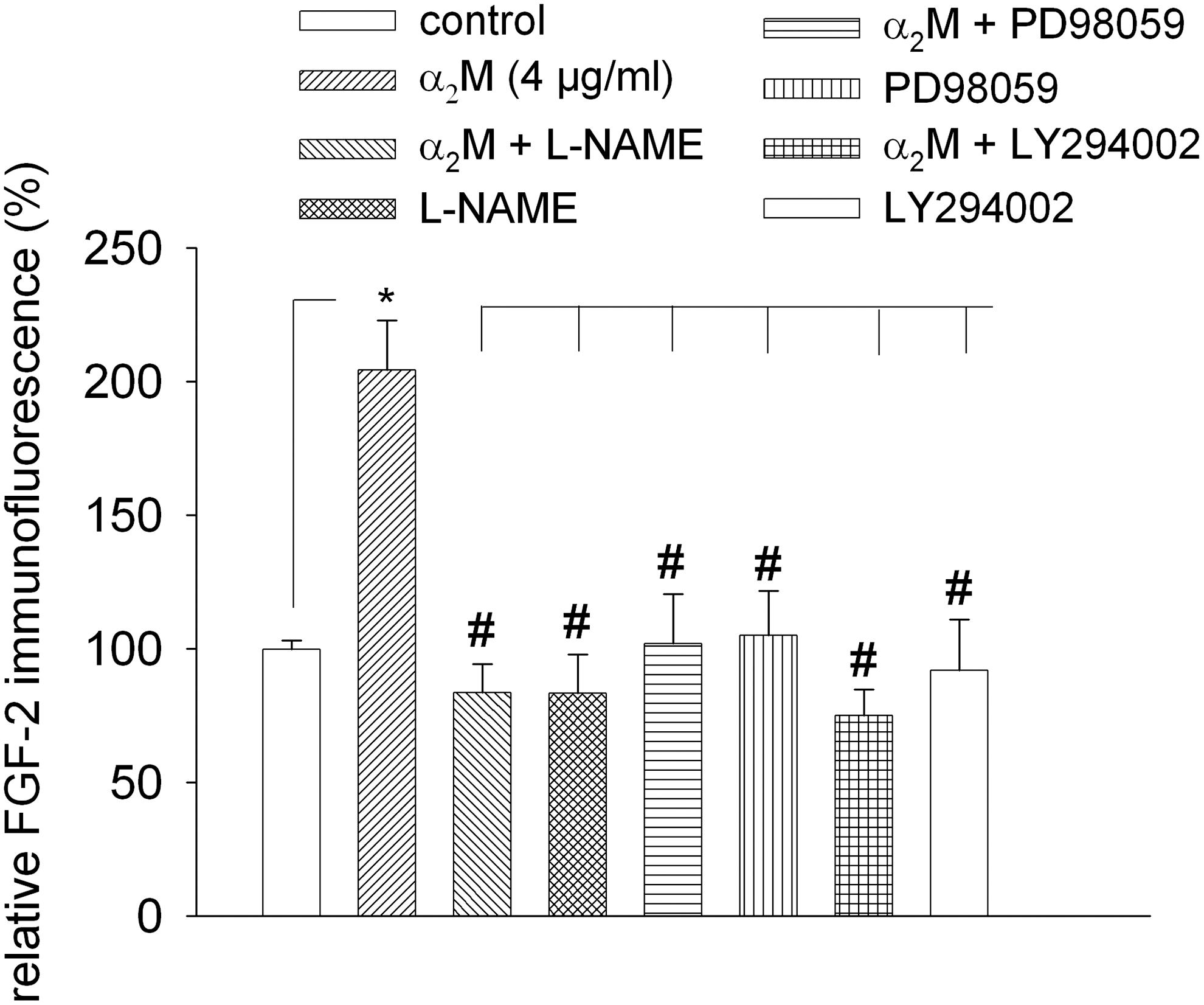
Effect of inhibition of NOS, PI3K, and ERK1/2 on FGF-2 expression upon treatment with α2M. Four-day-old embryoid bodies were treated with either α2M alone or with α2M in the presence of the NOS inhibitor L-NAME (100 μM), the PI3K inhibitor LY294002 (20 μM), or the ERK1/2 inhibitor PD98059 (40 μM). FGF-2 expression was analyzed after 24 h of incubation by semiquantitative immunofluorescence. Under these experimental conditions, the α2M-induced stimulation of FGF-2 expression was totally inhibited. Note that incubation with inhibitors alone did not change FGF-2 expression levels. *,# P<0.05, significantly different as indicated.
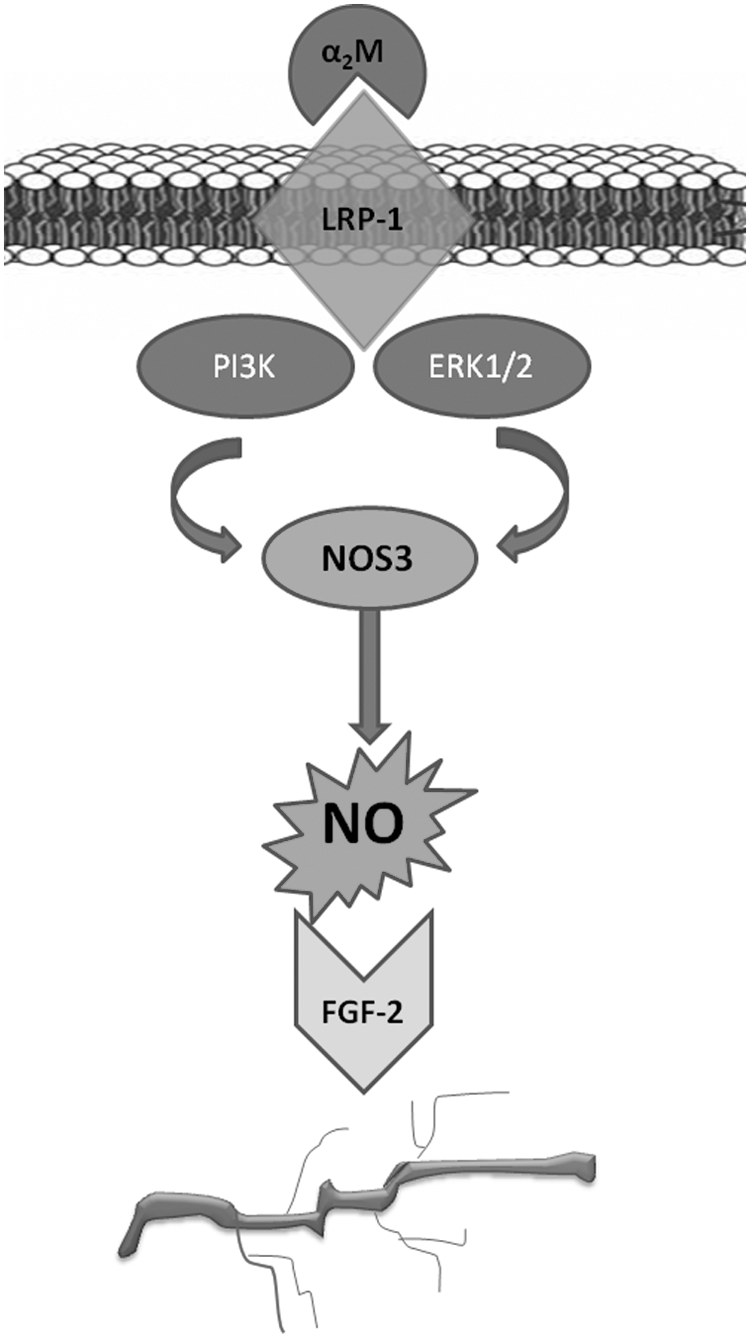
Model of the α2M downstream signaling cascade. Upon binding of α2M to the LRP-1 receptor, PI3K and ERK1/2 are phosphorylated. This stimulates NOS3 activity and NO generation. NO signaling results in upregulation of FGF-2, which is a potent proangiogenic growth factor in the ES cell system.
Discussion
Numerous studies have been performed on the action of α2M as a proteinase inhibitor and an early immediate response factor released upon various diseases and insults. However, the impact α2M as a stimulator of vasculogenesis/angiogenesis of stem cells has not been investigated, and the literature on the intracellular signaling cascades elicited by α2M is still narrow. In the present study, it is demonstrated that α2M stimulated vasculogenesis/angiogenesis of mouse ES cells and vascular growth in the in vivo CAM assay. This observation is of clinical importance in light of a possible biological effect of α2M in wound healing. Indeed, accumulation of α2M has been previously demonstrated in wounds [24] and skin burns [25,26], which suggests involvement in healing processes, including angiogenesis and stem cell activation. Furthermore, early endothelial progenitor cells display high expression levels of α2M [27], suggesting a role in endothelial progenitor cell maturation.
The data of the present study demonstrate that α2M dose dependently stimulated vasculogenesis/angiogenesis and stimulated protein expression of the early cardiovascular markers isl-1, Nkx-2.5, and flk-1, which indicates that α2M indeed initiated differentiation processes rather than proliferation of already existing endothelial cells. This assumption was supported by the observation that α2M did not elicit proliferation and sprouting of mature HUVEC cells, but did stimulate vascular growth in the in vivo CAM assay, which represents an embryonic system comparable to the ES cell assay.
Interestingly, expression of the smooth muscle marker α-smooth muscle actin was downregulated, indicating that the vascular structures were capillaries that are devoid of a tunica media of smooth muscle cells.
Since the stimulation of vasculogenesis/angiogenesis was abolished in the presence of the LRP-1 antagonist RAP or an antibody directed against LRP-1, an involvement of LRP-1 signaling was strongly assumed. LRP-1 may be involved in tissue modeling by regulation of the extracellular matrix composition. In this respect, LRP-1 was shown to regulate mRNA expression of collagen type III, a fibrillar collagen and the second most abundant collagen in the body, linked intimately to the blood vessel structure; the pigment epithelium-derived factor, a collagen-associated member of the Serpin family that lacks the proteinase inhibitory activity, but functions as a potent antiangiogenesis factor; and uPAR-AP/Endo-180, a member of the macrophage mannose receptor family that serves as a catabolic receptor for multiple collagens [28]. The upregulation of LRP-1 during differentiation of ES cells observed in the present study points toward a role in cell commitment processes. However, expression also in undifferentiated ES cells suggests that LRP-1 may be necessary for stem cell function. The proangiogenic effect of α2M was apparently mediated by an elevation of NO generation and phosphorylation of NOS3. Consequently, inhibition of the NOS activity abolished the effect of α2M on vascular cell differentiation. Although NO generation upon α2M has not yet been shown, it was recently demonstrated that the urokinase-type Plasminogen Activator (uPA) induced pulmonary microvascular endothelial permeability through LRP-1-dependent activation of NOS3 [29]. This suggests that NO generation may represent a general principle of LRP-1 activation. NO generation downstream the PI3K/AKT pathway has been demonstrated in a variety of preparations [30], and the role of endothelial NOS in both angiogenesis and vasculogenesis has been discussed [31]. Recently, the involvement of protein kinase C-α/PI3K signaling pathways was shown to regulate LRP-1-mediated astrocytoma invasion [32], suggesting that PI3K is an important effector of LRP-1 signaling. Indeed, the data of the present study demonstrate activation of PI3K upon treatment of ES cells with α2M. Consequently, NO generation and the stimulatory effect of α2M on vasculogenesis/angiogenesis were abolished in the presence of the PI3K inhibitor LY294002. NO generation and vasculogenesis/angiogenesis of ES cells were likewise blunted in the presence of the ERK1/2 inhibitor PD98059, thus corroborating our previous studies that demonstrated a pivotal role of PI3K and ERK1/2 for vascular cell differentiation from ES cells [19,20,33]. Vasculogenesis/angiogenesis of ES cells involves different proangiogenic growth factors, including the vascular endothelial growth factor (VEGF) [34], platelet-derived growth factor-BB [35], and FGF-2 [36]. Angioblasts (endothelial cell precursors) appear to be induced by FGF-2, whereas VEGF appears to be critical for growth and morphogenesis of angioblasts into the initial vascular pattern [23]. Furthermore, FGF-2 is among other growth factors strongly involved in the mesoderm commitment [37]. In the present study, treatment with α2M dose dependently increased FGF-2 expression. This was apparently mediated via LRP-1 signaling since RAP significantly inhibited this effect. Furthermore, the stimulation of vasculogenesis/angiogenesis by α2M was completely abolished in the presence of the FGF-2 receptor antagonist SU5402. Interestingly, this compound also strongly inhibited vasculogenesis/angiogenesis of ES cells in the absence of α2M, which points to the notion that FGF-2 plays a primordial role for endogenous vasculogenesis in the ES cell system. Furthermore, the increase in FGF-2 expression upon treatment with α2M was abolished upon coincubation with L-NAME, LY294002, and PD98059, which suggests that α2M regulates FGF-2 expression via NO, PI3K, and ERK1/2 (see Fig. 7). It may be speculated that α2M initiates the differentiation toward cardiovascular progenitor cells through FGF-2 upregulation, whereas sprouting and tubulomorphogenesis are coordinated by endogenous VEGF levels, which are increased in avascular embryoid bodies derived from ES cells before the initiation of vascular sprout formation [38].
It is well known that α2M orchestrates the activity of a number of growth factors by reversible binding and inhibition of receptor interactions. Regulation of FGF-2 expression by α2M has so far not been reported; however, it has been shown that α2M is binding FGF-2 [39]. Activated α2M inhibited FGF-2-induced proliferation of fetal bovine heart endothelial cells and HUVECs in a dose-dependent manner, but did not inhibit FGF-2-induced endothelial cell tubule formation in Matrigel® or in type I collagen gels [40]. FGFs are secreted from cells and bind in the pericellular space to heparan sulfate proteoglycans (HSPGs) present on the cell surface and the extracellular matrix. It has been discussed that FGF-2 effects on angiogenesis may be regulated by the ability of FGF-2 to distribute between activated α2M and HSPGs [12]. This differential distribution of FGF-2 may be supported by α2M-mediated LRP-1 stimulation and initiation of a NO-regulated signaling cascade, which regulates FGF-2 expression and vascular commitment of ES cells.
Footnotes
Acknowledgments
We gratefully acknowledge the technical assistance of Barbara Arnold. This work was supported by the International Research Training Group 1566 (PROMISE), the Excellence Cluster Cardiopulmonary System (ECCPS), and the von Behring-Röntgen-Foundation.
Author Disclosure Statement
None declared.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.