
Abstract
ARS-Interacting Multi-functional Protein 1 (AIMP1) is a cytokine that is involved in the regulation of angiogenesis, immune activation, and fibroblast proliferation. In this study, fibroblast growth factor receptor 2 (FGFR2) was isolated as a binding partner of AIMP peptide (amino acids 6–46) in affinity purification using human bone marrow-derived mesenchymal stem cells (BMMSCs). AIMP1 peptide induced the proliferation of adult BMMSCs by activating Akt, inhibiting glycogen synthase kinase-3β, and thereby increasing the level of β-catenin. In addition, AIMP1 peptide induced the translocation of β-catenin to the nucleus and increased the transcription of c-myc and cyclin D1 by activating the β-catenin/T-cell factor (TCF) complex. By contrast, transfection of dominant negative TCF abolished the effect of AIMP1. The inhibition of Akt, using LY294002, abolished the accumulation and nuclear translocation of β-catenin induced by AIMP1, leading to a decrease in c-myc and cyclin D1 expression, which decreased the proliferation of BMMSCs. An intraperitoneal injection of AIMP1 peptide into C57/BL6 mice increased the colony formation of fibroblast-like cells. Fluorescence activated cell sorting analysis showed that the colony-forming cells were CD29+/CD44+/CD90+/CD105+/CD34−/CD45−, which is characteristic of MSCs. In addition, the fibroblast-like cells differentiated into adipocytes, chondrocytes, and osteocytes. Taken together, these data suggest that AIMP1 peptide promotes the proliferation of BMMSCs by activating the β-catenin/TCF complex via FGFR2-mediated activation of Akt, which leads to an increase in MSCs in peripheral blood.
Introduction
M
ARS-interacting multifunctional protein 1(AIMP1) was originally identified as a member of the mammalian multi-ARS complex [18]. AIMP1 is secreted in response to hypoxia and cytokine stimulation; it functions as a cytokine with various target cells including endothelial cells, monocyte/macrophage cells, dendritic cells, and pancreatic α cells [19 –26]. Recently, macrophages were shown to secrete on stimulation with tumor necrosis factor α in wound lesions, and AIMP1 was shown to enhance wound healing, which was mediated by fibroblast proliferation and collagen synthesis via ERK activation [20]. Deletion mapping analysis showed that the N-terminal domain (amino acids 6–46) of AIMP1 was responsible for the stimulation of fibroblast proliferation [27]. Since the proliferation of MSCs is critical to providing a reservoir of cells or back up for the repair or regeneration of damaged tissues, we studied AIMP1 to determine whether it could promote the proliferation of BMMSCs. The results of this study showed that AIMP1 peptide increased the expression of cyclin D1 and c-myc by stabilizing β-catenin via FGF receptor 2 (FGFR2)-mediated activation of Akt. This promoted the proliferation of BMMSCs without affecting their differentiation into adipocytes, chondrocytes, and osteocytes.
Materials and Methods
Cell culture and proliferation assay
BMMSCs were purchased from PromoCell and were maintained in low-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin/penicillin. Cells between passages 4 and 7 were used for this study. For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, human BMMSCs (1×104 cells) were seeded onto 96-well plates and cultured for 24 h. After serum starvation for 4 h with low-glucose DMEM containing 0.5% FBS, the BMMSCs were treated with different concentrations of AIMP1 peptide (amino acids 6–46) in the presence or absence of LY294002 (10 μM) (Calbiochem) and U0126 (10 μM)(Calbiochem). BmMSCs were cultured for 24 h. Subsequently, 10 μL of Ez-Cytox (DaeilLab) was added to each well, and the cells were cultured for another 4 h. At the end of the incubation, we evaluated cell viability by measuring the optical density at 450 nm. For the cell counting assay, human BMMSCs (1.2×104 cells) were seeded onto 24-well plates and cultured in low-glucose DMEM supplemented with 10% FBS and 1% streptomycin/penicillin for 12 h. After serum starvation for 4 h with low-glucose DMEM containing 0.5% FBS, human BMMSCs were treated with different concentrations of AIMP1 peptide for the indicated time. The cells were stained with tryphan blue and counted using a hemacytometer under a light microscope.
Immunoblot
Human BMMSCs (2×105 cells) were seeded onto six-well plates and cultured for 12 h. After serum starvation for 4 h with low-glucose DMEM containing 0.5% FBS, the BMMSCs were treated with AIMP1 peptide (100 ng/mL) for the indicated time. Protein extracts of human BMMSCs were prepared with lysis buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1 mM EDTA, 10 mM sodium fluoride, 10 mM β-glycerophosphate, 0.5 mM sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride). Forty micrograms of protein extract per lane was subjected to electrophoresis, transferred to a polyvinyldene fluoride membrane (Millipore), and blotted with their specific antibodies. Antibodies against p-glycogen synthase kinase-3β (GSK3β), GSK3β, pAkt, Akt, pERK, and ERK were purchased from Cell Signaling Technology. Antibodies against β-catenin, c-myc, HSP90, YY1, and cyclin D1 were purchased from Santa Cruz Biotechnology. Anti-tubulin antibody was purchased from Sigma-Aldrich. Antibody against FGFR2 was purchased from Abcam. To separate the nucleus and the cytoplasm, cells were lysed with hypotonic solution (10 mM HEPES, pH 7.4, 10 mM KCl, 0.5 NP40, 1.5 mM MgCl2, 0.5 mM EGTA, 12 mM β-glycerophosphate, 10 mM NaF, 1 mM Na3VO4, 1 mM phenylmethane sulfonylfluoride (PMSF), and 5 μg/mL aprotinin) to obtain the cytoplasmic proteins. The nucleus was then lysed with 10 mM HEPES, pH 7.4, 0.3 M KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 12 mM β-glycerophosphate, 10 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and 5 μg/mL aprotinin.
Immunofluorescence staining
Human BMMSCs (5×104 cells) were seeded onto a cover slip (9×9 mm) and cultured for 12 h. After serum starvation for 6 h with low-glucose DMEM containing 0.5% FBS, the BMMSCs were treated with AIMP1 peptide (100 ng/mL) for 2 h. The cells were fixed with 4% paraformaldehyde at room temperature (R.T) for 10 min and washed thrice with 1X phosphate-buffered saline (PBS). The cells were permeabilized with 0.1% Triton X-100 at R.T for 5 min and washed thrice with 1X PBS. The cells were blocked with 1X PBS containing 2% bovine serum albumin (BSA) at R.T for 1 h and incubated with anti-β-catenin antibody diluted 1:100 at R.T for 2 h. Thereafter, the cells were washed thrice with 1X PBS and then reacted with Alexa Fluor-488-conjugated secondary antibody (Invitrogen) at R.T for 1 h. The cell nuclei were counterstained with 4′-6′ diamidino-2-phenylindole (Invitrogen).
Luciferase assay
BMMSCs (3×104 cells) were seeded onto a 12-well plate and cultured in low-glucose DMEM supplemented with 10% FBS and 1% streptomycin/penicillin for 24 h. BMMSCs were then transfected with TOPflash plasmid and Renilla luciferase vector using Lipofectamine 2000 (Invitrogen) for 12 h. After serum starvation for 12 h with low-glucose DMEM containing 0.5% FBS, the cells were treated with AIMP1 peptide (100 ng/mL) in the presence or absence of LY294002 and U0126 for 24 h. To assess the dominant negative (DN) effect of T-cell factor (TCF), 0.1, 0.2, and 0.5 μg of TOPflash plasmid expressing DN-TCF (kindly provided by Barry M. Gumbiner, University of Virginia) was transfected into BMMSCs with the TOPflash plasmid and the Renilla luciferase vector and expressed for 12 h. The transfected cells were treated with AIMP1 peptide (100 ng/mL) for 24 h. Luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions.
RT-polymerase chain reaction
Human BMMSCs (3×105 cells) were seeded onto a six-well plate and cultured in low-glucose DMEM containing 10% FBS and 1% streptomycin/penicillin for 12 h. After serum starvation for 4 h with low-glucose DMEM containing 0.5% FBS and 1% streptomycin/penicillin, the cells were treated with AIMP1 peptide (100 ng/mL) in the presence or absence of LY294002 and U0126 for 8 h. Total RNA was extracted using Easy-spin (iNtRON Biotechnology) according to the manufacturer's instructions. cDNA was synthesized by reverse transcription with 0.5 μg of total RNA, and each gene transcript was then amplified by quantitative reverse transcription-polymerase chain reaction (PCR) with specific primers. The primer sequences are summarized in Table 1.
Colony formation assay
C57/BL6 (male) mice were purchased from Orient Bio at 6 weeks of age and maintained until 7 weeks of age. The CHA Animal Care and Use Committee approved all animal studies (IACUC110013), and the investigation conformed to the Guide for the Care and Use of Laboratory Animals, published by the United States National Institutes of Health. AIMP1 peptide was diluted in sterile PBS and intraperitoneally injected as indicated. Mice were euthanized with isoflurane (Hana Pharm), and blood was harvested. Mononuclear cells were harvested using a Ficoll–Paque cushion (GE Healthcare). Mononuclear cells (5×106/well) were seeded into 0.1% gelatin-coated 12-well plates in DMEM with 10% FBS and 1% penicillin/streptomycin and maintained in a humidified chamber of 5% CO2. Non-adherent cells were removed, and the medium was replaced every 3 days.
Flow cytometry
For the phenotypic analysis of fibroblast-like cells after passage 3, cells were resuspended in PBS containing 2% BSA and fixed with 2% paraform–aldehyde for 10 min. The cells were washed with PBS containing 2% BSA and incubated for 1 h at 4°C with the following antibodies: CD34-FITC (BD Biosciences), CD45-adenomatous polyposis coli (APC) (Miltenyi Biotec), CD105-APC (R&D Systems), CD44-APC (BD Biosciences), CD29-FITC (AbD Serotec), and CD90-APC (BioLegend). After washing twice with PBS containing 2% BSA, the cells were analyzed by flowcytometry using an FACScalibur flow cytometer (BD Immunocytometry Systems).
Differentiation of MSCs
Adipogenic differentiation was induced at a cell density of 1×104 cells/cm2 in adipogenic medium (StemPro® Adipogenesis Differentiation Kit, Gibco BRL) for 14 days. Differentiated MSCs were fixed with 10% formaldehyde for 15 min at R.T, and washed once with 60% isopropanol. Cells were then stained with 0.6% (w/v) oil red O solution for 45 min and washed repeatedly with 60% isopropanol. Cells were washed twice with distilled water and examined under a light microscope. For osteogenic inductions, cells were cultured at 1×104 cells/cm2 in DMEM containing 10% FBS, 1% GlutaMax (Gibco BRL), 0.2 mM ascorbic acid, 10 mM glycerol 2-phosphate (Sigma), 1% antibiotics, and 0.1 μM dexamethasone for 21 days. The differentiated osteogenic cells were fixed with 10% formaldehyde for 15 min and washed thrice with deionized water. Cells were then incubated in 5% silver nitrate and exposed to strong light for 1 h. The reaction was stopped by treatment with sodium thiosulfate, and cells were examined under a light microscope. To induce chondrogenic differentiation, cells were cultured at 1×104 cells/cm2 for 21 days in DMEM containing 10% FBS, 1% antibiotics, 1% Insulin-Transferrin-Selenium-A Supplement (Gibco BRL), 50 μM ascorbic acid, 100 nM dexamethasone (Sigma), and 10 ng/mL transforming growth factor-beta 1 (ProSpec-Tany TechnoGene Ltd.). The differentiated chondrogenic cells were fixed for 10 min with 4% paraformaldehyde. Cells were stained with 0.5% Alcian blue in 0.1 N HCl (pH 1.0) overnight, and examined under a light microscope.
Affinity purification
AIMP1 peptide was coupled to Affi-Gel 15 (Bio-Rad) following the manufacturer's instructions. Briefly, Affi-Gel 15 beads were washed thrice with cold deionized water. The beads were incubated with AIMP1 peptide in 10 mM HEPES buffer for 4 h at 4°C. To block non-coupled beads, the beads were further incubated with 1 M ethanolamine-HCl (pH 8.0) for 1 h. The beads were transferred to a column and washed with 10 mM HEPES buffer. To prepare the plasma membrane fraction of BMMSCs, cells were homogenized with 20 strokes of a Dounce homogenizer in Tris-HCl buffer (25 mM Tris-HCl (pH 7.4), 1 mM EDTA, 0.5 mM EGTA, 10 mM NaCl, and protease inhibitor cocktails). One-tenth volume Tris-HCl buffer with 2.5 M sucrose was added to the homogenates. After centrifugation at 1,000g for 10 min, the supernatant was carefully harvested and further centrifuged at 15,000g for 15 min. The pellet was resuspended in Tris-HCl buffer containing 3 mM MgCl2 and 0.25M sucrose and was designated the plasma membrane fraction. The plasma membrane fraction was then incubated with AIMP1 peptide-coupled bead for 4 h at 4°C and washed thrice with Tris-HCl buffer containing 3 mM MgCl2 and 0.25M sucrose buffer. The precipitates were then dissolved in SDS sample buffer and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Bands of interest were in-gel digested with trypsin (Promega). For NanoLC-MS/MS analysis, the peptides were loaded on an Agilent 1100 Series nano-LC and LTQ-mass spectrometer (Thermo Electron). The mobile phase A for LC separation was 0.1% formic acid in deionized water, and the mobile phase B was 0.1% formic acid in acetonitrile. The chromatography gradient was set up to give a linear increase. Mass spectra were acquired using data-dependent acquisition with a full mass scan (400–1800 m/z) followed by MS/MS scans. Each MS/MS scan acquired was an average of one microscan on the LTQ. The temperature of the ion transfer tube was maintained at 200°C, and the spray was 1.5.0–2.0 kV. The normalized collision energy was set at 35% for MS/MS. The database search criteria were as follows: taxonomy, homo sapiens; fixed modification; carboxyamidomethylated (+57) at cysteine residues; variable modification, oxidized (+16) at methionine residues; maximum allowed missed cleavage, 1; and MS tolerance, 100 ppm. Typical contaminants such as trypsin (used for proteolysis) and keratin were excluded.
Modeling of AIMP1 peptide structure and AIMP1 peptide-FGFR2 docking
The AIMP1 peptide structure was predicted using the Rosetta method that was implemented with the Monte Carlo minimization [28]. The AIMP1 peptide was docked into the binding site of the FGFR2 structure using ClusPro [29 –33]. The X-ray structure of FGFR2 (PDB code 1IIL) was used to define the binding site for the molecular docking studies. Energy minimization was conducted for the AIMP1 peptide and FGFR2 with the Amber force field [34] with UCSF Chimera [35]. The standard set parameters were used in all calculations. For energy assessment of the AIMP1 peptide-FGFR2 interaction, FireDock [36,37] with an energy-based score was used. During energy assessment, all values were set to the default values. The APBS program [38] and Coulombic Surface Coloring method with UCSF Chimera were used to calculate the electrostatic surface potentials of the protein-peptide complex model. Electrostatic potential maps were calculated by numerically solving the Poisson–Boltzmann equation based on molecular mechanics. The AIMP1 peptide-FGFR2 complex structure was analyzed and displayed with the UCSF Chimera software [35].
Statistical analysis
SPSS, version 10.0 (SPSS, Inc., Chicago, IL) was used for statistical analysis. The data are expressed as mean±SD. The differences between the means were calculated using the Mann–Whitney U-test. A P-value<0.05 indicated a statistically significant difference.
Results
AIMP1 peptide induces the phosphorylation of GSK3β, Akt, and ERK via FGFR2 in BMMSCs
In a previous report, we showed that the N-terminus of AIMP1 increased the proliferation of fibroblast via ERK activation to enhance wound repair [20]. To determine whether AIMP1 could activate signaling molecules related to proliferation, a peptide corresponding to the N-terminal residues (amino acids 6–46) of human AIMP1 was added to BMMSCs for various times. AIMP1 induced the phosphorylation of Akt within 5 min, after the phosphorylation of ERK and GSK3β (Fig. 1A). Next, we used affinity purification with the cytoplasmic membrane fraction of BMMSCs to isolate the cognate receptor for AIMP1, as described in the Materials and Methods section. NanoLC-MS/MS analysis showed that AIMP1 peptide bound to FGFR2 (Table 2). We then investigated whether the treatment of AIMP1 peptide to BMMSCs could induce phosphorylation of FGFR2. As shown in Fig. 1B, AIMP1 peptide induced phosphorylation at tyrosine residue of FGFR2. In addition, whether the knock-down of FGFR2 using specific si-RNA could abolish the activation of Akt and ERK by AIMP1 peptide was examined. As shown in Fig. 1C, knock-down of FGFR2 abolished Akt and ERK activation by AIMP1 peptide, which suggests that AIMP1 peptide activates Akt and ERK via FGFR2. To predict the potential interaction between AIMP1 peptide and FGFR2, various in silico molecular models were generated using the FGFR2 crystal structure previously reported [39], and the best structure was selected based on the energy-based score. In this model, AIMP1 peptide was located on the inner space of FGFR2 that was composed of four subunits, through electrostatic interactions (Supplementary Fig. S1A, B; Supplementary Data are available online at
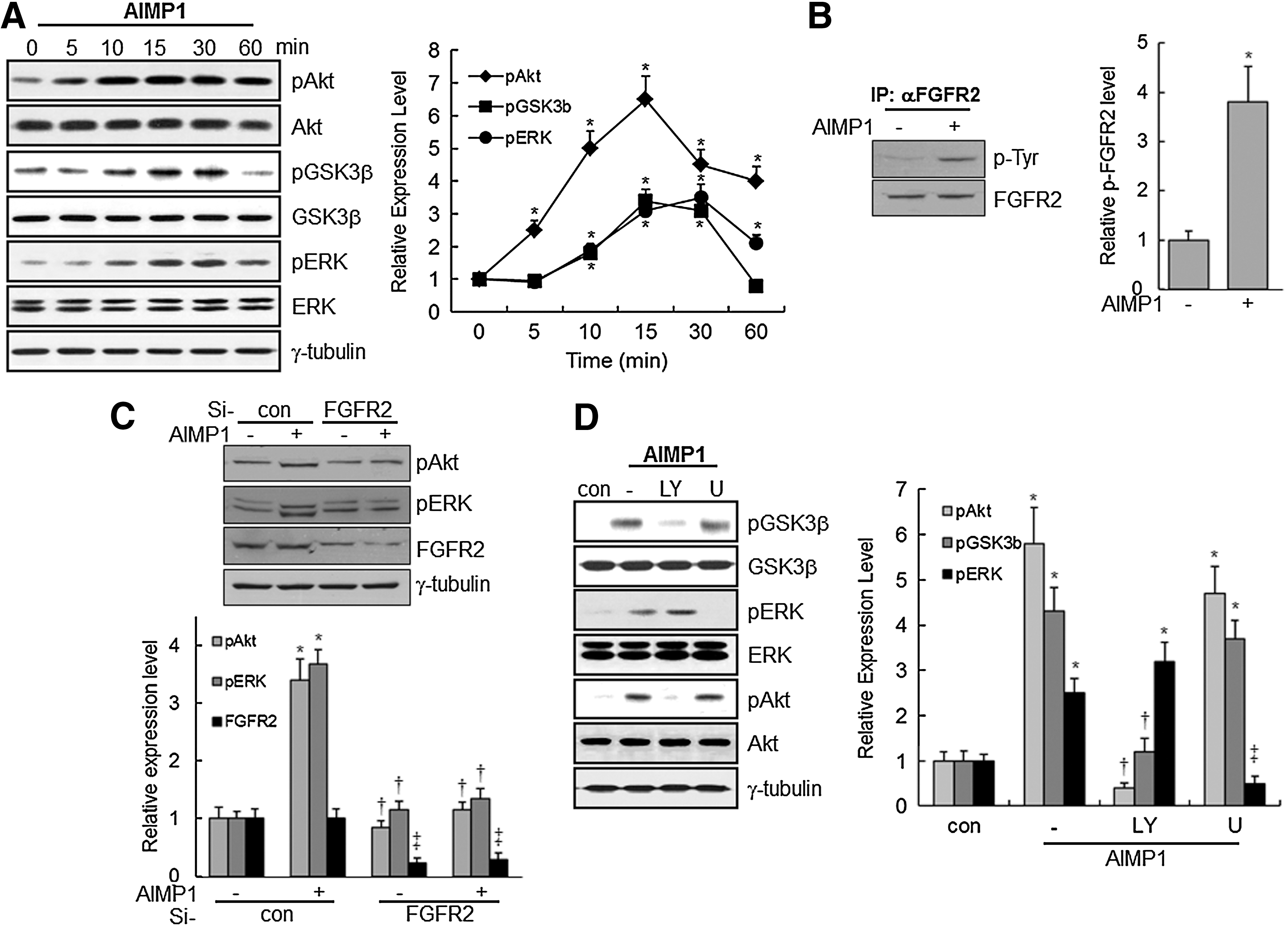
ARS-Interacting Multi-functional Protein 1 (AIMP1) induces the phosphorylation of Akt, ERK, and glycogen synthase kinase-3β (GSK3β) via fibroblast growth factor receptor 2 (FGFR2) in human bone marrow-derived mesenchymal stem cells (BMMSCs).
Score is −10×Log(P), where P is the probability that the observed match is a random event; it is based on NCBInr database using the MASCOT searching program as LC-MS data.
GSK3β is a cytoplasmic serine/threonine kinase that regulates Wnt/β-catenin, Hedgehog, Notch, and insulin signaling [40]. Moreover, the phosphorylation of Ser9 in GSK3β by phosphorylated Akt decreases the activity of GSK3β [41]. Therefore, the relationship between the phosphorylation of GSK3β and Akt activation was examined. Pretreatment with LY294002, a PI3K inhibitor, abolished the phosphorylation of Akt and GSK3β; whereas pre-treatment with U1026, an ERK inhibitor, did not eliminate the phosphorylation of Akt and GSK3β. These findings suggest that activation of Akt by AIMP1 peptide is prerequisite for the phosphorylation of GSK3β (Fig. 1D).
AIMP1 peptide enhances the protein stability and nuclear translocation of β-catenin
GSK3β phosphorylates β-catenin in the Wnt/β-catenin signaling pathway, leading to the ubiquitination and proteasomal-dependent degradation of β-catenin [42]. Since the phosphorylation of GSK3β by Akt activation prevents the proteasomal degradation of β-catenin, we evaluated whether AIMP1 peptide could induce the accumulation of non-phosphorylated β-catenin via Akt in BMMSCs. Immunoblot analysis clearly showed that AIMP1 increased the β-catenin level in a dose-dependent manner; treatment with LY294002 abolished this increase (Fig. 2A, B). In addition, we examined whether β-catenin translocated into the nucleus in response to AIMP1. Protein extracts were fractionated into cytoplasmic and nuclear fractions, and β-catenin was detected with a specific antibody. As shown in Fig. 2C, β-catenin accumulated in the nucleus after treatment with AIMP1 peptide. Furthermore, immunofluorescence staining of β-catenin clearly showed that AIMP1 peptide induced the translocation of β-catenin into the nucleus (Fig. 2D). Pretreatment with LY294002, but not U0126, inhibited the nuclear accumulation of β-catenin by AIMP1, further confirming that the nuclear translocation of β-catenin induced by AIMP1 peptide was mediated by the activation of Akt (Fig. 2E).
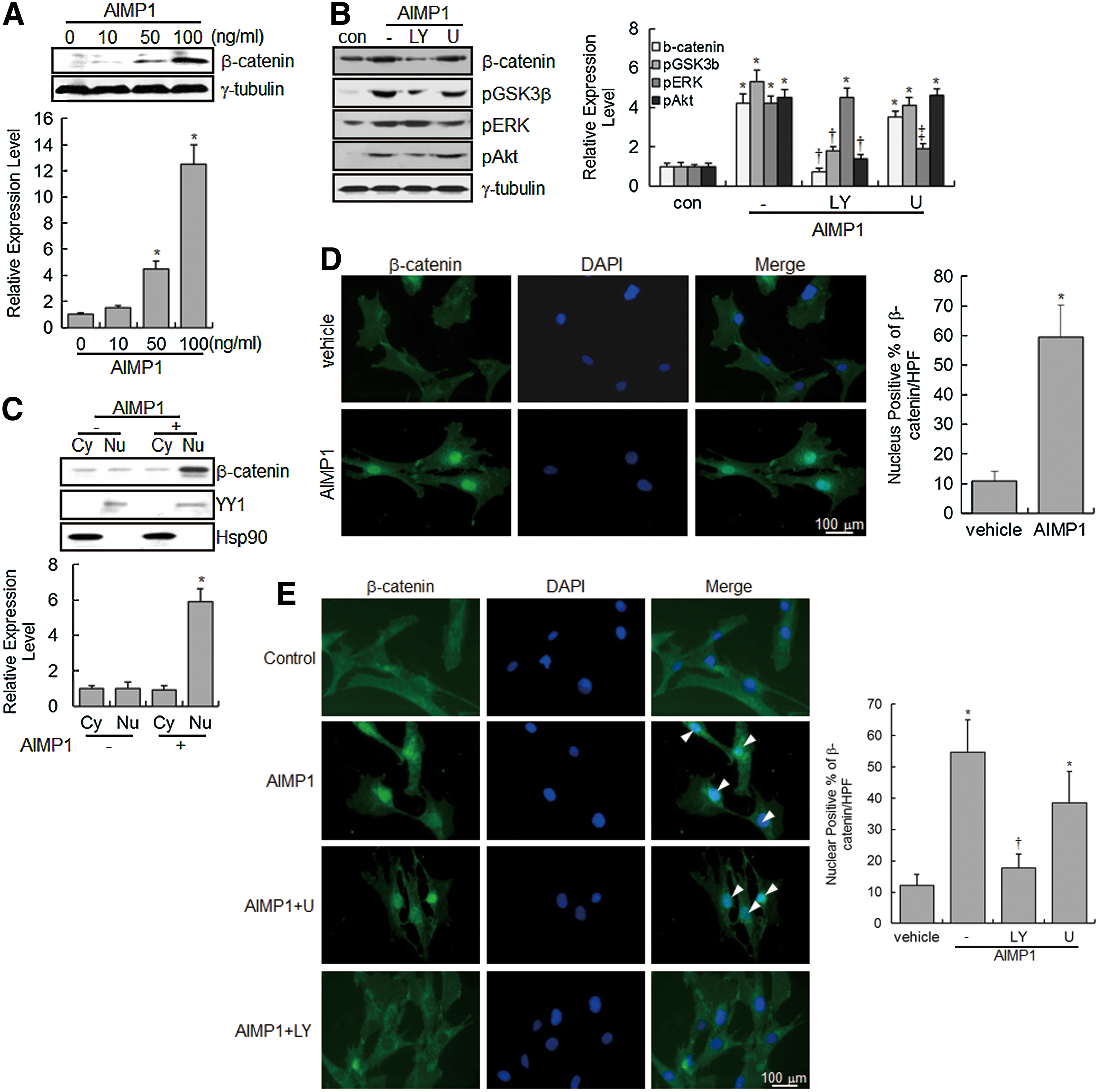
AIMP1 induces the stabilization and nuclear translocation of β-catenin in human BMMSCs.
AIMP1 peptide increases cyclin D1 and c-myc expression via the β-catenin/TCF complex
Nuclear accumulation of β-catenin stimulates the activation of the lymphoid-enhancer factor (LEF)/TCF complex, leading to the transcriptional up-regulation of downstream target genes, including cyclin D1 and c-myc, which are critical for cell proliferation and development [43,44]. We investigated whether the nuclear accumulation of β-catenin, induced by AIMP1 peptide, activated the LEF/TCF complex. First, a luciferase assay was performed using a TOPflash vector with LEF/TCF-binding sites. AIMP1 peptide significantly increased the luciferase activity about four-fold compared with the control treatment (Fig. 3A). Western blot and qRT-PCR analyses showed that AIMP1 peptide increased the expression of cyclin D1 and c-myc at the transcriptional level (Fig. 3B, C). In addition, the correlation between the activation of Akt, ERK, and the LEF/TCF complex was investigated. Pre-treatment with U1026 and LY294002 suppressed the increase in luciferase activity induced by AIMP1 peptide. These results suggested that the activation of Akt and ERK by AIMP1 peptide was critical to the increased activity of the LEF/TCF complex (Fig. 3D). Western blot and qRT-PCR analyses supported that the increase in cyclin D1 and c-myc expression induced by AIMP1 peptide was regulated at the transcriptional level via Akt and ERK activation (Fig. 3E, F). We knocked down β-catenin using specific si-RNA and examined whether the increases in luciferase activity and in cyclin D1 and c-myc expression are mediated via the stabilization of β-catenin induced by AIMP1 peptide. As shown in Fig. 4A, knock-down of β-catenin abrogated the increase in luciferase activity induced by AIMP1 peptide. Western blot and qRT-PCR analyses showed that the increase in cyclin D1 and c-myc expression induced by AIMP1 peptide is mediated by β-catenin (Fig. 4B, C). In addition, DN TCF inhibited the increase in TOPflash luciferase activity induced by AIMP1 peptide in a dose-dependent manner (Fig. 4D). Western blot and qRT-PCR analyses further supported that the activation of the LEF/TCF complex by AIMP1 peptide played a critical role in increasing the expression of cyclin D1 and c-myc (Fig. 4E, F).
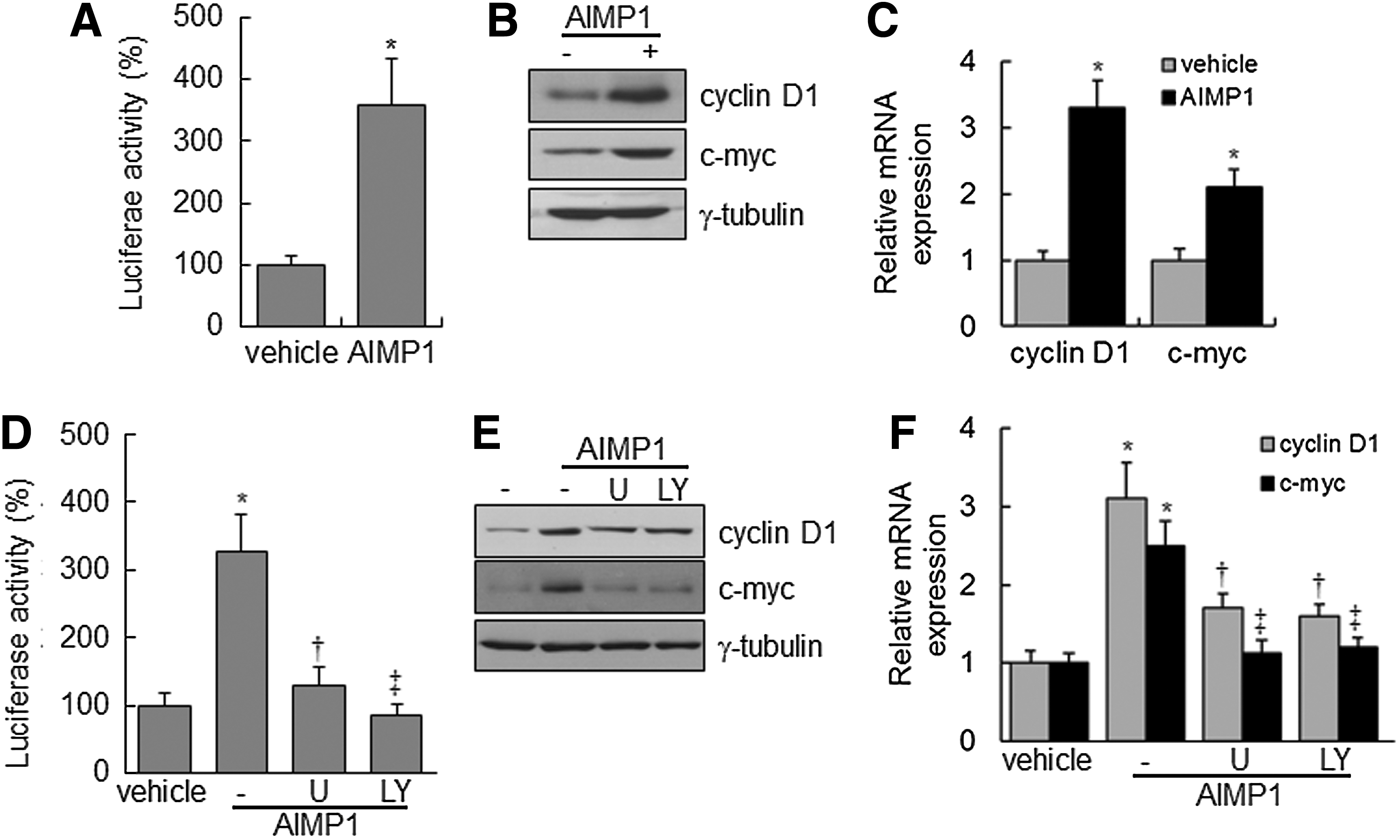
AIMP1 increases the transcriptional activity of T-cell factor (TCF) in human BMMSCs. BMMSCs expressing TOPflash vector were treated with AIMP1 peptide (100 ng/mL) and harvested.
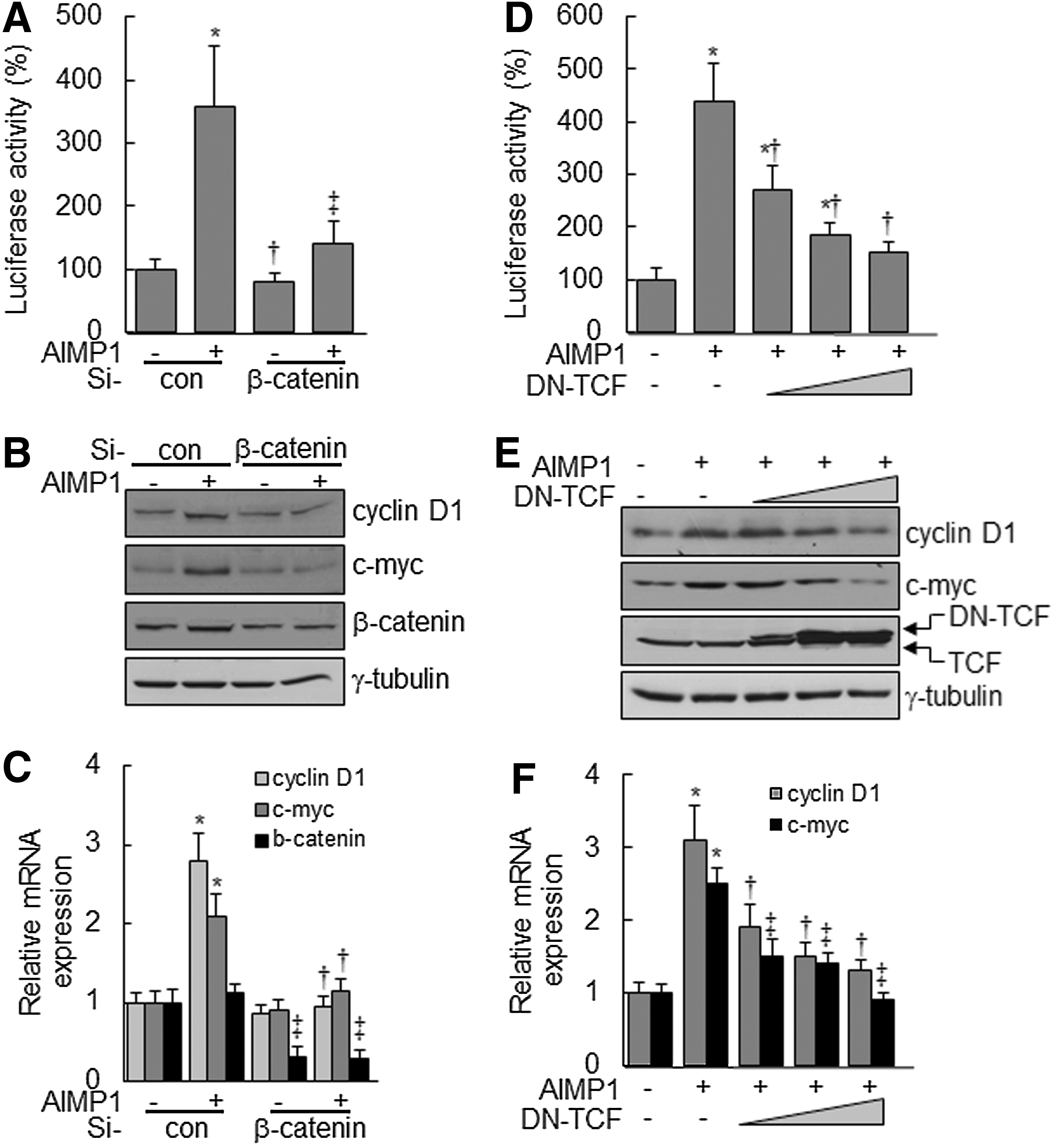
AIMP1 increases the expression of cyclin D1 and c-myc via β-catenin and TCF.
AIMP1 increases the proliferation of human BMMSCs
The induction of cyclin D1 and c-myc genes by the activation of the LEF/TCF complex is critical for cell cycle progression [43,45]. Therefore, we investigated whether the increase in cyclin D1 and c-myc expression induced by AIMP1 peptide could lead to promoting the proliferation of BMMSCs. First, AIMP1 peptide was treated to MSCs for 24 h, and the MTT assay was performed. AIMP1 peptide stimulated the proliferation of BMMSCs in a dose-dependent manner (Fig. 5A). In addition, the proliferation-inducing activity of AIMP1 peptide was examined to determine whether it could lead to an increase in the number of cells. AIMP1 peptide was added to BMMSCs in a time-dependent manner, and the number of cells was counted at the times indicated. AIMP1 peptide significantly increased the proliferation of BMMSCs in a time-dependent manner compared with control treatments (Fig. 5B). The knock-down of β-catenin using specific si-RNA inhibited the proliferation of BMMSCs induced by AIMP1 peptide in both the MTT assay and the cell counting assays (Fig. 5C, D). DN TCF also suppressed the proliferation of BMMSCs induced by AIMP1 peptide, further confirming that AIMP1 peptide induced the proliferation of BMMSCs through the LEF/TCF complex via the stabilization of β-catenin (Fig. 5E, F). We further examined whether AIMP1 peptide would affect the differentiation of human BMMSCs into adipocytes, chondrocytes, and osteoblasts. However, the differentiation of BMMSCs into each phenotype was not affected by treatment with AIMP1 peptide (data not shown).
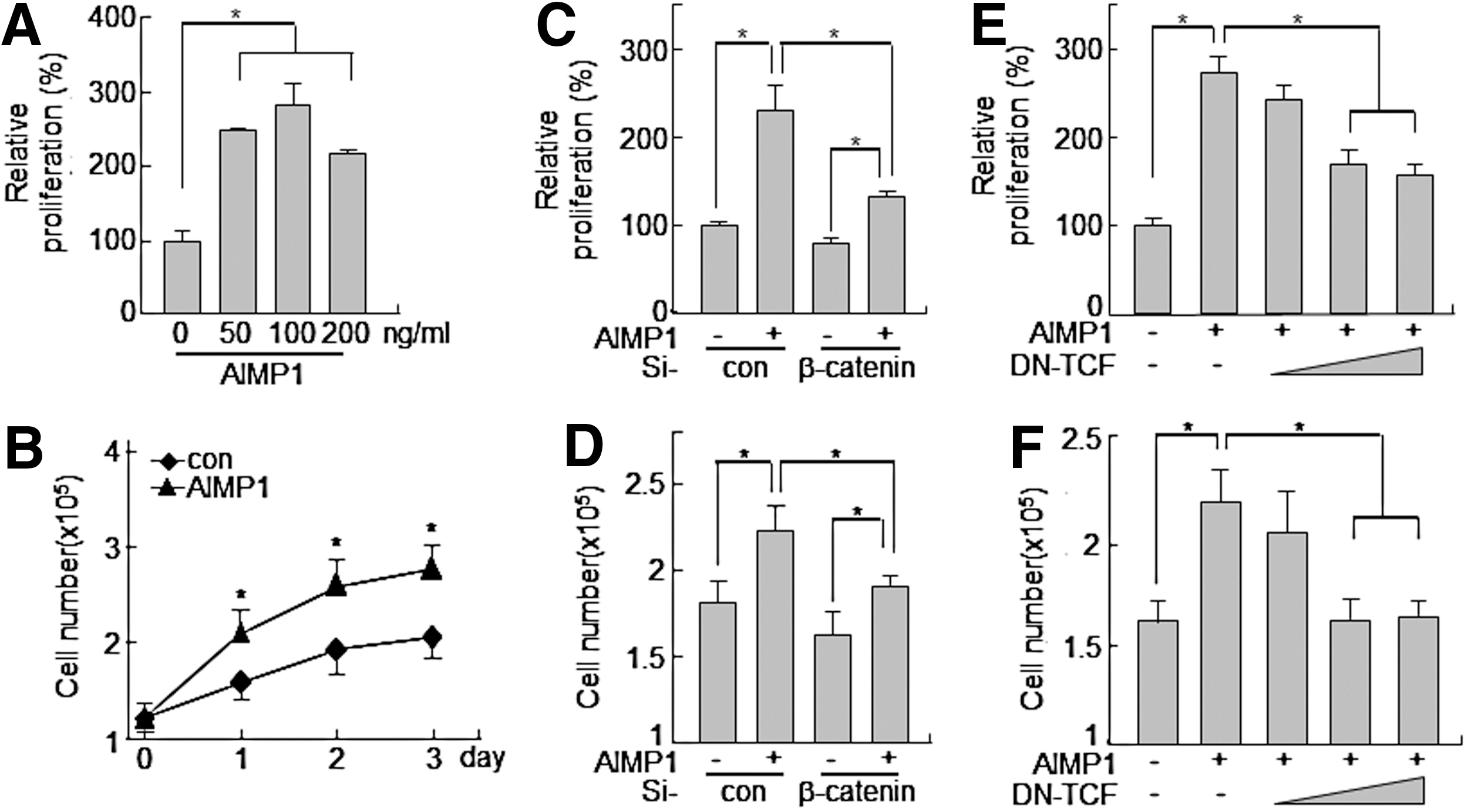
AIMP1 peptide enhances the proliferation of human BMMSCs.
AIMP1 peptide increases colony formation of MSCs
We injected AIMP1 peptide into the intraperitonium of mouse and analyzed colony-forming ability using mononuclear cells isolated from peripheral blood. AIMP1 peptide significantly increased fibroblast-like colony formation in a dose- and time-dependent manner (Fig. 6A, B). To determine whether the fibroblast-like cells were MSCs, flow cytometry was performed using a variety of MSC markers. The cultured fibroblast-like cells were positive for CD105, CD44, CD90, and CD29, but negative for CD34 and CD45, which is characteristic of MSCs (Fig. 6C). Therefore, we next examined whether the isolated fibroblast-like cells could differentiate into other cells such as adipocytes, osteocytes, and chondrocytes. As shown in Fig. 6D, fibroblast-like cells efficiently differentiated into adipocytes, osteocytes, and chondrocytes, confirming that the colonies of fibroblast-like cells induced by AIMP1 peptide were MSCs.
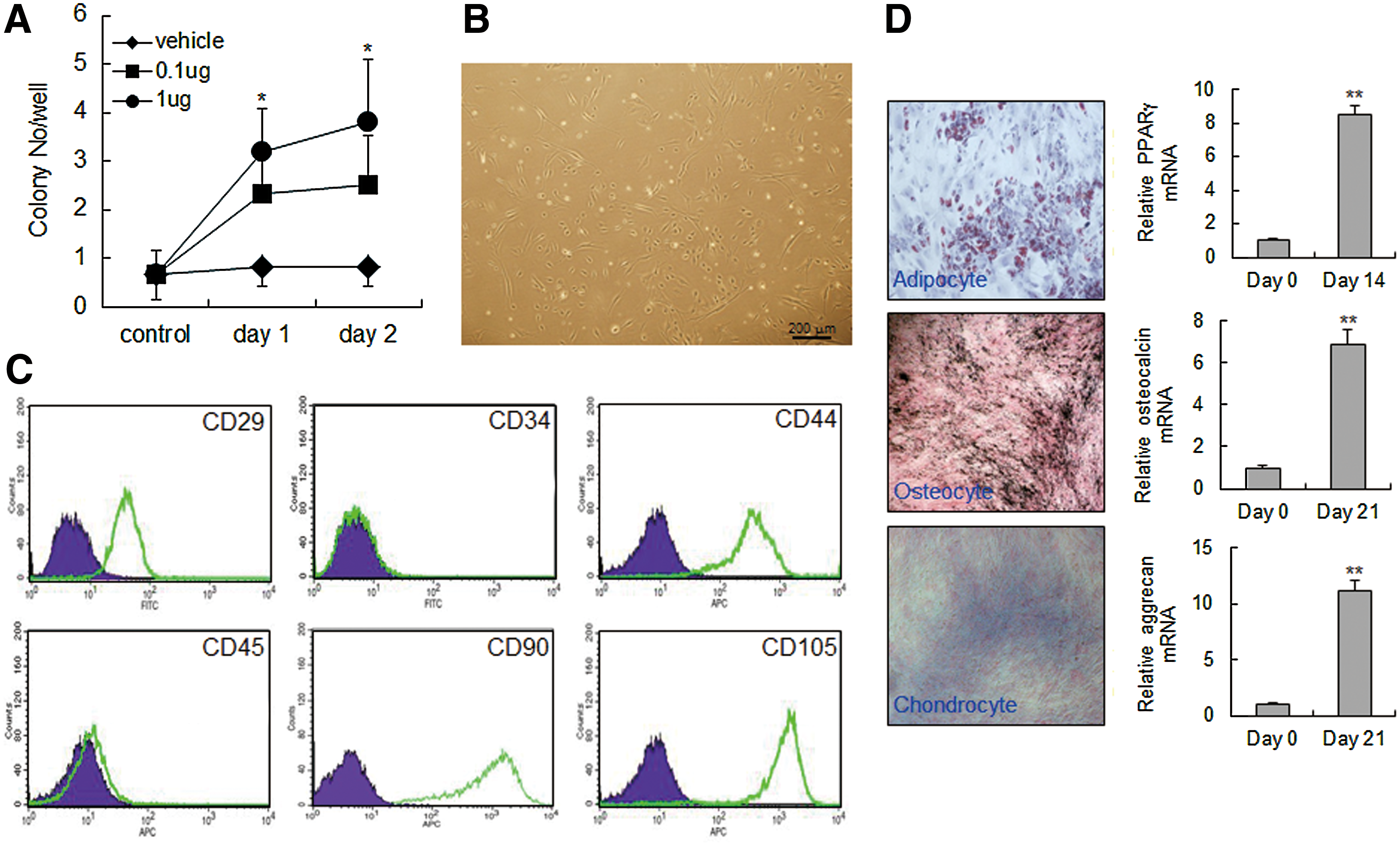
AIMP1 peptide stimulates colony formation of mouse MSCs.
Discussion
BMMSCs have been studied as a potential source of material for cell therapy. However, it is difficult to obtain an adequate supply of MSCs from a single patient. Moreover, methods for the ex vivo manipulation of MSCs are poorly developed due to the animal source for cell manipulation. Thus, the development of an in vitro expansion method that preserves the differentiation and proliferative potential of MSCs has been suggested. Recently, S1P, EGF, and FGF were shown to induce the proliferation of MSCs without affecting their multipotency [14 –17]. In our previous report, we suggested that AIMP1 could function as a growth factor to induce fibroblast proliferation [20]. Therefore, we examined whether AIMP1 peptide could induce the proliferation of MSCs. First, the ability of AIMP1 peptide to activate the signaling mediators, including Akt, ERK, JNK, and p38 MAPK, was examined. AIMP1 was previously shown to activate a variety of signaling molecules in various cell lines [20,23,25,46]. Consistently, in our study, AIMP1 peptide activated Akt in BMMSCs, leading to the inhibition of GSK3β. Since GSK3β plays a critical role in the regulation of β-catenin stability when Wnt is stimulated, we investigated whether AIMP1 peptide, similar to Wnt, could inhibit GSK3β via the activation of APC complex. However, AIMP1 peptide did not induce the phosphorylation of disheveled (Dvl) or the interaction of axin with low-density lipoprotein receptor-related protein 5/6, indicating that AIMP1 peptide did not activate the canonical Wnt signaling pathway (data not shown). In addition, inhibition of Akt using LY294002 abolished the accumulation and the nuclear translocation of β-catenin after stimulation with AIMP1 peptide. This suggests that AIMP1 peptide promoted the stabilization of β-catenin via the activation of Akt in a Wnt-independent manner.
In this study, AIMP1 peptide was shown to activate ERK (Fig. 1); treatment with U0126 inhibited the phosphorylation of ERK but not Akt. In addition, the inhibition of Akt activity using LY294002 did not suppress ERK phosphorylation. The findings suggest that AIMP1 peptide induced the phosphorylation of ERK independent of Akt activation: AIMP1 activated Akt and ERK via FGFR2 as shown in Fig. 1B, and Supplementary Fig. S1. FGFR downstream signaling occurs through two main pathways: the Ras-dependent MAPK pathway and the Ras-independent PI3K-Akt pathway [47,48]. Interestingly, inhibition of ERK phosphorylation suppressed the expression of LEF/TCF target genes induced by AIMP1 peptide (Fig. 3D); whereas AIMP1 peptide-induced β-catenin accumulation and nuclear translocation were not affected (Fig. 2B). Considering that phosphorylated ERK accumulates in the nucleus and activates TCF/Elk-1 for the expression of downstream target genes [49 –51], it appears that AIMP1 peptide increased the proliferation of BMMSCs by inducing the expression of TCF target genes via the activation of ERK as well as Akt via FGFR. In addition, the down-regulation of β-catenin using si-RNA and DN TCF abrogated the increase in cell proliferation induced by AIMP1 peptide, further supporting the fact that AIMP1 peptide induced cell proliferation via sequential stimulation of β-catenin and the TCF complex through the activation of Akt and ERK.
AIMP1 peptide stimulated the colony-forming ability of MSCs in peripheral blood, indicating that AIMP peptide increased the population of MSC precursor cells. However, it is not clear whether the increased population of peripheral blood MSCs induced by AIMP1 peptide was a result of increased proliferation in peripheral blood or mobilization from BM. Further studies are required to unveil the exact mechanism. Taken together, the results of this study suggest that AIMP1 peptide induces the expression of cyclin D1 and c-myc by activating the β-catenin/TCF complex through activation of ERK and Akt via FGFR2, leading to the proliferation of BMMSCs. Since AIMP1 peptide has FGF-like activity in promoting the proliferation of MSCs, AIMP1 peptide might be a useful tool with which to manipulate MSCs in vitro.
Footnotes
Acknowledgments
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MEST) (2012M3A9C6049719).
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.