
Abstract
Thrombocytopenia (TTP) is a blood disease common to canines and human beings. Currently, there is no valid therapy for this disease except blood transfusion. In this study, we report the generation of canine induced pluripotent stem cells (ciPSCs) from canine embryonic fibroblasts, and a novel protocol for creating mature megakaryocytes (MKs) and functional platelets from ciPSCs. The ciPSCs were generated using lentiviral vectors, and differentiated into MKs and platelets on OP9 stromal cells supplemented with growth factors. Our ciPSCs presented in a tightly domed shape and showed expression of a critical pluripotency marker, REX1, and normal karyotype. Additionally, ciPSCs differentiated into cells derived from three germ layers via the formation of an embryoid body. The MKs derived from ciPSCs had hyperploidy and transformed into proplatelets. The proplatelets released platelets early on that expressed specific MK and platelet marker CD41/61. Interestingly, these platelets, when activated with adenosine diphosphate or thrombin, bind to fibrinogen. Moreover, electron microscopy showed that the platelets had the same ultrastructure as peripheral platelets. Thus, we have demonstrated for the first time the generation of ciPSCs that are capable of differentiating into MKs and release functional platelets in vitro. Our system for differentiating ciPSCs into MKs and platelets promises a critical therapy for canine TTP and appears to be extensible in principle to resolve human TTP.
Introduction
T
Dogs are an unrivalled application model for human diseases compared to traditional model organisms, for well-known reasons. Unlike other test animals, dogs have lived with humans for a long time and share a common environment, so that differing environmental factors between dogs and humans do not arise. Second, dogs have a long life span unlike other traditional animal models, so that medical surveillance can be conducted over a long life span. Third, about 58% of dog genetic diseases are known to have equivalent human diseases, which show similar clinical traits [14]. Further, because modern dog breeds have originated out of a small population, genetic diversity has been restricted. Specific inherited diseases have consequently been identified, which is helpful in modeling human genetic studies [15]. In clinical regenerative medicine, the formation of cancer from transplanted cells is a critical issue. Dogs display a similar biological behavior and histopathological characteristics to tumors that occur in humans. Therefore, platelets derived from ciPSCs may not only serve as an excellent solution for canine disorders, but could also be a promising regenerative medicine model for human medicine.
In the present study, we generated ciPSCs from canine embryonic fibroblasts (CEFs) and differentiated the ciPSCs into MKs and platelets. Our ciPSCs adapted a spherically, tightly packed shape, and expressed critical pluripotency markers such as REX1, the same as in canine ESCs (cESCs), and differentiated into the cells derived from three germ layers in vitro. The platelets derived from our ciPSCs were generated through the formation of ciPSC-derived sacs (iPS-sacs) using a coculture system in vitro. Further, the platelets could be activated so as to bind to fibrinogen, and the electron microscope examination showed that the platelets had the same ultrastructure as peripheral platelets. We do not demonstrate teratoma formation, but show that our ciPSCs differentiate into MKs and platelets, whereas others have differentiated their ciPSCs into other cell types [16 –20]. Platelets derived from our ciPSCs have a potential to be a first regenerative therapy for canine TTP, and perhaps ultimately for human TTP.
Materials and Methods
All use of animals, and tissues obtained from animals, was approved and conducted according to the regulations of the Institutional Animal Care and Use Committee.
Cells
Mouse embryonic fibroblasts (MEFs) for feeder cell coculture with ciPSCs were generated using day-14.5 mouse embryos (Jcl:ICR [Institute for Cancer Research], Japan; SLC, Shizuoka, Japan), according to a previous report [21]. CEFs were generated from a fetus contained in an uterus isolated from a 30-day pregnant mixed canine in the same way as MEFs. The OP9 stromal cell line was obtained from the RIKEN cell bank (Ibaraki, Japan) and was maintained according to previous report [22]. The 293FT cell line was obtained from Invitrogen (Grand Island, NY).
Antibodies and cytokines
The primary antibodies (Abs) used and their respective dilutions are as follows: mouse anti-human stage-specific embryonic antigen (SSEA)-4 (NG1938439; Millipore, Billerica, MA), 1:1,000; mouse anti-human β-III-TUBULIN (MAB1637; Millipore), 1:1,000; goat anti-human NANOG (ab77095; Abcam, Cambridge, MA), 1:1,000; mouse anti-human TRA-1-60 (ab129000; Abcam), 1:1,000; mouse anti-human SOX17 (ab84990; Abcam), 1:1,000; rabbit anti-human DESMIN (ab8592; Abcam), 1:1,000; mouse anti-sheep CD41/61 (MCA1095; AbD Serotec, Oxford, UK), 1:1,000; biotin-conjugated mouse anti-canine CD34 (550427; BD Pharmingen, San Jose, CA), 1:1,000; and PerCP-conjugated streptavidin (554064; BD Pharmingen). The secondary Abs used were as follows: Alexa Fluor® 488-conjugated goat anti-mouse IgG (A-11001; Invitrogen), Alexa 546-conjugated goat anti-rabbit IgG (A-11010; Invitrogen), PE-conjugated donkey anti-goat IgG (NB7590; Novus, Littleton, CO), and Cy3-conjugated goat anti-mouse IgM (AP128C; Millipore). Canine vascular endothelial growth factor (VEGF) and canine stem cell factor (SCF) were from R&D (Abingdon, UK). Human thrombopoietin (TPO) was from Sigma (St Louis, MO), and heparin was from Wako (Osaka, Japan).
Cell culture
ciPSCs were maintained on feeder layers of mitomycin C-treated MEFs at a density of 3.6×105 cells per 35-mm dish in Dulbecco's modified Eagle's medium/F12 (DMEM/F12; Sigma), supplemented with 20% knockout serum replacement (KSR; Gibco, Grand Island, NY), 2 mM
Lentiviral infection
One day before transduction, 293FT cells were seeded at 2×106 cells per 100-mm dish. Next day, the four lentiviral vectors CSIV-CMV-hOCT3/4, hKLF4, hSOX2, and hC-MYC-IRES2-Venus (RIKEN BioResource Center) were introduced into 293FT cells using OPTI-MEM1 (Gibco), Virapower packaging mix, and Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Sixteen hours after transduction, the medium was changed to DMEM high-glucose containing 10% FBS and 1× penicillin/streptomycin, and the supernatants containing lentivirus were filtered at 24 h through a 0.45-μm cellulose acetate filter (Whatman, Kent, ME, UK). One day before infection, CEFs at passage 3 were seeded at 2.85×105 cells per 60-mm dish. The next day, CEFs were incubated in 50% virus-containing supernatants for 16–18 h and then placed again in a fresh medium. A multiplicity of infection of 20 was determined by using lentiviral vectors carrying a green fluorescent protein cassette. At 6 days after infection, the transduced cells were reseeded at 2.75×104 cells per 35-mm dish onto feeder layers of mitomycin C-treated MEFs.
Alkaline phosphatase staining and karyotype analysis
The Alkaline Phosphatase Staining Kit II (Stemgent, Cambridge, MA) was used for alkaline phosphatase (ALP) staining according to the manufacturer's instructions.
The karyotype ciPSCs was analyzed using a protocol described below. ciPSCs were transferred to another dish without dividing at passage 16. Next day, 0.1 μg/mL colcemid (Gibco) was added in the dish for 2 h, and the medium was washed by PBS. The cells were isolated with trypsin and centrifuged, and 0.075 M potassium chloride was added in the sediment at 37°C for 15 min, and then, carnoy fluid was added. After centrifugation, the procedure an indicated above was performed twice. After final centrifugation, the sediment was dropped to glass slide, and the number of chromosomes by Giemsa staining was analyzed. Fifteen G-banded metaphase spreads were examined for each ciPSC line.
Immunocytochemical analysis
ciPSCs were washed in PBS and then fixed with 4% paraformaldehyde for 5 min and permeabilized with 0.05% Tween 20 in PBS for 5 min at room temperature. The cells were blocked with 10% bovine serum albumin for 30 min, and then incubated with primary and secondary Abs. The Abs were used according to the manufacturer's recommendation. Information on the Abs used in this study is contained in the Antibodies and cytokines section.
Reverse transcriptase–polymerase chain reaction
Total RNA was recovered using an RNeasy microkit (Qiagen, Tokyo, Japan). Reverse transcription was performed with oligo-dT and ReverTra Ace (Toyobo, Osaka, Japan), and polymerase chain reaction (PCR) was conducted with Blend Taq
™, and PCR products were resolved on 2% agarose gels containing ethidium bromide, and were then photographed. All primers are listed in Supplementary Table S1 (Supplementary Data are available online at
In vitro differentiation of ciPSCs
The colonies of ciPSCs were mechanically isolated and transferred to 100-mm dishes containing the culture medium without LIF or bFGF to produce embryoid bodies (EBs). After 5 days, EBs were transferred to tissue culture dishes coated with 0.1% gelatin (Wako), and incubated in the same culture medium supplemented with 20% FBS instead of KSR. Half of the volume of the culture medium was changed every 2 days. After 14–21 days, the differentiated cells were analyzed by immunofluorescence staining and reverse transcriptase–PCR (RT-PCR). All Abs used in this study are listed in the Antibodies and cytokines section. All primers are listed in Supplementary Table S1.
ciPSC differentiation on OP9 stromal cells
OP9 stromal cells were treated with mitomycin C, as with the MEFs. The mitomycin C-treated OP9 cells were seeded at 2×105 cells per 35-mm dish before plating of undifferentiated ciPSCs as small clumps. Two hours before the ciPSCs were transferred, the medium was changed to an embryonic stem cell differentiation medium supplemented with 20 ng/mL VEGF, and the medium was refreshed every 2 days according to a previous method [23]. On days 14–15, the iPS-sacs were trypsinized, isolated, and passed onto fresh OP9 cells; they were maintained in a differentiated medium with 10 ng/mL TPO, 25 U/mL heparin and 50 ng/mL SCF, and the medium was changed every 2 days. Floating cells were collected and analyzed by Giemsa staining, immunofluorescence staining, and flow cytometry during culture. All Abs used in this study are contained in the Antibodies and cytokines section.
Flow cytometry
FACSCalibur (BD Pharmingen) was used for flow cytometric analysis after the following incubations.
Canine hematopoietic progenitor cells were detected by incubation with biotinized anti-canine CD34 monoclonal Ab (mAb), followed by incubation with PerCP-conjugated streptavidin (BD Pharmingen). Canine megakaryocytic cells and platelets were detected by incubation with anti-CD41/61 mAb (AbD Serotec), followed by incubation with Fluor 488-conjugated anti-mouse goat IgG (Invitrogen). All incubations described above were performed on ice for 30 min.
The nuclear ploidy content of megakaryocytic cells was detected according to a previous method [24] after immunostaining for CD41/61 and propidium iodide (Sigma).
To perform functional analysis for the platelet, platelet-like particles in the culture supernatant were carefully collected, and a one-ninth volume of acid citrate dextrose (Sigma) was added. The medium containing particles was then centrifuged at 200g for 20 min to eliminate any large nucleated cells. The supernatant was transferred to a new tube, and 2 μM prostaglandin E1 and 2 U/mL apyrase were added to prevent platelet activation, after which the mixture was centrifuged at 3,500 rpm for 10 min to collect the platelet pellets. The pellets were then resuspended in an appropriate volume of PBS, including 2% FBS. The prepared medium containing platelet-like particles was incubated for 10 min at 37°C with 100 mg/mL fluorescein isothiocyanate (FITC)-labeled dog fibrinogen in the absence or presence of 2 U/mL human thrombin or 40 μM adenosine diphosphate (ADP). Binding of fibrinogen was quantified using flow cytometry. Nonspecific binding was determined in the presence of 5 mM ethylenediaminetetraacetic acid (EDTA).
Electron microscopy of platelets derived from ciPSCs
Differentiated platelets derived from ciPSCs were collected as described above. Canine control peripheral blood was obtained from a mature female beagle. The blood was collected in a tube containing PBS, and lymphocyte separation solution (Nacalai Tesque, Kyoto, Japan) was added according to the manufacturer's recommendation so as to isolate platelet-rich plasma (PRP). PRP was centrifuged at 200g for 20 min at room temperature. The supernatant was stored in 2.5% glutaraldehyde in 0.1 M phosphate buffer (PB; pH 7.4) overnight at room temperature, followed by washing with 0.1 M PB. Samples were then postfixed with 1% osmium tetroxide for 2 h at room temperature and were embedded in an epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate and were examined with a Hitachi H-7500 electron microscope (Hitachi, Tokyo, Japan).
Results
Generation of ciPSCs and the pluripotency markers
We have successfully reprogrammed CEFs into ciPSCs by using lentiviral vectors containing four human factors (OCT3/4, SOX2, KLF4, and C-MYC). Eight days after transduction, the first ciPSC colonies were appeared (Fig. 1A) and had similar characteristics to cESCs with a domed and tightly packed shape (Fig. 1A). From day 10 after transduction, stem cell-like colonies had grown large enough to be isolated mechanically and transferred onto fresh mitomycin C-inactivated MEF layers (Fig. 1A). A total of 13 primary ciPSC clones were isolated from a total of 2.75×104 transduced CEFs, giving an efficiency of reprogramming ∼0.047% based on the morphology. Almost all clones were maintained until 10–12 passages. After approximately passage 10, three clones that showed intense positive ALP activity at passage 5 were fully characterized and expressed the pluripotency markers NANOG, SSEA4, and TRA-1-60 by immunofluorescence staining, and maintained their characteristics beyond passage 25 (Fig. 1A, B). Clones incubated with a buffer rather than the primary Ab confirmed the specificity of the secondary Abs (Fig. 1B).
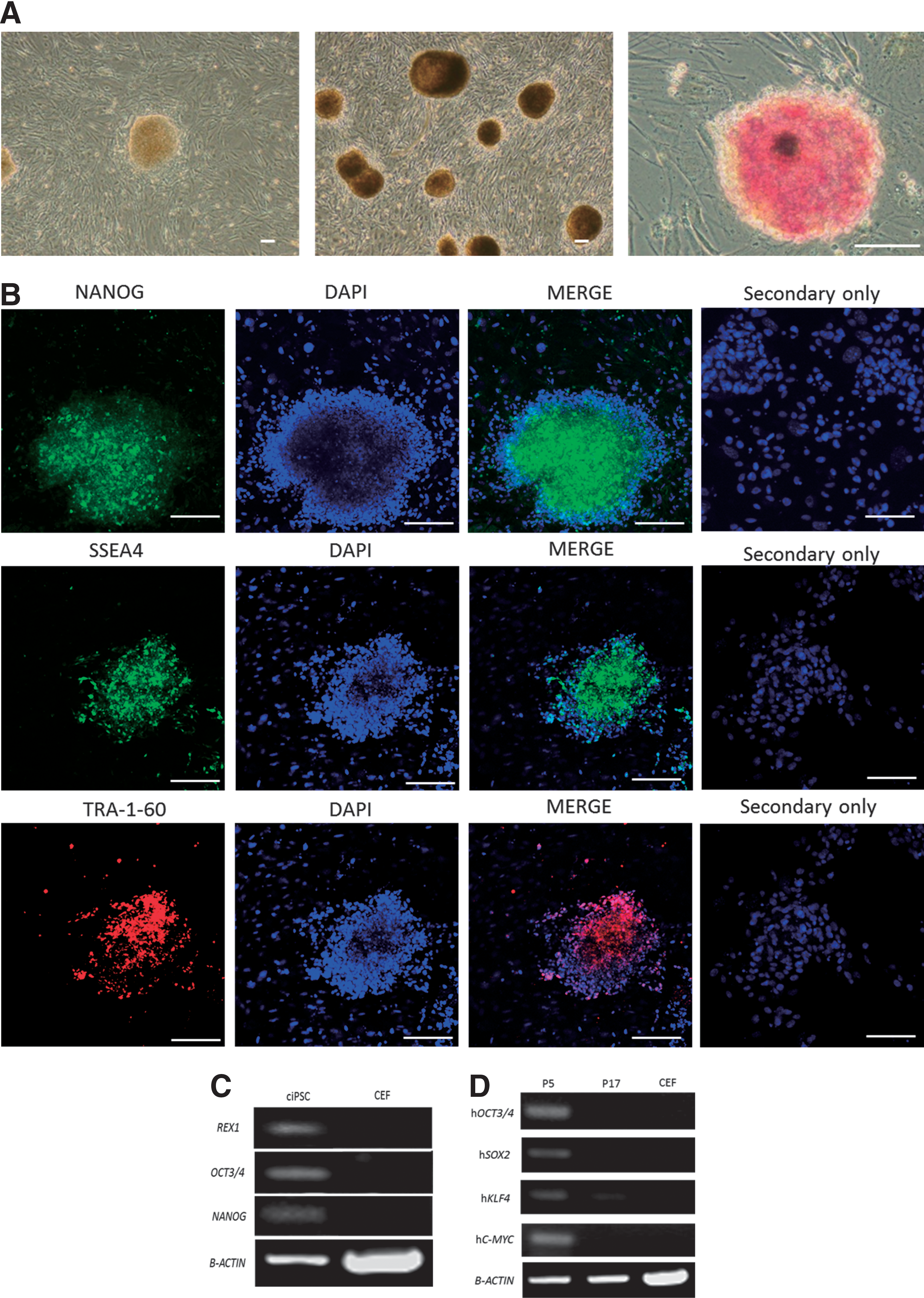
Characteristics of canine induced pluripotent stem cells (ciPSCs).
RT-PCR analysis of the three clones for further characterization demonstrated the expression of endogenous canine OCT3/4, NANOG, and ESC-specific gene (REX1) at passage 17, but not in control CEFs, from which ciPSCs were generated (Fig. 1C). RT-PCR analysis confirmed that all exogenous human transgenes were still transcribed in all three clones at passage 5 and were barely detectable or silenced at passage 17 (Fig. 1D).
In vitro differentiation and karyotyping of ciPSCs
In vitro differentiation capacity of three ciPSC clones at passage 10 was evaluated by immunohistochemical staining of EBs with Abs β-III-TUBLIN, DESMIN, and SOX17 (Fig. 2A). Cells incubated with a buffer rather than a primary Ab confirmed the specificity of the secondary Abs (Fig. 2A). RT-PCR was also conducted with the ectoderm marker β-III-TUBLIN, mesoderm marker DESMIN, and endoderm marker AFP to confirm the differentiation of ciPSCs into all three germ layers (Fig. 2B). Karyotype analysis revealed that all three clones that expressed pluripotency markers had a normal 78, XY karyotype, with 38 matched pairs of autosomes at passage 16, with no gross structural rearrangements identifiable (Fig. 2C). Fifteen cells were analyzed for each ciPSC line, and all cells displayed an euploid male karyotype.

Differentiation of ciPSCs into the cells derived from embryoid bodies (EBs) and karyotype.
Differentiation of ciPSCs into MKs and platelets in vitro
In our next experiment, one of three ciPSC clones was differentiated at passage 5 by using a coculture system with VEGF, and these cells formed flat, stretched colonies that showed clear contrast with ciPSC colonies, and began to produce individually recognizable cells from the center of the colony into the culture supernatant from 2 days after culture (Fig. 3A). Giemsa staining showed that the cells collected from the culture supernatant displayed morphologic features of immature MKs, and the cells attached to the dish expressed CD41/61 antigen by immunofluorescence staining (Fig. 3B). Cells incubated with the buffer rather than a primary Ab confirmed the specificity of the secondary Abs (Fig. 3B). Inflated sac-like structures appeared in the culture medium on days 12–14 (Fig. 3A), and these structures contained round hematopoietic-like cells that migrated to and outward the periphery of the colonies (Fig. 3A). On day 15, the spherical cells were reseeded onto fresh OP9 layers with several cytokines, human TPO, canine SCF, and heparin instead of canine VEGF so as to produce mature MKs and platelets. A day after the passage, we found large round cells appeared in the supernatant and expressed CD34, indicating that HSCs were produced from our ciPSCs. MK-like cells were detected by Giemsa staining and expressed CD41/61 antigen, indicating that we successfully generated MKs from our ciPSCs (Figs. 3C and 4A, B). Moreover, flow cytometric analysis demonstrated that DNA ploidy of MKs was more than 8N. These results show that we generated mature MKs from ciPSCs (Fig. 4C). We also found morphologically transformed cells with long-beaded figures, the so-called proplatelets, from which platelets were produced (Fig. 4A).

Differentiation of ciPSCs on OP9 stromal cells.
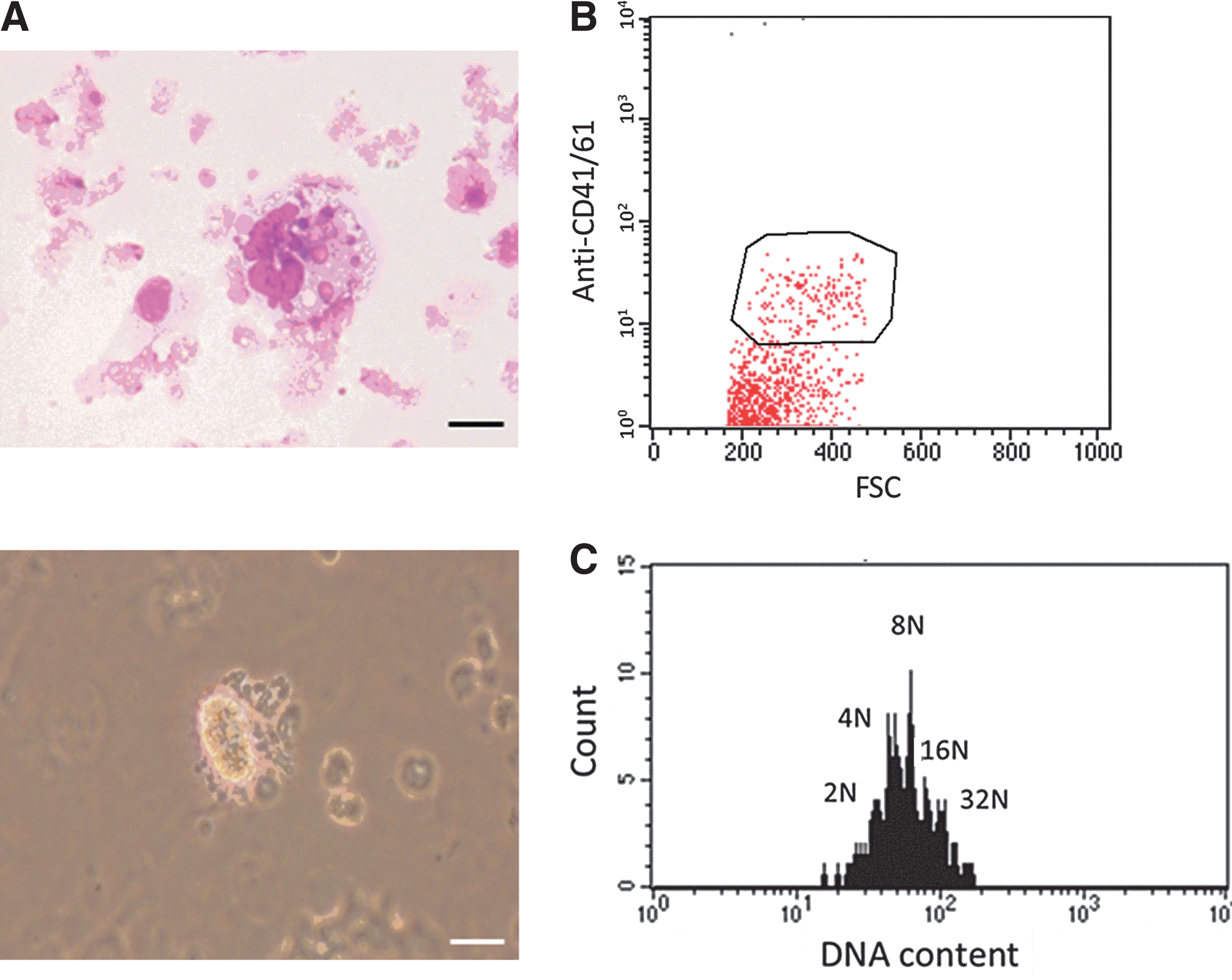
Mature megakaryocytes (MK) and proplatelets.
Platelet release from mature MKs
In flow cytometry, the forward and side-scatter (FSC and SSC) profile of platelet-like particles derived from ciPSCs was similar to fresh peripheral platelets. Moreover, they expressed CD41/61 (GP IIb/IIIa), a specific marker for platelet and MKs (Fig. 5A, B). We conclude that the megakaryocytic cells derived from ciPSCs released platelets into the culture supernatant. The ratio of peripheral platelets that was positive for CD41/61 Abs was 29%, and the ratio of platelets from culture supernatant was 17% (Fig. 5B). Platelets that expressed CD41/61 were detected within the first 2 days after transfer of ciPSCs to the ES differentiation culture. Electron microscope examination revealed that the platelets were oval in shape, with ultrastructures such as open canalicular system (OCS) and α-granules, which were specific to platelets, and crucially were similar to the structure of canine peripheral platelets (Fig. 5C).
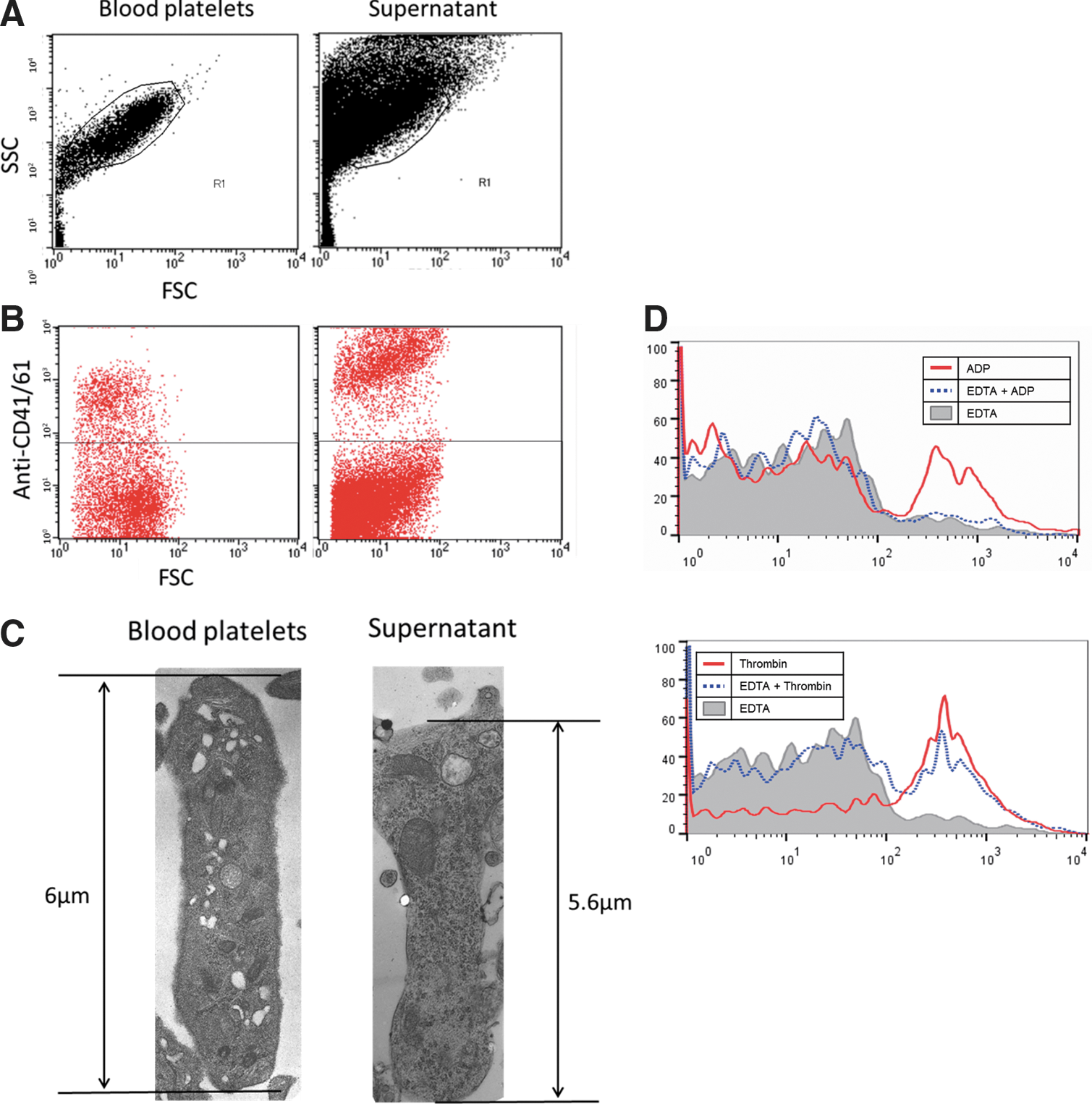
Functional assays and electron microscopy of platelets derived from ciPSCs.
Functional platelets derived from ciPSCs
We next investigated the functional properties of the platelets that were mobilized. Platelet-activated agonists, thrombin, and ADP were added to the solution containing platelets so as to activate the αIIbβIII receptor required for platelet aggregation (inside-out signaling). EDTA was also added to inhibit the conformation of platelets, as a control. We then examined whether determined the platelets derived from ciPSCs could bind to canine FITC–fibrinogen using flow cytometry. Platelets with ADP bound weakly to FITC–fibrinogen, but the platelets with both ADP and EDTA rarely bound to fibrinogen. In contrast, the platelets activated with thrombin bound strongly to fibrinogen, and further, thrombin was activated even in the presence of EDTA (Fig. 5D).
Discussion
In this report, our ciPSCs formed a spherical domed colony similar to the cESCs established by Vaags et al. [25] and Wilcox et al. [26]. Some previous studies have demonstrated the generation of cESCs that have flattened colonies [27 –29]. Only Vaags et al. [25] and Wilcox et al. [26] generated domed cESC colonies. It has been reported that PSC colonies have two typical morphologies, a domed colony [21] and a flat colony, which have been called naïve or primed state, respectively [30]. Naïve PSCs are in a less-differentiated state than primed PSCs, and the rate of proliferation is very high [31]. Theunissen et al. [32] have demonstrated that primed mouse iPSCs are able to be reprogrammed to naïve state by using glycogen synthase kinase-3-beta (GSK3β) inhibitor and mitogen-activated protein kinase (MEK) inhibitor. Although some researchers have reported the generation of ciPSCs using these inhibitors, the colonies were flat in shape [16,20], like other cESC colonies [27 –29]. Whitworth et al. [19] generated slightly domed ciPSC colonies using six transgenes with GSK3β inhibitor, MEK inhibitor, and transforming growth factor-beta antagonist. In comparison to previous reports, however, we were able to generate domed ciPSC colonies onto MEF feeder layers at a high cell density (3.7×104/cm2) with no inhibitors. MEFs secrete various factors, for example, LIF, bFGF, and SCF, which enhance the canine embryonic development [33]. Analysis of the secretion factors in feeder cells will help to identify small molecules that generate naïve ciPSCs.
Our ciPSCs intensively expressed ALP, and also expressed the pluripotent markers NANOG, OCT3/4, SSEA4, TRA-1-60, and REX1, similar to ciPSCs reported by Vaags et al. [25]. REX1 is a marker specific to ESCs and is expressed only in cESCs [25], which have an ability to form teratomas. The complete silencing of transgene expression was detected in OCT3/4, SOX2, and C-MYC, and KLF4 was detected distantly at passage 17. Incomplete transgene silencing has also been described in some ciPSCs [16,17,19]. Our ciPSCs differentiated into the cells derived from three germ layers, and the expression of differentiation markers was confirmed by RT-PCR. Although we have yet to perform teratoma assays, our results suggest that our ciPSCs are pluripotent. Our ciPSCs were maintained a stable karyotype of 78 chromosomes, XY types at passage 16, and which means that the proliferation of the cells have not been from tumor generation.
In this study, we have demonstrated for the first time the generation of platelets from ciPSCs with the HSCs differentiating to MKs using a coculture system in vitro. At first, we produced sac-like structures on days 12–14, with similar features to the iPS-sac reported previously using the OP9 layers with VEGF [12]. We used VEGF to form the iPS-sacs, because the cytokine related to the formation of blood vessels and blast colony in mouse and rat embryos [33]. HSCs and platelet-releasing mature MKs were generated through the formation of iPS-sacs with TPO, SCF, and heparin. Such iPS-sacs generate a large number of HSCs from progenitor cells inside the structures, and the HSCs differentiated into platelet-releasing MKs with response to TPO [23,34]. Our HSCs were also generated from iPS-sacs, and the platelet-releasing MKs derived from HSCs had hyperploidy, implying that the MKs were mature and similar to normal MKs. Although canine HSCs have been generated from cESCs [25], the generation of HSCs from sac-like structures has not been reported previously. This platelet-releasing system has been found only in human PSCs before, and we have shown that ciPSCs also have the potential to form sac-like structures. Our method of generating functional platelets from ciPSCs via iPS-sacs represents a contribution to the hiPSC differentiation technology. It has been reported that hiPSCs are efficiently differentiated into platelets by manipulating the expression of C-MYC [12]. In our ciPSC culture system, average 9.8×105 platelets were generated from 3×104 ciPSCs. We are now attempting to modify the culture system to provide sufficient amounts of platelets for clinical applications.
Platelets derived from our ciPSCs expressed CD41/61 antigen, and the ratio was almost the same as to platelets from peripheral blood. The platelets were activated with ADP or thrombin and bound to fibrinogen. Platelet activators, ADP or thrombin, stimulate platelets and cause the αIIbβIII receptor, activated, to bind fibrinogen. After fibrinogen binding, the internal αIIbβIII receptors from α-granules are exposed so as to bind another fibrinogen, and larger aggregates are formed [35]. These results indicate that we have produced successfully functional platelets from ciPSCs in vitro. It is known that thrombin is a stronger activator than ADP. Our platelets bind to fibrinogen more with thrombin than with ADP, and even in the presence of EDTA, some platelets bound to fibrinogen upon adding thrombin. This also implies that our platelets were activated with each activator functionally. Also, the platelets derived from our ciPSCs had the same ultrastructures, OCS, and α-granules as peripheral platelets when examined by electron microscopy. OCS has the functions of spreading and transforming platelets into an activated form for hemostasis. Alpha-granules contain fibrinogen, Willebrand factor, and αIIbβIII receptor; after activation, these proteins are released and also activate the platelets [36,37]. These results show that our platelets have functional structures, and it is possible that the platelets also function properly in vivo. ciPSCs are generated from somatic cells and have a self-renewal system. By using this technology to generate functional platelets from ciPSCs, we may produce unlimited amounts of platelets that function properly and facilitate a new therapy for canine TTP in place of blood infusion. Functional platelets have so far been derived only from hiPSCs, [12], and no study has been published on generating iPSC-derived functional platelets in large-animal models. Large-animal models are useful for many human research areas because of their size and life span close to humans.
There are two main obstacles to overcome before the iPSC technology can be utilized as cell therapy. One is overexpression of transgenes, and the other is the purity of transplanted cells. It is possible that overexpression of transgenes causes generation of tumors inside the body of transplanted patients. Also, the contamination of undifferentiated cells in transplanted cells could cause the formation of teratomas or other tumors. Nonetheless, platelets have anuclear structures, so that the overexpression of transgenes does not cause tumor generation. Moreover, they can be irradiated before transfusion to eliminate residual ciPSCs or other differentiated nucleated cells.
Canines have some characteristics similar to humans: genetically, environmentally, and biologically [14,15]. iPSCs have unlimited self-renewal systems and could be used for autografts of cells and tissues. Generation of functional platelets from ciPSCs through patient somatic cells provides not only a sustainable source but also an immune-compatible one. This regenerative technology facilitates innovative medical research, promising cure for many human ailments.
In conclusion, we have generated ciPSCs that formed domed colonies with a critical pluripotent marker (REX1) and differentiated into three germ layers in vitro. Our ciPSCs differentiated into platelets via HSCs to MKs. The resulting MKs had hyperploidy and were induced to reach maturity. Interestingly, our platelets were activated to bind to fibrinogen with ADP or thrombin, and were suppressed by EDTA. Further, our platelets have functional ultrastructures, α-granules, and OCS similar to peripheral platelet. These results indicate that our platelets have morphological and functional properties similar to peripheral platelets, and we believe that they may behave similarly in vivo, leading to a new therapy for TTP.
Footnotes
Acknowledgments
We appreciate the provision of Lentiviral vectors from Miyoshi H, RIKEN BioResource Center. This work was supported in part by a Grant-in-Aid for Scientific Research (B) no. 24380172 and for Challenging Exploratory Research no. 24658270 from the Japan Society for the Promotion of Science, and by the Science Research Promotion Fund of the Promotion and Mutual Aid Corporation for Private School of Japan.
Author Disclosure Statement
The authors each declare that there are no actual or potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.