
Abstract
Tightly regulated trophoblast invasion and immunomodulation at the feto-maternal interface is important during implantation and fetal development. Although trophoblasts as a pregnancy-specific cell has been reported to be a key factor capable of regulating certain events during implantation, however, its regulatory mechanisms are still unclear. In this study, we analyzed the effects of chorionic plate-derived mesenchymal stem cells (CP-MSCs) and human placenta extract (hPE) isolated from human normal placentas on trophoblasts invasion and immune responses. We investigated the effects of CP-MSCs, hPE treatment, and their combination on trophoblasts invasion and on T-cells suppression through human leukocyte antigen-G (HLA-G) expression. Trophoblasts invasion was significantly increased by co-culture of CP-MSCs or by hPE treatment (P<0.05), and enhanced by the combination of CP-MSCs and hPE treatment (P<0.05). The proliferation of T-cells was decreased by co-culture of CP-MSCs and hPE treatment, whereas the population of regulatory T-cells was increased (P<0.05). Also, the dynamics alterations of multiple cytokines were observed in the culture supernatants of trophoblasts and T-cells depending on CP-MSCs co-culture and hPE treatment. Interestingly, the concentration of soluble HLA-G was increased by CP-MSCs co-culture, by hPE treatment and by combination of them on trophoblasts and activated T-cells (P<0.05). These findings suggested that CP-MSCs and hPE can regulate trophoblasts invasion and T-cell by alteration of HLA-G expression. These results will provide understandings of trophoblasts invasion and the immunological network at the feto-maternal interface during pregnancy and contribute to the foundation of a new treatment strategy for pregnancy disorders.
Introduction
S
During invasion, trophoblasts recognized as foreign escape the maternal immune responses through the mechanism of placental immune privilege. Although the immunologic function of trophoblasts is assumed to contribute to the development and protection of the feto-placental unit through maternal immune effector cells such as decidual cells, decidual natural killer (dNK) cells, and macrophages, the regulatory mechanisms of the trophoblasts are not completely understood. Recently, Samstein et al. determined that a subset of Treg cells that induce tolerance to self-antigens and play a key role in immune homeostasis also play an important role in maternal-fetal immune tolerance [5]. In pregnant mice and humans, Treg cells were increased [6], and women with repeated spontaneous abortions and pre-eclampsia had decreased numbers of CD25+CD4+ Treg cells [7,8]. Further, several cytokines regulate Treg cell proliferation and suppressive functions [9,10]. It is known that placental hormones and cytokines derived from trophoblasts orchestrate the composition and regulatory functions of maternal immune cells by autocrine or paracrine mechanisms.
In addition, the HLA-G antigen presented by trophoblasts as a molecule specific for pregnancy is reported to be a tolerogenic molecule that acts on cells involved in both innate and adaptive immunity. Generally, trophoblasts lack HLA-A and -B antigens, resulting in NK cell cytolysis at the feto-maternal interface. However, trophoblast cells express the minimally polymorphic HLA-C antigen and other nonclassical HLA molecules that are recognized by dNK cells so that dNK cells fail to transport perforin-containing granules to the surface, accounting for their lack of cytotoxicity [11]. The expression of HLA-G in trophoblasts is thought to play a key role in implantation by controlling trophoblast invasion [12,13]. Soluble HLA-G (sHLA-G) is found in the plasma and amniotic fluid and also is associated with trophoblasts in the placenta [14]. However, the carefully organized mechanisms of implantation and maintenance of a successful pregnancy are still not fully understood, and further studies on trophoblast invasion and immunomodulation are required.
Mesenchymal stem cells (MSCs) have received considerable attention because they have demonstrated a great potential for clinical use due to their capacity to differentiate into multiple cell types [15,16]. Additionally, MSCs have self-renewal and immunosuppressive abilities [17]. Further, MSCs are known to secrete a variety of cytokines with many functions, such as regulating the survival, growth, and death of cells, immune responses, and cell invasion and the migration process [18,19]. The mechanisms of the immunosuppressive effects of MSCs have been well described in a number of in vitro and in vivo studies of refractory graft-versus-host disease and Crohn's disease. These mechanisms include the suppression of T-cell proliferation and inhibition of dendritic cell differentiation [20,21]. Previously, we reported that chorionic plate-derived MSCs (CP-MSCs), a subset of placenta-derived MSCs, have immunomodulatory effects and that their immunomodulatory effects were stronger than those of MSCs derived from other tissues (eg, bone marrow, adipose-derived MSCs); in addition, we observed that the expression level of HLA-G in CP-MSCs was higher than others MSCs [22,23]. Further, an aqueous human placenta extract (hPE) contained many immune regulatory cytokines, and hPE had an immunosuppressive effect [24 –26].
Therefore, the objectives of this study were to demonstrate the effects of CP-MSCs and hPE derived from term placenta, and their combination effects on the invasion ability of trophoblasts and on the suppression of T-cell proliferation through alteration of HLA-G expression.
Materials and Methods
Isolation of primary trophoblast cells
Primary trophoblast (primary TB) cells were isolated from human term placenta. In brief, placental villi tissues were chopped and washed in cold Hank's balanced salt solution. Then, they were centrifuged at room temperature for 5 min at 1,000 rpm. And then, the tissue pellet was incubated with enzyme solution supplemented with 40 mg/mL of trypsin, 20 mg/mL of DNase I, and 24 U/mL dispase for 20 min at room temperature. The suspended supernatant was centrifuged at room temperature for 5 min at 1,000 rpm. The cell pellets were washed with culture medium supplemented with 1% Pen/Strep (Invitrogen), 10% fetal bovine serum (FBS; Invitrogen), 2 mM
Cell culture
The collection of samples and their use in research was approved by the Institutional Review Board of CHA General Hospital, Seoul, Korea. All participants provided written, informed consent before the collection of the samples. Full-term normal human placentas were collected from women who were free of medical, obstetrical, and surgical complications (≥37 gestational weeks) on caesarean section deliveries. CP-MSCs were harvested and characterized their mesenchymal features as described previously [27]. Briefly, the cells were separated from the inner side of the chorioamniotic membrane of the placenta by treatment of 0.5% collagenase IV (Sigma-Aldrich) for 30 min at 37°C. The harvested cells were cultured in Ham's F-12 medium/Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 1% penicillin/streptomycin (Pen/Strep; Invitrogen), 10% FBS (Invitrogen), 25 ng/mL FGF-4 (Peprotech, Inc.), and 1 μg/mL heparin (Sigma-Aldrich). The normal fibroblast cell line WI-38 (ATCC) was cultured in alpha-minimum essential medium (Gibco-Invitrogen), containing 1% P/S, and 10% FBS (Gibco-Invitrogen). HTR-8/SVneo trophoblasts (provided by Dr. Charles H. Graham, Queen's University) were cultured in RPMI-1640 medium (Gibco-Invitrogen) with 1% P/S and 10% FBS. All cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Preparation of hPE
The hPE was prepared as described previously [28]. Briefly, aqueous hPE was prepared as follows. The washed placental tissue was chopped using sterile scissors. The tissue was snap-frozen in liquid nitrogen for additional grinding, thawed, and resuspended in 30 mL of homogenizing buffer (DMEM high glucose; 0.32 M sucrose; 2 mM EDTA). After centrifugation, the total protein content of the hPE was measured using a BCA™ protein assay kit (Pierce Chemical). The components of hPE, including cytokines, chemokines, and growth factors, were analyzed using a multiplex supernatant cytokine assay [24].
Isolation and activation of T-cells
Mononuclear cells (MNCs) were isolated from human umbilical cord blood (UCB) and the decidual layer of placentas by centrifugation on a Ficoll-Hypaque density gradient. UCB and deciduas from normal-term deliveries were donated by CHA General Hospital, Seoul, Korea. To isolate MNC from UCB, 30 mL of UCB was added to 20 mL of Ficoll-Hypaque, centrifuged at 400g for 35 min, and the MNC layer was collected and washed. To isolate MNCs from the deciduas, the decidual layer was separated from the maternal side of the placenta, chopped and treated with DNase I (00.05 mg/mL; Sigma-Aldrich), dispase (1.2 U/mL; Invitrogen), and collagenase IV (1 mg/mL; Sigma-Aldrich) for 30 min at 37°C, and separated on a Ficoll-Hypaque density gradient method as mentioned above. Aliquots of 2×105 MNCs isolated from the UCB or deciduas were co-cultured with 2×103, 1×104, and 2×104 CP-MSCs or WI-38 cells and/or 0–80 μg/mL hPE per well in a 96-well culture plate with or without 1 μg/mL anti-CD3 and anti-CD28 T-cell-activating mAbs (eBioscience, Inc.) for 72 h at 37°C in a humidified atmosphere with 5% CO2.
Invasion analysis using the transwell system
The invasion of primary TB and HTR-8/SVneo trophoblast cells was analyzed by using a 24-transwell insert system (8 μm pore size; BD Biosciences). In the upper chamber of the insert, 2.5×104 primary TB and HTR-8/SVneo cells per cell were seeded in FBS-free medium. Culture medium supplemented with 5% FBS and hPE at various concentrations (0–120 μg/mL) was added to the lower chamber with or without the CP-MSCs and WI-38 co-culture cells at a concentration of 2.5×104, 5×104, or 1×105 cells/well, for 24 h of incubation. After incubation, the primary TB and HTR-8/SVneo cells remaining in the upper side of insert were removed with a swab, and the primary TB and HTR-8/SVneo cells that had infiltrated the lower side of the insert were fixed with 100% methanol and stained with Mayer's hematoxylin (Dako, Produktionsnej, Glostrup). The stained nuclei were counted. All experiments were performed in triplicate.
T-cell proliferation analysis
To assess the ability of MSCs to suppress T-cell proliferation, the MSCs were first treated with 50 μg/mL of mitomycin C for 50 min to suppress their proliferation. Next, 2×105 MNCs from UCB or the decidual layer were co-cultured with 2×103, 1×104, or 2×104 CP-MSCs or WI-38 cells per well in a 96-well culture plate in medium supplemented 3% FBS, with or without 1 μg/mL of anti-CD3 and anti-CD28 T-cell-activating monoclonal antibodies (mAbs; eBioscience) and hPE at various concentrations (0–120 μg/mL) for 72 h. To analyze the suppression of the clonal expansion proliferative response of T-cells, clusters of T-cells were examined in bright field and the BrdU ELISA (Roche) assay was performed according to the manufacturer's protocol at 72 h of cultivation. All experiments were repeated three times.
Zymography
Zymography was performed to analyze the MMP-2 activity in primary TB and HTR-8/SVneo trophoblasts were co-cultured with/without CP-MSCs or WI-38 cells and/or treated with the indicated concentration of hPE (0–80 μg/mL). The supernatants from the HTR-8/SVneo cells or primary TBs co-cultured with/without CP-MSCs or WI-38 cells and/or treated with the indicated concentration of hPE (0–80 μg/mL) were collected and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis containing 0.5% gelatin (Sigma-Aldrich). The MMP-2 activities were assessed using 20 μL of supernatant per sample. Following electrophoresis, the gels were washed for 30 min using renaturation buffer (Bio-Rad) and incubated overnight in a buffer containing 50 mM Tris-HCl (pH 7.4), 0.2 M NaCl, 5 mM CaCl2, and 1% Triton X-100 at 37°C. Enzyme expression was revealed by staining with 0.1% Coomassie blue in a 1:3:6 acetic acid:methanol:water mixture. All experiments were performed in triplicate.
Real-time PCR
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen). Reverse transcription was performed with 1 μg of total RNA and Superscript III reverse transcriptase (Invitrogen). cDNA were amplified by PCR. Real-time PCR was performed using SYBR EX taq (Roche) and Exicycler TM96 quantitative thermal block (Bioneer). PCR reaction conditions were as follows: denaturation at 95°C for 5 min, 40 cycles of 95°C for 30 20 s, annealing temperature at 60°C for 15 min, extension at 70°C for 15 min, and final extension at 72°C for 7 min. Expression values of specific marker genes were normalized to the expression level of 18rRNA (Bioneer). The used primer sequences were as follows: HLA-G forward primer; 5′-GCG GCT ACT ACA ACC AGA GC-3′; HLA-G reverse primer; 5′-GCA CAT GGC ACG TGT ATC TC-3′, 18 sRNA forward primer; 5′-GTA ACC CGT TGA ACC CCA TT-3′; 18 sRNA reverse primer; 5′-CCA TCC AAT CGG TAG TAG CG-3′.
ELISA assay
To analyze the activity of MMP-2, we performed the MMP-2 Qunatikine ELISA assay (R&D Systems) by manufacturer's instruction. Briefly, 50 μL of diluent solution was added to each well and 50 μL of supernatants collected from experimented transwell were added and mixed gently, and incubated at room temperature (RT) for 2 h. Following incubation, the wells were washed four times with 200 μL of provided wash buffer. And then, 200 μL of MMP-2 conjugate was added to each well and incubated at RT for 2 h. Following incubation, the wells were washed with 200 μL of provided wash buffer four times and incubated with provided 200 μL of substrate solution at RT for 30 min. Following incubation, 200 μL of stop solution was added to each well. Activity of MMP-2 was measured using microplate reader (BioTek) at 450 nm. All experiments were performed in triplicate.
Flow cytometry analysis
For detection of the CD4+CD25+ Treg cell population, flow cytometry analysis was performed. MNCs with T-cell-activating antibodies that had been co-cultured with CP-MSCs or WI-38 cells were harvested and washed with phosphate-buffered saline (PBS). They were incubated with isotype control IgG (diluted 1:200; BD Biosciences) or with CD4 (PE conjugated, diluted 1:200; BD Biosciences) and CD25 (FITC conjugated, diluted 1:200; BD Biosciences) antibodies for 30 min. Flow cytometry analysis was performed using a vantage FACSCalibur flow cytometer (BD Biosciences). For detection of the HLA-G-positive cell population, primary TB were prestained for co-culture with CP-MSCs or WI-38 cells using PKH26 and the cells were harvested and stained with FITC conjugated HLA-G antibody (diluted 1:100; eBioscence) and analyzed using a vantage FACSCalibur flow cytometer (BD Biosciences).
Multiplex supernatant cytokine assay (Luminex)
The supernatants were collected after 72 h of cultivation under the following conditions: MNCs seeded at 2×105 with anti-CD3 and anti-CD28 and co-cultured with/without CP-MSCs or WI-38 cells seeded at 2×103, 1×104, or 2×104 cells in 96-well plates. Each 50 μL supernatant sample was combined with coated beads from a MILLIPLEX MAP kit (Millipore Corp.). After the final wash, the beads from the 96-well microtiter plates were resuspended in the 125-μL cuvette of a Luminex instrument. An acquisition gate was set between 7,500 and 13,500 for a doublet discriminator, the sample volume was 75 μL, and 100 events/region were acquired. The raw data (mean fluorescence intensity) from all the bead combinations tested were analyzed with MasterPlex QT30.0 quantification software (MiraiBio, Inc.) to obtain the concentration values. All samples were assayed in duplicate and analyzed with a Luminex 200 LabMAP system (Luminex). Data analyses were performed using Bio-Plex Manager software version 4.1.1 (Bio-Rad Laboratories). All experiments were performed in triplicate.
Detection of sHLA-G concentrations
The concentrations of sHLA-G in the culture supernatants from the HTR-8/SVneo cells (2×104 cells/well) cultured with/without CP-MSCs (2×104 cells/well) or WI-38 cells (2×104 cells/well) with/without hPE (40 or 80 μg/mL) treatment or from the T-cells cultured with/without CP-MSCs and WI-38 cells with/without 40 μg/mL hPE were measured using a sHLA-G detection kit (BioVendor, Inc.). The assay was performed and calculated according to the manufacturer's protocol. All experiments were performed in triplicate.
Immunofluorescence
To investigate the expression of HLA-G in HTR-8/SVneo trophoblast cells cultured with/without CP-MSCs or WI-38 cells, HTR-8/SVneo cells (5×104 cells/well) were seeded onto glass coverslips at the bottom of 24-well plates and CP-MSCs or WI-38 cells (2.5×104 cells/well) were grown in the upper chamber of 24-well transwell inserts for 24 h. After removing the inserts, the cells on the coverslips were washed with PBS and incubated with serum-free protein blocking buffer (Dako) at 37°C for 30 min and incubated with mouse anti-human HLA-G (1:50; Santa Cruz Biotechnology) overnight at 4°C. After washing, the cells were incubated with a secondary antibody conjugated to Alexa568 (1:500; Invitrogen) for 40 min at RT. The nuclei were counter stained with 4′, 6-diaminino-2-phenlindole. The cells were mounted over fluorescence mounting medium (Dako) and observed using a confocal laser scanning microscope imaging system (LSM510; Carl Zeiss, Inc.).
Human chemokines array
To investigate the concentration of chemokines in the culture supernatants from the primary TB (2×104 cells/well) cultured with/without CP-MSCs (2×104 cells/well) or primary TB cells (2×104 cells/well) combined with/without CP-MSCs and 40 μg/mL hPE were measured using a human chemokine array (Ray Biotech). The assay was performed and calculated according to the manufacturer's protocol. Briefly, the slide was dried for 2 h at room temperature and incubated with 100 μL blocking buffer at room temperature for 30 min. After adding the blocking solution from each sub array, 70 μL of media sample was added and incubated overnight at 4°C. After washing, the glass chip was incubated with biotin-conjugated anti-cytokine antibodies for 2 h at room temperature, and then it was washed with 1×wash solution I at room temperature. Diluted streptavidin-Fluor (1:1,500) was added onto the glass chip and incubated for 2 h at room temperature. Then, the slide was rinsed with de-inoized water and centrifuged at 1,000 rpm for 3 min. The slide was analyzed using GenPix 4000B scanner (Axon Instrument) and quantified with GenePix Software (Axon Instrument).
Statistical analysis
The results are presented as the mean±SE. The statistical significance was tested using the t-test with a significance level of P<0.05.
Results
Co-culture with CP-MSCs and hPE stimulates the invasive ability of trophoblasts via activation of MMP-2
To investigate the effects of CP-MSCs and hPE on the invasive ability primary TB, the cells were cultured with CP-MSCs or WI-38 cells present in the lower chamber of the 24-transwell system, and hPE was added to the lower chamber at 20, 40, or 80 μg/mL. Invasive ability of primary TB cultured with WI-38 was increased at 5×104 and 1×105 compared with co-free group (
*P<0.05; Fig. 1A, B). In addition, invasive ability of primary TB cultured with CP-MSCs was increased at 2.5×104, 5×104, and 1×105 compared with co-free group (
*P<0.05). Interestingly, invasive ability of primary TB cultured with CP-MSC was significantly increased at 2.5×104, 5×104, and 1×105 compared with primary TB cultured with WI-38 (
 P<0.05). (Fig. 1A, B).
P<0.05). (Fig. 1A, B).

Effect of CP-MSCs and hPE on invasion of primary TB.
represents significant differences between CP-MSCs and WI-38 cells at each dose of cells (P<0.05). #represents significant differences between hPE- and nontreated cells at each dose of cells (P<0.05). Enzyme activities of MMP-2 co-cultured with CP-MSCs or WI-38 cells (upper) and hPE treated (lower) trophoblasts by
To analyze activities of MMPs, it is well known that MMPs are a key factor in the regulation of trophoblast invasion, we performed zymography. There is no difference the activity of MMP-9 between in each groups (data not shown). Activity of MMP-2 was significantly increased at 2.5×104, 5×104, and 1×105 cultured with CP-MSCs and WI-38 compared with co-free group (
*P<0.05). Also, activity of MMP-2 cultured with CP-MSCs was significantly increased at 1×105 compared with primary TB cultured with WI-38 at same dose (
 P<0.05; Fig. 1C, D). Activity of MMP-2 was significantly increased at 2.5×104 and 5×104 cultured with CP-MSCs or WI-38 with hPE compared to only those cultured with CP-MSCs or WI-38 (#P<0.05) and was enhanced at 5×105 combined with CP-MSCs and hPE compared with MMP-2 of WI-38 treated with hPE (
P<0.05; Fig. 1C, D). Activity of MMP-2 was significantly increased at 2.5×104 and 5×104 cultured with CP-MSCs or WI-38 with hPE compared to only those cultured with CP-MSCs or WI-38 (#P<0.05) and was enhanced at 5×105 combined with CP-MSCs and hPE compared with MMP-2 of WI-38 treated with hPE (
 P<0.05; Fig. 1C, D).
P<0.05; Fig. 1C, D).
Next, we investigated the combination effects of the presence of CP-MSCs and co-treatment of hPE on primary TB invasion. Invasive ability of primary TB cultured with CP-MSCs and WI-38 with hPE treatment was significantly increased compared with only co-culture group (
#P<0.05). In addition, invasive ability of primary TB was significantly increased in combined with CP-MSCs and hPE compared with ability of WI-38 with hPE (
 P<0.05; Fig. 1A, B). Further, we profiled the chemokine activities of primary TB co-cultured with/without CP-MSCs and treated with/without hPE to confirm the effect of CP-MSCs and hPE on chemokine activities. Growth-regulated oncogene (GRO) activity was increased by 12-fold with CP-MSCs compared without CP-MSCs. In addition, activity of GRO was increased by 8-fold and 11-fold with hPE or combined with CP-MSCs and hPE, respectively. Also, activity of monocyte chemattractant protein-1 (MCP-1) was increased by 3.2, 3, and 2.8-fold with CP-MSCs or with hPE or combined with CP-MSCs and hPE compared without CP-MSCs, respectively. Further, activity of RANTES was increased by 39- and 19-fold with CP-MSCs or combined with CP-MSCs and hPE compared without CP-MSCs, respectively. However, activity of regulated on activation, normal T cell expressed and secreted (RANTES) was not difference with hPE compared without hPE. In case of macrophage inflammatory protein-1 beta (MIP-1β), activity was increased in a dose-dependent manner with CP-MSCs, hPE, or combined with CP-MSCs and hPE compared without CP-MSCs and hPE. In addition, activity of interleukin (IL)-8 was increased by 19, 4, and 17-fold with CP-MSCs, hPE, or combined with CP-MSCs and hPE compared without CP-MSCs and hPE, respectively (Supplementary Fig. S1; Supplementary Data are available online at
P<0.05; Fig. 1A, B). Further, we profiled the chemokine activities of primary TB co-cultured with/without CP-MSCs and treated with/without hPE to confirm the effect of CP-MSCs and hPE on chemokine activities. Growth-regulated oncogene (GRO) activity was increased by 12-fold with CP-MSCs compared without CP-MSCs. In addition, activity of GRO was increased by 8-fold and 11-fold with hPE or combined with CP-MSCs and hPE, respectively. Also, activity of monocyte chemattractant protein-1 (MCP-1) was increased by 3.2, 3, and 2.8-fold with CP-MSCs or with hPE or combined with CP-MSCs and hPE compared without CP-MSCs, respectively. Further, activity of RANTES was increased by 39- and 19-fold with CP-MSCs or combined with CP-MSCs and hPE compared without CP-MSCs, respectively. However, activity of regulated on activation, normal T cell expressed and secreted (RANTES) was not difference with hPE compared without hPE. In case of macrophage inflammatory protein-1 beta (MIP-1β), activity was increased in a dose-dependent manner with CP-MSCs, hPE, or combined with CP-MSCs and hPE compared without CP-MSCs and hPE. In addition, activity of interleukin (IL)-8 was increased by 19, 4, and 17-fold with CP-MSCs, hPE, or combined with CP-MSCs and hPE compared without CP-MSCs and hPE, respectively (Supplementary Fig. S1; Supplementary Data are available online at
Next, we performed similar experiments with HTR-8/SVneo trophoblast cell line to confirm the effect of CP-MSCs and hPE on trophoblast. Similar to data shown on primary TB, the number of invasive HTR-8/SVneo cells was significantly increased when CP-MSCs were co-cultured, compared with co-free cultures (*P<0.05). In addition, hPE treatment enhanced the invasive activity of HTR-8/SVneo cells (#
P<0.05; Supplementary Fig. S2A). In zymography and ELISA assay, the activity of MMP-2 was significantly increased in co-cultures with both CP-MSCs and WI-38 cells compared with that of the co-free group, and the MMP-2 expression level in the supernatant from trophoblasts co-cultured with CP-MSCs was significantly increased compared with WI-38 cells at 5×104 and 1×105 of CP-MSCs (Supplementary Fig. S2B, C). However, there were no differences in the MMP-9 activity following co-cultivation with CP-MSCs or WI-38 cells (data not shown). Finally, we investigated the combination effects of the presence of CP-MSCs and co-treatment of hPE on trophoblasts invasion. Similar to data of primary TB, the invasive ability of trophoblasts combined with CP-MSCs and hPE was significantly enhanced compared with those of WI-38 combined with WI-38 and hPE (
 P<0.05). Also, the MMP-2 activity in the medium of HTR-8/SVneo cells co-cultured with CP-MSCs and hPE (40 μg/mL) was more significantly increased compared with those co-cultured with CP-MSCs without hPE (
#P<0.05) and co-cultured with WI-38 with hPE (
P<0.05). Also, the MMP-2 activity in the medium of HTR-8/SVneo cells co-cultured with CP-MSCs and hPE (40 μg/mL) was more significantly increased compared with those co-cultured with CP-MSCs without hPE (
#P<0.05) and co-cultured with WI-38 with hPE (
 P<0.05) (Supplementary Fig. S2B, C). These findings demonstrate that invasion abilities of trophoblast including primary TB and trophoblast cell line were stimulated by co-culture with CP-MSCs and by hPE treatment. Additionally, the combination of CP-MSCs and hPE has a positive combination effect on trophoblast invasion by controlling the activity of MMP-2.
P<0.05) (Supplementary Fig. S2B, C). These findings demonstrate that invasion abilities of trophoblast including primary TB and trophoblast cell line were stimulated by co-culture with CP-MSCs and by hPE treatment. Additionally, the combination of CP-MSCs and hPE has a positive combination effect on trophoblast invasion by controlling the activity of MMP-2.
Effects of CP-MSCs co-culture and hPE treatment on UCB-derived T-cell response
In previous reports, we demonstrated that CP-MSCs have an immunomodulatory effect [22,23]. Therefore, we investigated whether co-culture with CP-MSCs could suppress the proliferation of activated T-cells, enhance the Treg cell population, and/or alter the immune-related cytokines to evaluate the effect of CP-MSCs on T-cells. MNCs isolated from UCB were activated by T-cell-activating mAbs and then co-cultured with/without CP-MSCs or WI-38 cells. The activated T-cell clustering was suppressed (Fig. 2A), and T-cell proliferation was significantly reduced in co-cultures with CP-MSCs compared to the controls (*P<0.05) and co-cultures with WI-38 (# P<0.05) (Fig. 2B). We also analyzed whether CP-MSCs affected Treg cell populations derived from UCB. As shown in Fig. 2C, there were no differences in the CD4+CD25+Treg cell population in the co-culture systems with CP-MSCs and WI-38 cells (Fig. 2C).

Immune responses on UCB-derived T-cells depending on CP-MSCs co-culture.
represents significant differences between CP-MSCs and WI-38 at each dose of cells (P<0.05). All experiments were performed in triplicate. The values were expressed as the fold increase as mean±S.E. CD−, not treated with anti-CD3 and anti-CD28 nontreated; CD+, treated with 1 μg/mL of anti-CD3 and anti-CD28 treated; UCB, umbilical cord blood; MNCs, mononuclear cells; CP, CP-MSCs; WI, WI-38 cells; IL, interleukin; IFN, interferon; TNF, tumor necrosis factor.
In the multiplex supernatant cytokine assay, there were dynamic changes in the levels of several cytokines depending on the co-culture conditions. The expression levels of IL-5 and IL-10 were increased more in a dose-dependent manner by co-culturing with CP-MSCs than in co-culturing with WI-38 cells. In contrast, the expression levels of interferon (IFN)-γ and tumor necrosis factor (TNF)-α in the co-cultures with CP-MSCs were decreased. The expression levels of IL-4 was increased only at high dose of co-culture with CP-MSCs and at low, middle dose of co-culture with WI-38 cells (Fig. 2D). These data suggested that CP-MSCs had suppressive effects on proliferation of activated T-cell via alteration of cytokines, but they had no effect on the regulation of the Treg population. Further, we investigated whether hPE treatment could suppress activated T-cell proliferation and/or alter the levels of any cytokines.
We had previously reported that hPE contains an abundance of molecules with biological activity such as cytokines, chemokines, and growth factors that are known to be essential factors in cell–cell interactions [24]. The formation of T-cell clusters and T-cell proliferation were significantly suppressed by treatment with 0–80 μg/mL of hPE in MNCs derived from UCB and activated by T-cell-activating mAbs (*P<0.05; Fig. 3A, B). Based on the results of the supernatant cytokine assay, IL-5 was increased and IFN-γ, TNF-α, IL-4, and IL-10 were decreased in the supernatant from the activated T-cell culture as a result of treatment with 40 μg/mL hPE (*P<0.05; Fig. 3C). Interestingly, the expression patterns for IFN- γ, TNF-α, and IL-5 in the activated T-cells treated with hPE were similar to those of the activated T-cells co-cultured with CP-MSCs. These data suggested that hPE suppress proliferation of T-cells via the alteration of cytokine levels,
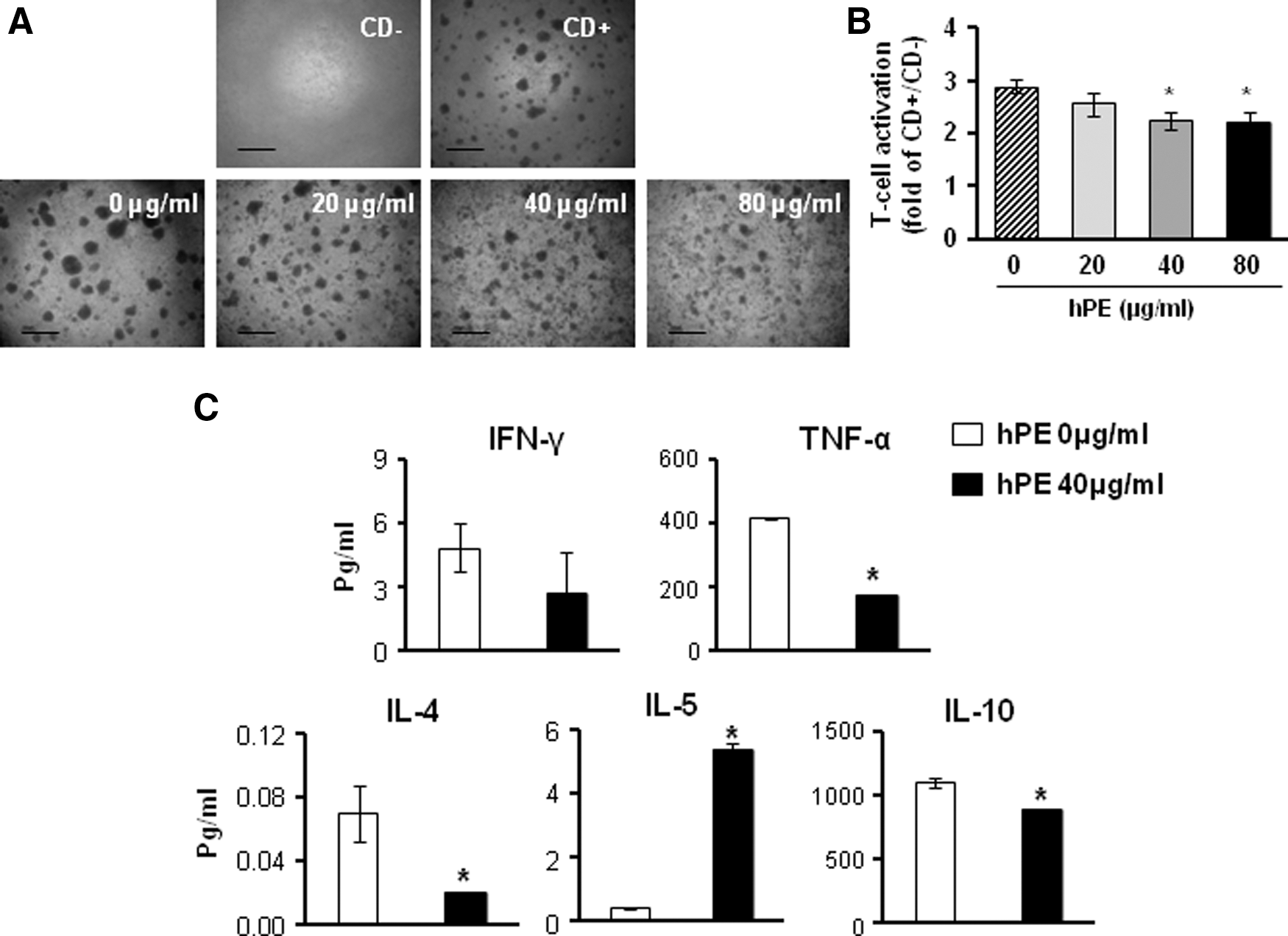
Immunoregulatory effects of hPE on activated T-cells isolated from UCB.
Combination effect of CP-MSCs and hPE treatment on activated T-cells derived from UCB
We confirmed that CP-MSCs and hPE have suppressive effects on proliferation of activated T-cell derived from UCB although their mechanisms differ. Therefore, we investigated whether CP-MSCs and hPE have a combination effect on activated T-cells by exposing activated T-cells. As shown in Fig. 4, the inhibition of T-cell cluster formation was further decreased by co-culture with CP-MSCs combined with hPE treatment compared to co-culture with CP-MSCs without hPE (Fig. 4A). In addition, the inhibitory effect was shown to be enhanced dependent on the increased number of CP-MSCs. Further, the proliferation of T-cells was decreased in co-cultures with CP-MSCs combined with hPE treatment compared with co-cultures with CP-MSCs only (# P<0.05; Fig. 4B). Although there were no differences in co-cultures with WI-38 cells, normal fibroblasts, a significant suppression of T-cell proliferation was observed when hPE was included in the treatment (# P<0.05; Fig. 4B). In the multicytokine assay, IFN-γ, TNF-α, IL-4, and IL-5 were increased and IL-10 was decreased in the supernatants of cells that were co-cultured with CP-MSCs and treated with hPE (*P<0.05; Fig. 4C). These result suggested that the combination of CP-MSCs and hPE treatment had combination effects on the suppression of T-cell proliferation.
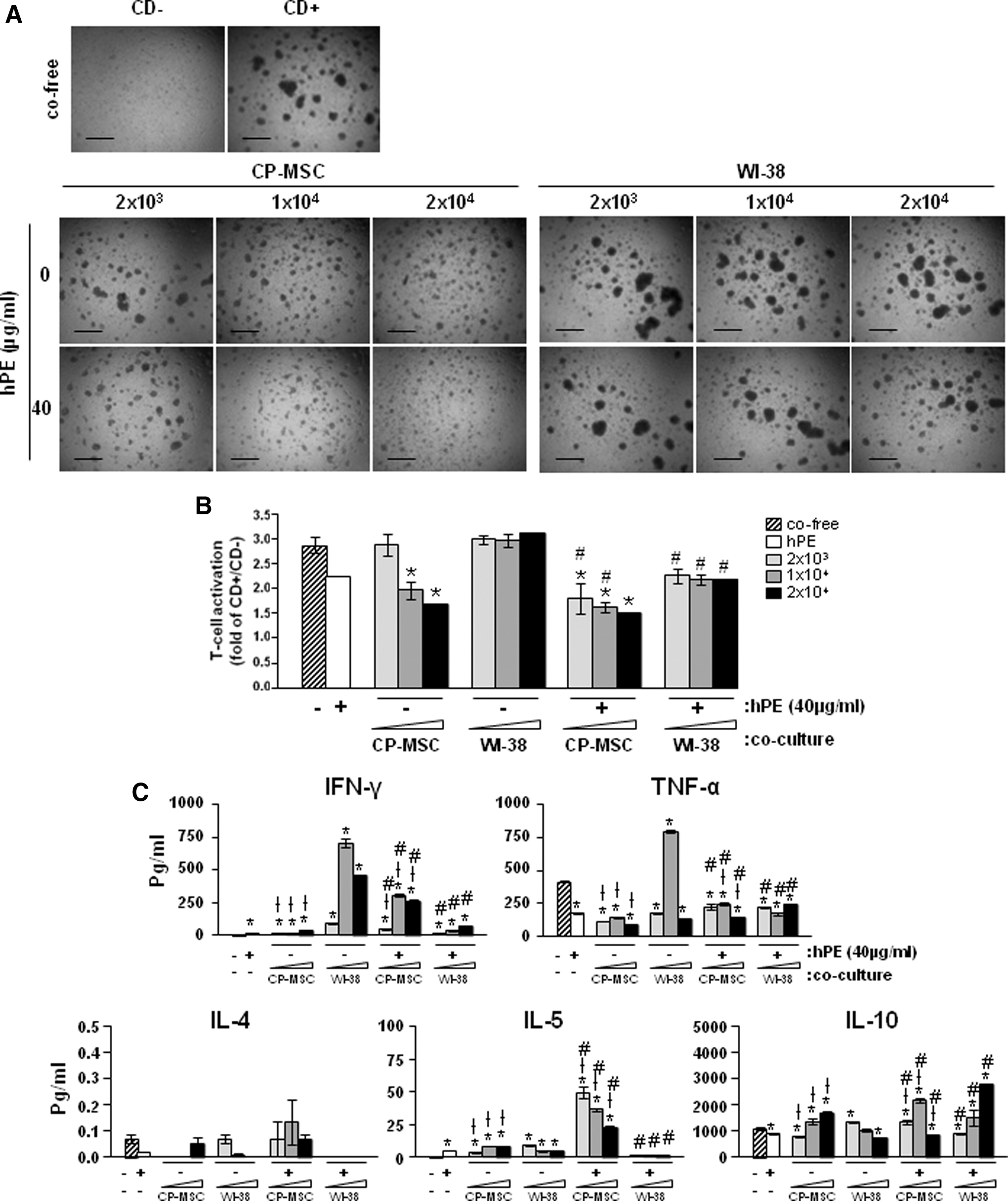
Combination effects of CP-MSCs co-culture and hPE treatment on activation of UCB-derived T-cells.
The combination of CP-MSCs co-culture and hPE treatment induced HLA-G expression on activated T-cells and trophoblasts
To investigate effect of CP-MSCs co-culture and hPE treatment on HLA-G expression, we analyzed the HLA-G expression using real-time PCR. Interestingly, HLA-G expression was significantly higher in primary TB co-cultured with CP-MSCs than co-cultured with Wi-38 cells, regardless of the hPE treatment (†P<0.05; Fig. 5A). Further, the HLA-G expression was significantly increased when primary TB were co-cultured with combination of CP-MSCs and hPE, but not with WI-38 cells (# P<0.05; Fig. 5A). Thus, it appears that co-culturing primary hepatocytes with combination of CP-MSCs and hPE increased the HLA-G expression, while WI-38 cells decreased it. The reasons underlying these apparently conflicting effects await further investigation. Notably, HLA-G expression was regulated by CP-MSCs co-cultured in primary TB. In addition, HLA-G expression in primary TB was significantly induced by CP-MSCs co-cultured (*P=0.05; Fig. 5B) in FACS analysis.
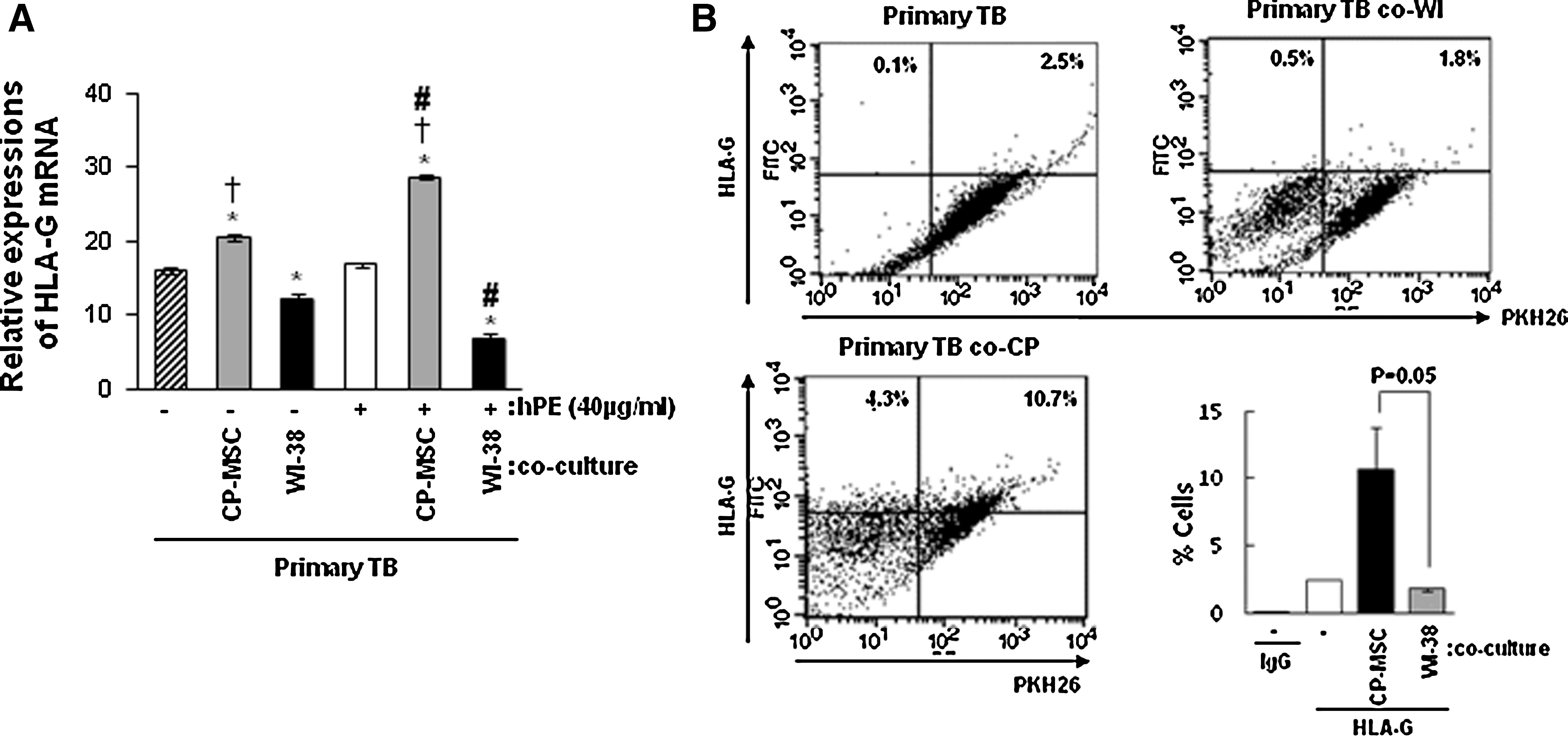
Human leukocyte antigen-G (HLA-G) expresssions in primary TB co-cultured with CP-MSCs and hPE treatment.
We next examined whether HLA-G was correlated with activities for T-cell and HTR-8/SVneo trophoblasts invasion by co-cultured CP-MSCs, a sHLA-G ELISA analysis was performed. sHLA-G concentration in trophoblast co-cultured with CP-MSCs was higher than that of those co-cultured with WI-38 cells (*P<0.05; Fig. 6A), however, sHLA-G concentration with hPE added was enhanced by more than 2-fold compared to that with HTR-8/SVneo cells alone (Fig. 6A). Interestingly, hPE treatment had an induction effect sHLA-G expression in co-cultured with WI-38 cells (# P<0.05; Fig. 6A). In addition, sHLA-G concentration in the activated T-cells co-cultured with CP-MSCs was significantly increased compared with those of the WI-38 cells and control groups (*P<0.05), but, there were no differences in activated T-cells co-cultured with or without WI-38 cells (Fig. 6B). However, hPE treatment induced the concentration of sHLA-G in T-cells and was shown to have a significant combination effect in the co-cultures with CP-MSCs (# P<0.05; Fig. 6B). Also, we found that co-culture of HTR-8/SVneo cells with CP-MSCs, but not with control fibroblast WI-38 cells, prominently induced HLA-G expression, as examined by HLA-G immunofluorescence (Fig. 6C). Taken together, our results strongly suggest that CP-MSCs co-culture induces HLA-G expression in trophoblasts.
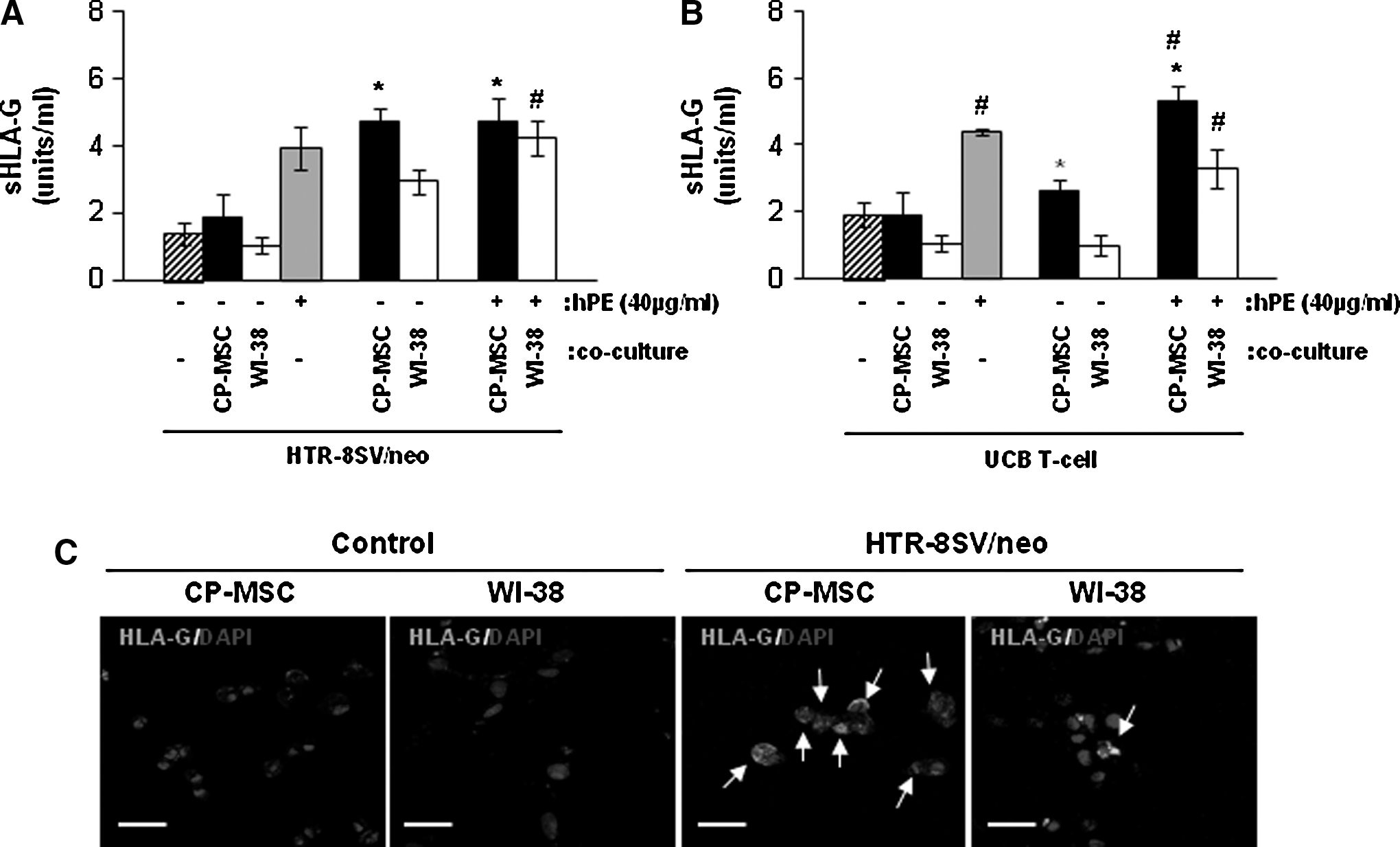
Concentration of HLA-G in the supernatant of HTR-8/SVneo cells or UCB-derived T-cells on CP-MSCs co-culture and hPE treatment.
Discussion
Several adult tissues-derived MSCs have received attention in regenerative medicine for their therapeutic potential. Recently, many researchers have suggested that MSCs have a therapeutic effect via immunomodulatory and paracrine mechanisms, although they have limited self-renewal activity and differentiation potential [29,30]. Depending on the microenvironment, several cytokines and growth factors secreted by MSCs induce their auto-activation via an autocrine mechanism, resulting in migration of the MSCs to sites of tissues damage and paracrine secretion of several bioactive molecules. Consequently, networking of cytokines regulated by MSCs may have therapeutic effects for the regeneration of injured tissues [31]. In addition, bioactive molecules secreted by MSCs play a role in enhancing the cellular activity of aged cells by decreasing oxidative stress and promoting migration via the expression of MMPs [32 –34]. This cascade of events may also improve the cellular processes that maintain the homeostasis of each organ component under optimal physiological conditions. Therefore, it is important to evaluate the therapeutic effects of several bioactive molecules derived from MSCs.
In a previous study, we reported that placenta-derived MSCs have the advantage of being capable of regeneration in injured liver tissue and an immunomodulatory effect on activated T-cells due to the secretion of various cytokines from placenta-derived MSCs [22,27]. Further, hPE extracted from term placenta using an aqueous solvent also contains many bioactive molecules when it is obtained without denaturation. This hPE extract can be used to culture several types of MSCs without FBS supplementation [24], and it induces hepatogenic differentiation of MSCs and has therapeutic effect in the early cirrhosis liver rat model created by CCl4 administration [28].
In the present study, we first demonstrated that CP-MSCs isolated from term placentas have the potential to enhance invasion of trophoblast including primary TB and trophoblast cell line via the expression of MMP-2 and to suppress proliferation of activated T-cell through alteration of several bioactive molecules including HLA-G using in vitro co-culture system. Further, hPE treatment in combination with the CP-MSCs enhanced the combination effect on trophoblasts invasion and on suppressive effects of activated T-cell proliferation derived from UCB or the decidual layers of the placenta (Supplementary Fig. S3). In particular, the combination of CP-MSCs and hPE significantly induced the expression of sHLA-G, a key factor for trophoblast invasion and immunomodulation.
Complex processes are required for the regulatory mechanisms involving cells of the mother and the embryo during embryo implantation, especially for the development of maternal immunotolerance to fetal cells (eg, trophoblasts). This tolerance is achieved through tightly controlled interactions that initiate the selective homing of immune cells to the feto-maternal site and regulate the proliferation and differentiation of a regulatory type of immune cell. Through this strict regulation, the entire immune system of the mother is switched to a tolerogenic state for the maintenance of pregnancy [35]. The failure of immunotolerance affects trophoblast invasion directly, resulting in poor invasive ability of trophoblasts and inducing pregnancy disorders such as pre-eclampsia. Therefore, overcoming the poor invasion of trophoblasts is an important issue in reproductive medicine. Although there have been many reports of products that regulate trophoblasts invasion, however, there have been no reports on whether MSCs can improve trophoblasts invasion. In the present study, we demonstrated that co-culture of CP-MSCs and treatment with hPE can promote trophoblasts invasion through induction of MMP-2 activity. In addition, CP-MSCs and hPE affect the invasion ability of HTR-8/SVneo cells and primary TB. The differences in the MMP-2 activity between the CP-MSCs co-culture and the hPE treatment group may be caused by different regulation of multiple cytokines to interact with the immune cells in the decidua and to aid trophoblasts invasion. Recently, it was reported IL-1β regulates cytotrophoblasts invasion by controlling MMP-2 and -9 [36] and the invasion ability of trophoblasts was negatively regulated by TNF-α [37] as shown on our data that TNF-α act as a negative regulator on trophoblast invasion.
Alteration of cytokines and chemokine also affects trophoblasts invasion and the development of immune cells during pregnancy. Recently, Tregs were suggested to play an important role in mammalian invasive placentation and their activity was regulated by several chemokines secreted from trophoblasts. Recently, MCP-1β, CCL-5, and CXCL8 secreted from trophoblast cells promote migration of Foxp3+ cell toward trophoblast cells [38]. Especially, RANTES induce to increase the expression Treg (CD4+CD25+Foxp3+) during interaction between maternal cells and trophoblasts [39]. In our data, the expressions of chemokine (eg, GRO, MCP-1, RANTES, and MIP-1β) in co-cultured with CP-MSCs were enhanced compared to those of co-free culturing (Supplementary Fig. S1). Further, naive CD4+CD25−T-cells can differentiate into a CD4+CD25+ suppressor T-cell phenotype Treg cells, and they express FoxP3 when TGF-β is present [40]. Recently, the secretion of TGF-β family proteins was reported to correlate with Treg fate [41,42]. The findings are consistent with our data (Supplementary Fig. S3E). In our data, IL-2 expression by CP-MSCs co-culturing was significantly greater than that of WI-38 cells co-culturing (*P<0.005; Supplementary Fig. S3). Interestingly, we showed that the Treg population was not altered in UCB T-cells, however, the CD4+CD25+Treg cell population was increased in activated T-cells from deciduas that were co-cultured with CP-MSCs (Supplementary Fig. S3C). These data mean that co-culture with CP-MSCs can induce Treg population of decidua-derived activated T-cells, which are considered more involved in the immune regulation at the feto-maternal interface than are UCB-derived activated T-cells. In our data, we elucidated that co-culture of CP-MSCs with T-cells from deciduas induced the population of CD4+CD25+ Treg cells, with dynamic changes in the levels of multiple cytokines, including IL-2, -4, -5, -6, -10, and all the TGF-β family members (TGF-β1, -β2, and -β3) (Supplementary Fig. S3E).
The nonclassical HLA-G molecule is a trophoblasts-specific molecule expressed mainly by human trophoblasts, cytotrophoblasts, and syncytiotrophoblasts and is present in almost every pregnancy [43,44]. HLA-G binds to and activates a number of cell surface receptors expressed by NK and endothelial cells. The binding of HLA-G to dNK cells leads to modulation of NK cell cytokine secretion [45]. In vitro studies indicated that soluble isoforms of HLA-G (sHLA-G1 or HLA-G5) induce apoptosis in activated endothelial cells, NK, and alloreactive cytotoxic T-cells [46 –48]. Further, McCormick et al. reported that sHLA-G regulated trophoblasts invasion [12]. Thus, we investigated the dual function of HLA-G for immune and invasion modulation in co-cultures with CP-MSCs. According to our data, co-culture with CP-MSCs increased invasion of HTR-8/SVneo cells and primary TB and suppressed T-cell proliferation through alteration of sHLA-G concentration in the in vitro models. However, the mechanism by which CP-MSCs regulate HLA-G requires further study.
Taken together, CP-MSCs can activate immunosuppression and trophoblasts invasion through expression of several bioactive molecules including HLA-G, although the function of the cytokines networks in the paracrine mechanisms of CP-MSCs should be further validated. In addition, hPE in combination with CP-MSCs can enhance the potential of CP-MSCs as a beneficial supplement. Therefore, the findings will provide an understanding of cell invasion and the immunological network at the feto-maternal interface during pregnancy and contribute to the foundation of a potential new treatment strategy to overcome pregnancy disorders.
Footnotes
Acknowledgments
We thank Dr. Charles H. Graham (Queen's University, Canada), who has provided HTR-8 SV/neo cell lines. This work was supported by the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A084923) and the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology, Republic of Korea (2012R1A1A3009776).
Author Disclosure Statement
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.