
Abstract
Calcium signals affect many developmental processes, including proliferation, migration, survival, and apoptosis, processes that are of particular importance in stem cells intended for cell replacement therapies. The mechanisms underlying Ca2+ signals, therefore, have a role in determining how stem cells respond to their environment, and how these responses might be controlled in vitro. In this study, we examined the spontaneous Ca2+ activity in human neural progenitor cells during proliferation and differentiation. Pharmacological characterization indicates that in proliferating cells, most activity is the result of transient receptor potential (TRP) channels that are sensitive to Gd3+ and La3+, with the more subtype selective antagonist Ruthenium red also reducing activity, suggesting the involvement of transient receptor potential vanilloid (TRPV) channels. In differentiating cells, Gd3+ and La3+-sensitive TRP channels also appear to underlie the spontaneous activity; however, no sub-type-specific antagonists had any effect. Protein levels of TRPV2 and TRPV3 decreased in differentiated cells, which is demonstrated by western blot. Thus, it appears that TRP channels represent the main route of Ca2+ entry in human neural progenitor cells (hNPCs), but the responsible channel types are subject to substitution under differentiating conditions. The level of spontaneous activity could be increased and decreased by lowering and raising the extracellular K+ concentration. Proliferating cells in low K+ slowed the cell cycle, with a disproportionate increased percentage of cells in G1 phase and a reduction in S phase. Taken together, these results suggest a link between external K+ concentration, spontaneous Ca2+ transients, and cell cycle distribution, which is able to influence the fate of stem and progenitor cells.
Introduction
T
Studies of Ca2+ signaling in stem cells so far have described a variety of different mechanisms, reflective of a combination of species, cell type, and developmental stage differences. In a study of human embryonic stem cell-derived neural epithelial cells, it appeared that transient receptor potential (TRP) channels were the predominant source of Ca2+ influx, and were also active, but to a lesser extent, in developing neurons derived from these cells [6]. In contrast, in human mesenchymal stem cells, Ca2+ signals largely or entirely originated from internal stores [7,8], though other sources also contribute [9]. This was also true of mouse embryonic stem cells (ESCs) [10], but in more lineage-restricted mouse epithelial cells, both intra- and extracellular sources were involved [11]. Interfering with Ca2+ signaling via InsP3 receptors [8] or via TRP channels [6] was found to impair proliferation.
During proliferation, Ca2+ is required for entry into and progression through G1, and for phase transition at the G1/M and G2/S phase boundaries [1,12 –14]. Growth factors promoting proliferation also act, in part, through Ca2+-sensitive mechanisms; it was found in rat neural stem cells (NSCs) that fibroblast growth factor (FGF)-2 induced Ca2+ influx via TRPC1 channels [15], and in mouse ESCs that epidermal growth factor (EGF) acted via mitogen-activated protein kinases (MAPK) and protein kinase C (PKC)-dependent pathways [16]. Ca2+ signals have also been implicated in neurogenesis, in mouse neural crest cells via IP3 receptors [17], and in primary cultures of mouse NSCs via voltage-gated Ca2+ channels (CaV) [18].
In this study, we have analyzed the pharmacological properties of spontaneous Ca2+ signals in a human fetal neural progenitor cell line, ReNcell VM (Millipore), identifying TRP channels as the dominant source in both proliferating and differentiating cells. We also sought to examine the effect of changing the level of Ca2+ signaling in culture on proliferation and differentiation.
Materials and Methods
Cell culture
The ReNcell VM cell line (Millipore) was used in this study [19
–21]. Cells were proliferated as neurospheres as described [19]. In short, cells were cultured for 7 days in uncoated 96-well plates with proliferation medium consisting of Dulbecco modified Eagle's minimal essential medium (DMEM)/F12, B27 media supplement, Glutamax (2 mM), gentamycin (50 μg/mL), and heparin (10 units/mL) and the growth factors EGF (20 μg/mL) and FGF-2 (10 μg/mL). Single neurospheres were plated on glass coverslips coated with laminin (10 μg/mL) and poly-
For altering the extracellular K+ concentration ([K+]E), a customized DMEM/F12 medium without KCl, NaCl, NaHCO3, NaH2PO4, Na2HPO4, and sodium pyruvate was used (PAA Laboratories). The control medium contained (in mM) the following: 4.16 KCl, 120.61 NaCl, 29.02 NaHCO3, 0.45 NaH2PO4, 0.50 Na2HPO4, and 0.50 sodium pyruvate; low [K+]E: 1 KCl, 123.77 NaCl, 29.02 NaHCO3, 0.45 NaH2PO4, 0.50 Na2HPO4, and 0.50 sodium pyruvate; and high [K+]E: 15 KCl, 109.77 NaCl, 29.02 NaHCO3, 0.45 NaH2PO4, 0.50 Na2HPO4, and 0.50 sodium pyruvate. For varying [K+]E during proliferation, all cells were plated in control media and allowed for 24 h to proliferate before changing to control, low, or high [K+]E media. The cells were subsequently differentiated in control media. For varying [K+]E during differentiation, cells were proliferated in control media, then differentiated for 24 h in control media to allow the cells to exit the cell cycle, so as not to conflate effects of [K+]E during proliferation with those during differentiation. After 24 h, the media were changed to the experimental conditions.
For analysis of cell cycle and proliferation, cells were cultured as a monolayer. Then, cells were seeded on laminin-coated cell culture flasks and allowed to proliferate for 3–4 days until they reached a confluence of ∼80%. Differentiation was induced by withdrawal of growth factors as described earlier.
Flow cytometry
Flow cytometry was used to quantify expression of βIII-tubulin (βIII-tub) or to determine the number of cells in different cell cycle states. The cells were cultured as described earlier. Cells were fixed in 1% paraformaldehyde (PFA) for 15 min at room temperature and then suspended in washing buffer [PBS+0.5% bovine serum albumin (BSA)+0.02% Na-azide]. To stain, cells were suspended in saponin buffer and incubated with the βIII tub antibody (1:100, Santa Cruz Biotechnology) for 2 h at room temperature. After washing, they were incubated with Alexa Fluor 647 (goat-anti-mouse, 1:1000, Molecular Probes) for a further hour, washed twice with saponin buffer, and afterward, re-suspended in washing buffer for analysis.
To analyse the cell cycle distribution, cells were grown as a monolayer culture. After plating, the cells were allowed for 24 h to settle in control medium before switching to the experimental conditions. The time of this medium change was taken as 0 h for the experiments. The cells were fixed in 70% ethanol for 1 h at −20°C, and stored in those conditions until use. RNase solution (1 mg/mL in HBS) was activated by heating to 37°C for 1 h. The cells were centrifuged for 10 min at 1300×g and 4°C, then suspended in RNase solution, and incubated for 30 min at 37°C. Cells were stained with propidium iodide (50 μg/mL) dissolved in HBSS for 30 min at 37°C. Experiments were performed using an FACSCalibur system (Becton Dickinson) in combination with Cell Quest Pro software.
Cell counting and viability
Cell proliferation and vitality was measured with a CASY Model TT (Roche). For CASY measurements, the cells were cultured as a monolayer. Each probe was measured thrice, and mean values were used for analysis.
Calcium imaging
For Ca2+ imaging experiments, human neural progenitor cells (hNPCs) were incubated in 5 μM Fura-2AM (Invitrogen) for 30 min at room temperature, then allowed a further 30 min after washing for the dye to de-esterise before imaging. The imaging system consisted of a Polychrome V light source and a PCO sensicam (Till Photonics) controlled by a PC running TillVision (v4.0, Till Photonics). Images were acquired at 1 Hz for 1,000 s with excitation wavelengths of 340 nm and 380 nm. Transients were analyzed offline using Mini Analysis (v6.0.7; Synaptosoft).
Western blotting
Western blots were performed to analyze the expression of a set of subtypes TRPV channels as they provide a common Ca2+ entry route in neurons. In short, protein concentrations in RIPA total cell lysates were estimated by the bicinchoninic acid assay (Pierce). Samples were diluted in 5×Sample buffer and boiled at 95°C for 5 min. 25 μg of proteins were loaded into gradient (4%–15%) Sodium dodecyl sulfate-polyacrylamide gels and separated electrophoretically (Criterion; Biorad). Prestained peqGOLD marker IV (PEQLAB) was used as a molecular weight marker. Proteins were transferred to nitrocellulose membranes (Hybond-ECL; GE Healthcare) by semi-dry blotting (Bio-Rad). After blocking with 2% BSA in Tris buffered saline with 0.1% Tween20 (TBST) for 1 h, the membranes were incubated with primary antibodies (TRPV1, rabbit polyclonal, alomone labs, 1:1000; TRPV2, rabbit polyclonal, 1:2000, LSBio; TRPV3, mouse monoclonal, 1:2000, abnova; GAPDH, mouse monoclonal, abcam, 1:10.000) overnight at 4°C. Blots were rinsed thrice with TBST and probed with fluorescent secondary antibodies (goat anti-rabbit Alexa Fluor 680, goat anti-mouse Alexa Fluor 680, and goat anti-mouse IRDye800, 1:10.000 each) in TBST with 0.1% BSA for 1 h. Visualization and quantification of proteins were performed using the Odyssey Infrared Imaging System (LI-COR Biosciences GmbH) in combination with Odyssee 1.2 software. Expression of GAPDH protein was used as a loading control to normalize the expression of the target proteins. Thus, relative expression levels of the target proteins were determined.
Statistical analysis
Analysis of the data was carried out with GraphPad Prism 5 (GraphPad Software, Inc.). Data are given as mean±SEM. Unless otherwise stated, unpaired t-tests were used to test for significance, with *P<0.05 and **P<0.01. Number of independent preparations is given by “N,” and number of cells measured is given by “n.”
Results
hNPCs generate spontaneous Ca2+ transients
Spontaneous Ca2+ transients were present in hNPCs throughout proliferation and differentiation (Fig. 1A, B). In proliferation, 54.5%±3.2% (N=3, n=19) of cells exhibited Ca2+ transients (Fig. 1A), where the frequency of transients in individual cells was generally low (2.9±0.2 mHz, Fig. 1D), but a small proportion of cells showed high-frequency oscillations (Fig. 1A). The level of activity in differentiating cells was lower (3 days differentiated (DD) 27.8%±2.4%, N=4, n=13; 5DD 25.6%±2.2%, N=2, n=4; 7DD 23.4%±2.2%, N=5, n=16; P<0.001, ANOVA with Tukey's multiple comparison test), with the exception of 4DD (44.6%±4.4% (N=4, n=8, P>0.05). In turn, the level of activity at 4DD was significantly higher than at 3 and 7DD (P<0.01 vs. 3DD, P<0.001 vs. 7DD). No high-frequency oscillations were seen in differentiating cells (Fig. 1B), and there was no difference in the frequency in individual cells (3DD n=118 cells, 4DD n=104, 7DD n=121, P>0.05, Kolmorogov–Smirnov 2 sample test). For further analysis, data from cells differentiated for 3, 5, and 7 days were pooled, excluding data of 4DD.
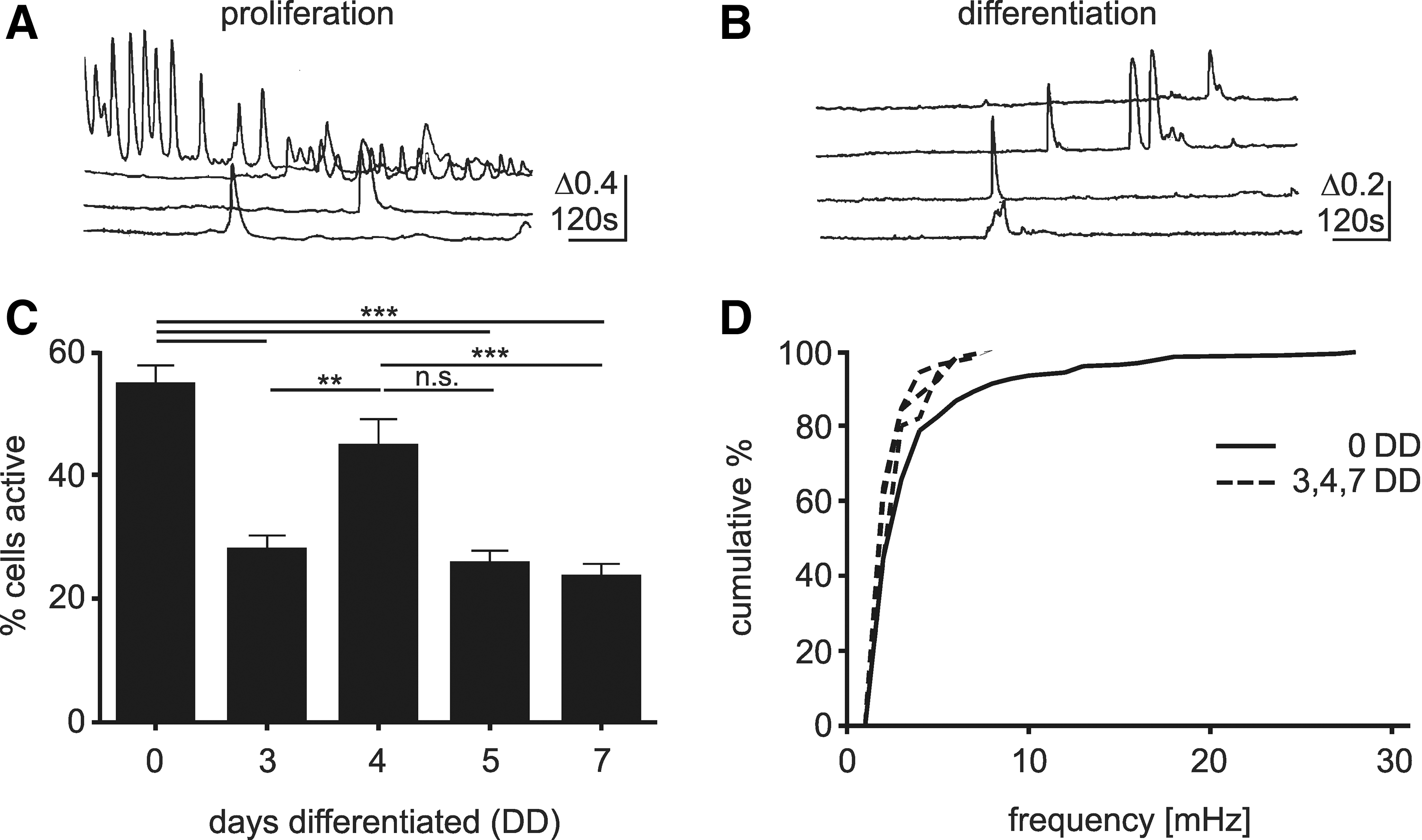
Spontaneous Ca2+ transients in hNPCs. Proliferating
To determine the source of the Ca2+ signals, recordings were made using a Ca2+-free extracellular solution, and from cells treated with thapsigargin, a SERCA pump inhibitor (Fig. 2). Removal of extracellular Ca2+ substantially reduced the number of active cells in both proliferating (control: 54.5%±3.2%, Ca2+-free: 7.9%±2.8%, N=2, n=10, P<0.001) and differentiating cells (control: 25.4%±1.5%; Ca2+-free: 0.5%±1.3%, N=3, n=7, P<0.001), indicating that the transients were almost entirely due to influx of Ca2+ from the extracellular space. In contrast, thapsigargin had no effect on differentiating cells (23.7%±3.8%, N=3, n=9, P>0.05 vs. control), but lowered the activity of proliferating cells (32.4%±6.1%, N=3, n=6, P<0.01 vs. control).
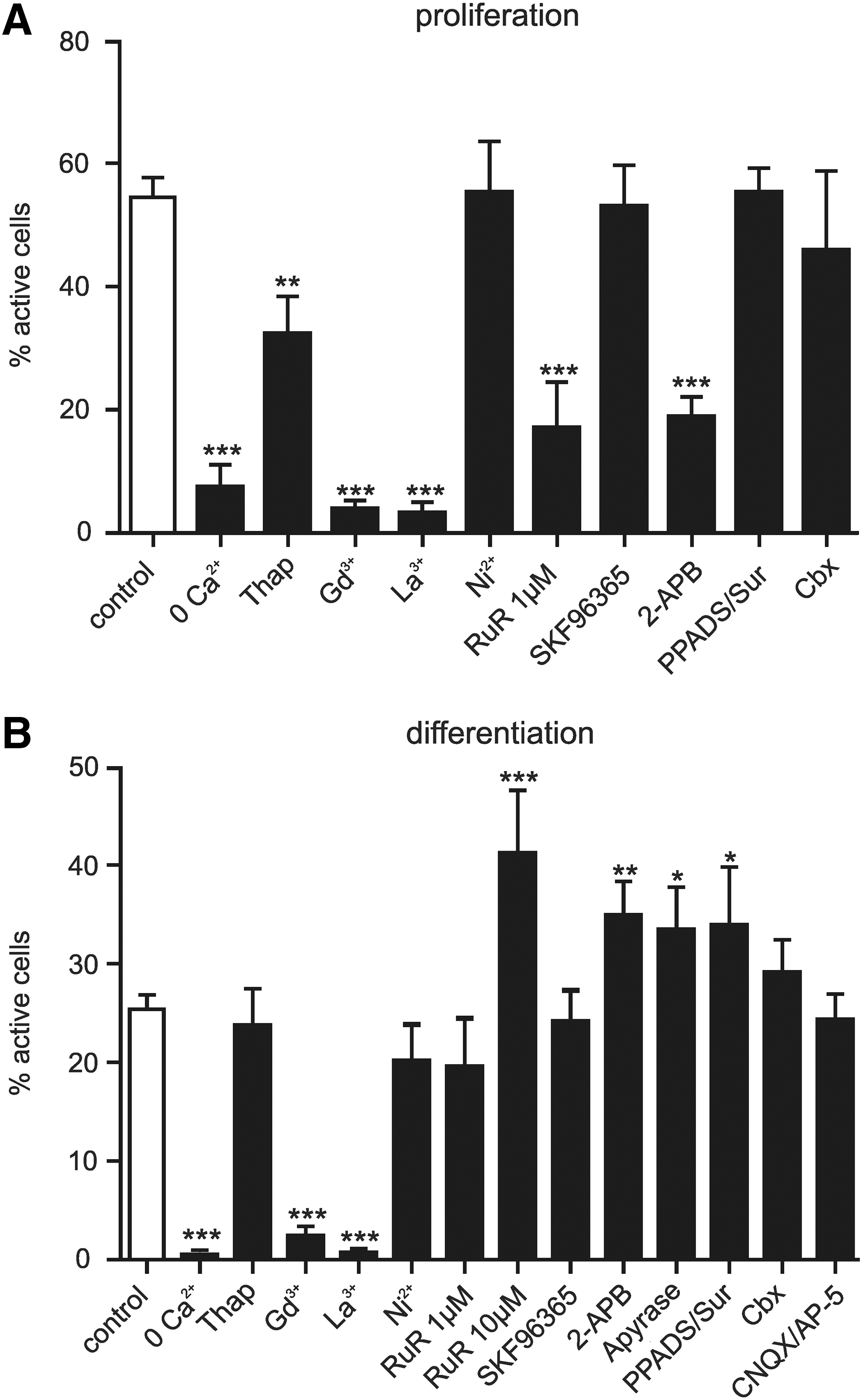
Pharmacological profile of spontaneous Ca2+ transients. To examine the mechanism underlying the transients, various Ca2+ channel antagonists were applied to proliferating
We next tested a range of antagonists of different types of plasma membrane Ca2+ channels (Fig. 2). The heavy metal ions GdCl3 or LaCl3 (100 μM) are broad spectrum antagonists, inhibiting Cav channels, P2×receptors, and many TRP channels [22 –24]. When applied to the hNPCs, they substantially reduced activity in both proliferating (GdCl3: 3.9%±1.4%, N=3, n=6, P<0.001 vs. control; LaCl3: 3.3%±1.8%, N=3, n=6, P<0.001 vs. control) and differentiating cells (GdCl3: 2.4%±2.8%, N=3, n=8, P<0.001 vs. control; LaCl3: 0.7%±1.2%, N=3, n=7, P<0.001 vs. control).
To narrow down the possible sources, we applied antagonists that are specific to each of the possible channel families. To block Cav channels, we applied a high concentration of NiCl2 (2.5 mM), which had no effect in proliferating (NiCl2: 55.3%±8.3%, N=3, n=6, P>0.05) or differentiating cells (NiCl2: 20.5%±3.0%, N=3, n=13, P>0.05). Combined application of P2X and P2Y receptor antagonists PPADS (50 μM) and suramin (100 μM) also had no effect during proliferation (PPADS/Sur: 55.3%±4.0%, N=3, n=9, P>0.05); while in differentiating cells, a slight significant increase was seen (PPADS/Sur: 34.0%±5.9%, N=3, n=7, P<0.05). This increase was reproduced by application of the ATP-degrading enzyme apyrase (10 U/mL) (apyrase: 33.4%±4.3%, N=2, n=8, P<0.05), suggesting that it is an effect of reduced puringeric signaling.
Following these findings, we then applied a variety of antagonists that have been shown to effect TRP channels. SKF 96365 (5 μM), frequently used as an inhibitor of TRPC channels, was shown to inhibit spontaneous Ca2+ transients in human ESC-derived neurons [6]; however, its application had no effect on proliferating (53.0%±6.6%, N=3, n=7, P>0.05) or differentiating cells (4.1%±3.2%, N=3, n=11, P>0.05). Ruthenium red (RuR) can block all TRPV channels depending on concentration, as well as TRPC3, TRPM6, and TRPA1. It is also known to inhibit ryanodine receptors and Cav channels. At 1 μM, it blocks TRPV1, 2, 3, 4, and 5; while TRPV6 is blocked by 10 μM [24]. Application of 1 μM RuR to proliferating cells reduced the level of spontaneous activity (17.1%±7.4%, N=3, n=6, P<0.001), whereas no effect was observed in differentiating cells (19.6%±4.9%, N=3, n=7, P>0.05). Unexpectedly, 10 μM RuR increased the level of activity in differentiating cells (RuR: 41.2%±6.8%, N=3, n=8, P>0.001). However, this effect is most likely an artefact that is related to the ability of RuR to bind to calmodulin [25], and potentially other Ca2+ binding proteins, which could act to reduce the endogenous Ca2+ buffering capabilities and increase the free Ca2+ to bind Fura-2. A third drug, 2-aminoethoxydiphenyl borate (2-APB), has diverging effects on TRP channels, inhibiting TRPC and TRPM channels, while acting as an agonist at TRPV1, TRPV2, and TRPV3 channels (but note reports that human TRPV2 does not respond to 2-APB [26]). Application of 2-APB (100 μM) to proliferating cells induced a robust, transient influx of Ca2+ (77.5%±7.8%, N=3; Fig. 3A, B), suggesting that TRPV1, TRPV2, and/or TRPV3 channels are present. In contrast, on wash-in to differentiating cells, a few exhibited a transient influx (11.6%±11.6%, N=3, Fig. 3A, B), suggesting that functional TRPV1 and/or TRPV3 channels are largely absent in differentiating cells, though many showed a small increase in their baseline Ca2+ level (Fig. 3A). Measurement of spontaneous activity in cells under 2-APB (pretreated for>15 min) showed decreased activity in proliferating cells (18.9%±3.2%, N=3, n=8, P<0.001); whereas in differentiating cells, activity levels increased (34.9%±3.4%, N=3, n=8, P<0.01). 2-APB inhibits a variety of Ca2+ signaling mechanisms, including release from internal stores and gap junction coupling via connexins [27]. Gap junction coupling can enable the movement of Ca2+ and InsP3 between cells, which may initiate Ca2+ transients in coupled cells, and unpaired connexins have been shown to initiate Ca2+ signals in the developing ventricular zone [28]. However, gap junction coupling did not appear to influence the Ca2+ signalling in the hNPCs, as application of the connexin antagonist carbenoxolone (100 μM) had no effect on proliferating (45.9%±12.8%, N=3, n=6, P>0.05) or differentiating cells (29.1%±3.3%, N=3, n=9, P>0.05).
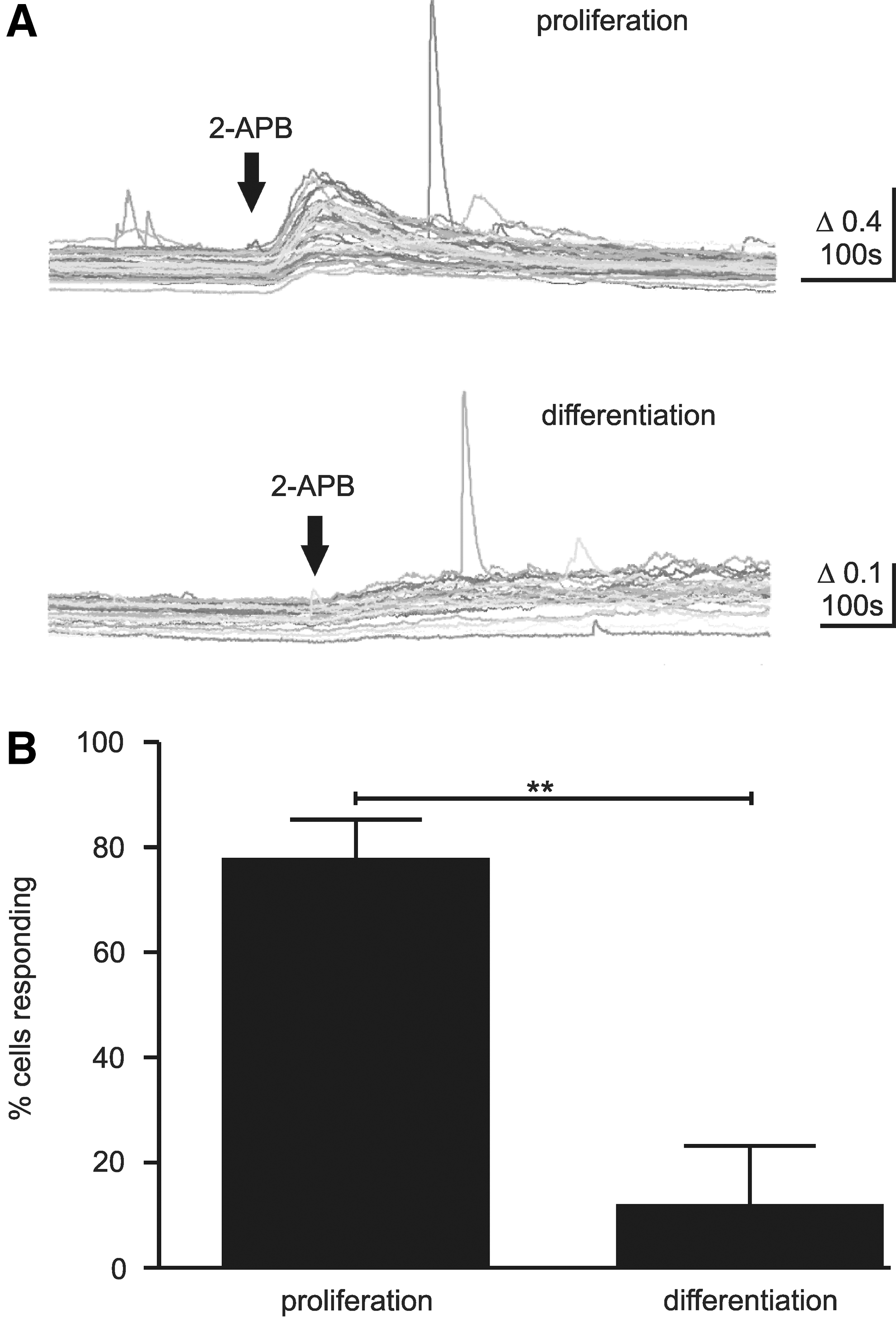
TRPV channel agonist induced Ca2+ increase in hNPCs. 2-APB was able to induce an increase in Ca2+ in proliferating (
Addition of the glutamate receptor antagonists AP5 (50 μM) and CNQX (20 μM) to differentiating cells had no effect on the level of activity (24.3%±2.6%, N=3, n=9, P>0.05).
TRPV channel expression is reduced after differentiation
The influx of Ca2+ in response to 2-APB in proliferating cells compared with differentiated cells suggested the presence of TRPV channels that were down regulated on differentiation. To find evidence for this, we performed western blot analysis for TRPV1, V2, and V3 channels (Fig. 4A–C, upper panel). The expression was analyzed in proliferating cells (0DD) and cells differentiated for 3 (3DD) and 7 (7DD days. TRPV1, V2, and V3 channels were present in proliferating cells and supporting the calcium imaging experiments, TRPV2 and V3 expression was reduced in cells differentiated for 3 days (51% and 58%, respectively; Fig. 4B) and 7 days (36% and 37%, respectively; Fig. 4C). In contrast, TRPV1 expression was slightly higher in differentiating cells, but this did not reach statistical significance (Fig. 4A).

Western blotting of TRPV1, TRPV2, and TRPV3 channel protein. Western blot experiments reveal expression of TRPV1, TRPV2, and TRPV3 channels in both proliferating (0DD) and differentiating (3DD and 7DD) hNPCs. Quantification of the corresponding signals revealed that TRPV2 and TRPV3 protein was significantly lower in differentiating cells (
Control of spontaneous Ca2+ transients via [K+]E
Given the influence of Ca2+ on many signaling pathways, control of Ca2+ signaling levels in cell culture might be a method to help control hNPC development. One basic method for regulating general levels of Ca2+ influx is given through adjustment of the extracellular K+ concentration ([K+]E), thereby changing the hNPC membrane potential and the driving force for Ca2+ entry, and which may also be used to activate CaV. With the exception of CaV channels, this method is nonspecific, potentiating or depressing all active Ca2+ signals. This method has been previously used to depolarize hNPCs during differentiation and was associated with an increase in neuronal development [29].
We, therefore, sought to examine the effect of increased and decreased [K+]E on the level of spontaneous Ca2+ signals. Prolonged exposure to increased [K+]E strongly decreased the levels of activity in proliferating and differentiating cells in comparison to control [K+]E (Fig. 5A, B), in both the percentage of cells active (proliferating: control 54.5%±3.2%, high K+ 3.6%±2.7%, differentiating: control 25.4%±1.5%, high K+ 3.1%±0.9%) and the frequency of transients per cell (proliferating: control 1.6±0.1 Hz, high K+ 0.2±0.05 Hz, P<0.01, differentiating: control 0.5±0.03 Hz, high K+ 0.2±0.08 Hz, P<0.01; Fig 5C). Lowering the KCl concentration had the opposite effect, inducing a strong increase in the level of activity, as a percentage of active cells (proliferating: control 54.5%±3.2%, low K+ 91.8%±1.9%, differentiating: control 25.4%±1.5%, low K+ 51.9%±3.8%), and frequency per cell (proliferating: 4.2±0.2 Hz, P<0.01 vs. control, differentiating: 3.2±0.2 Hz, P<0.01 vs. control; Fig, 5C). The effect of altered [K+]E was dependent on the concentration, with half maximal effective concentrations (EC50) of 2.5 and 1.2 mM calculated for proliferating and differentiating cells, respectively (Fig. 5D). The increase in activity resulting from removing extracellular K+ could be blocked by 100 μM LaCl3, indicating that the source of the transients was the same as that in control cells (proliferating 0 K+: 99.4%±0.6%, N=1, n=3 vs. 0 K++La3+ 1.3%±0.7% N=3, n=4, P<0.001; differentiating 0 K+: 93.1%±1.1%, N=5, n=18 vs. 0 K++La3+ 0.5±0.5, N=3, n=6, P<0.001).
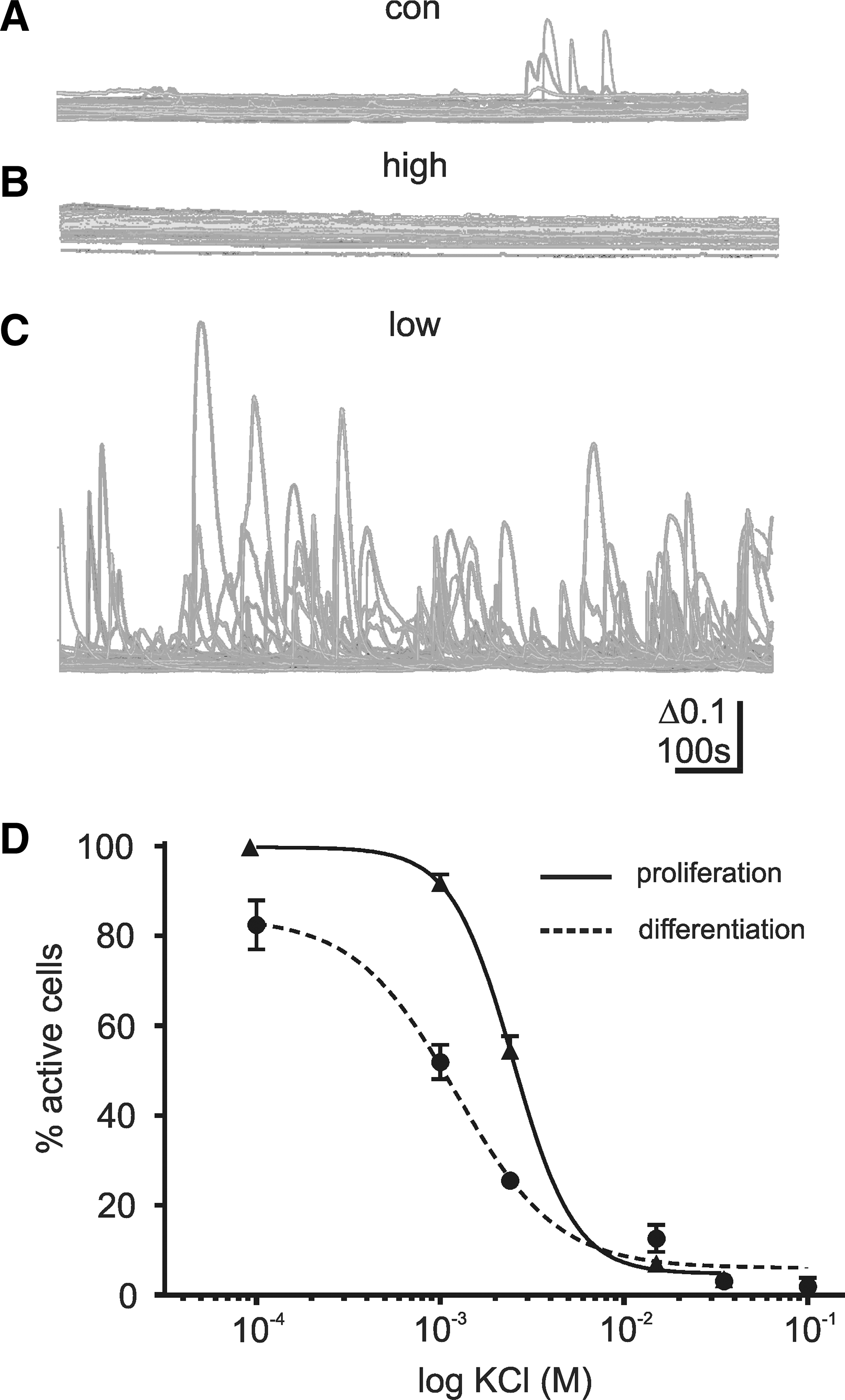
Spontaneous Ca2+ transients can be controlled by extracellular K concentration [K+]E. To investigate the influence of [K+]E, spontaneous Ca2+ transients were measured using external solution with altered [K+]E. In comparison to a solution with [K+]E.
Proliferation of hNPCs is attenuated by low concentration of K+
Since Ca2+ signals are a part of the cell cycle regulation, we examined how exposing the hNPCs to different K+ concentrations in culture affected their proliferation. Proliferation rates were calculated from cells exposed to control, low, and high [K+]E for 24, 48, and 72 h (Fig. 6A). Under low [K+]E conditions, the proliferation rate of the cells was slower than under conditions of control [K+]E or high [K+]E. (doubling time in hours: control: 17.4±2.2, N=5, low: 33.4±5.2, N=5, high: 17.2±1.7, N=3), and it resulted in significantly fewer cells at 72 h (Fig. 6C). There was no significant difference in the cell number between control and high [K+]E conditions. A small, but statistically significant reduction in the viability of cells under low [K+]E condition compared with control and high [K+]E condition was observed (P<0.05, ANOVA with Tukey's multiple comparison test), while there was no significant difference between high and control [K+]E condition (Fig. 6D).
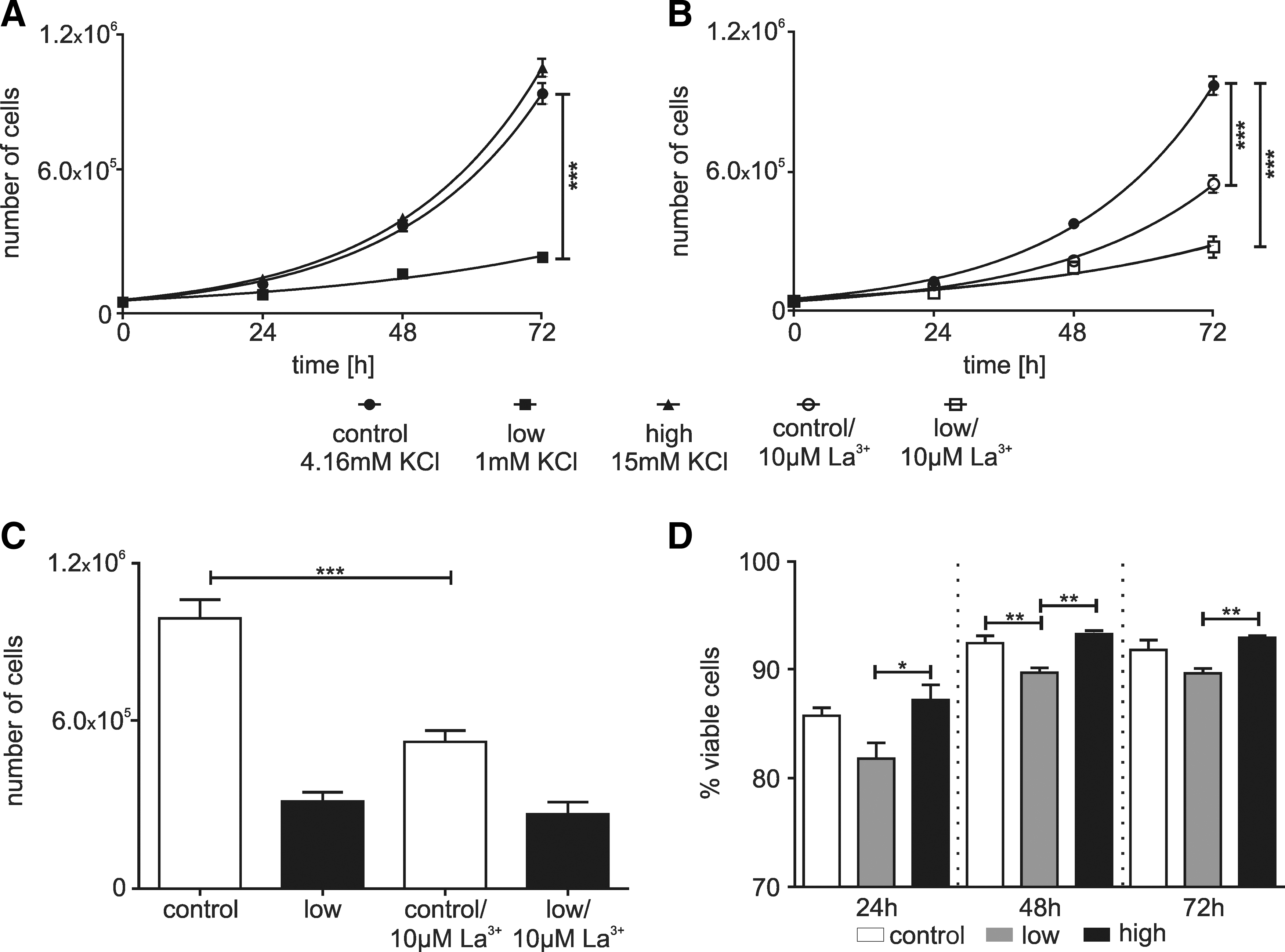
Proliferation of hNPCs is attenuated by high [K+]E.
To test the involvement of Ca2+ transients in the effect of low [K+]E, 10 μM La3+ was added to the control and low [K+]E conditions. In medium with control [K+]E, the addition of La3+ reduced the proliferation rate (Fig. 6B) and accordingly increased the doubling time (doubling time in hours: control: 16.9±1.7, N=7, 10 μM La3+: 19.2±3.1, N=3). The number of cells was significantly lower at 72 h (Fig. 6D). In contrast, addition of La3+ to low [K+]E condition had no effect on the doubling time (doubling time in hours: low: 29.4±4.6; N=7; 10 μM La3+: 28.8±10.6, N=3) or the total number of cells (Fig. 6D). There was a small reduction in viability under La3+ at 24 and 48 h, but this is insufficient to account for the reduction in proliferation rate under control conditions. Ca2+ signals and membrane potential are associated with transition between cell cycle phases and progression through G1 [1,30]. To determine whether lowering [K+]E was acting at a specific point in the cell cycle, distribution within the cycle was analyzed after 16, 28, and 40 h (Fig. 7A–C). Low [K+]E induced a slight increase of cells in G1/G0 phase compared with control medium (16 h N=6, 28 h N=4, and 40 h N=4; P<0.05, ANOVA with Tukey's multiple comparison test) and a reduction in the number of cells in S phase (P<0.05, ANOVA with Tukey's multiple comparison test; Fig. 7). This indicates, assuming cells progress uniformly through the cycle, a lengthening of G1 and a shortening of the S.
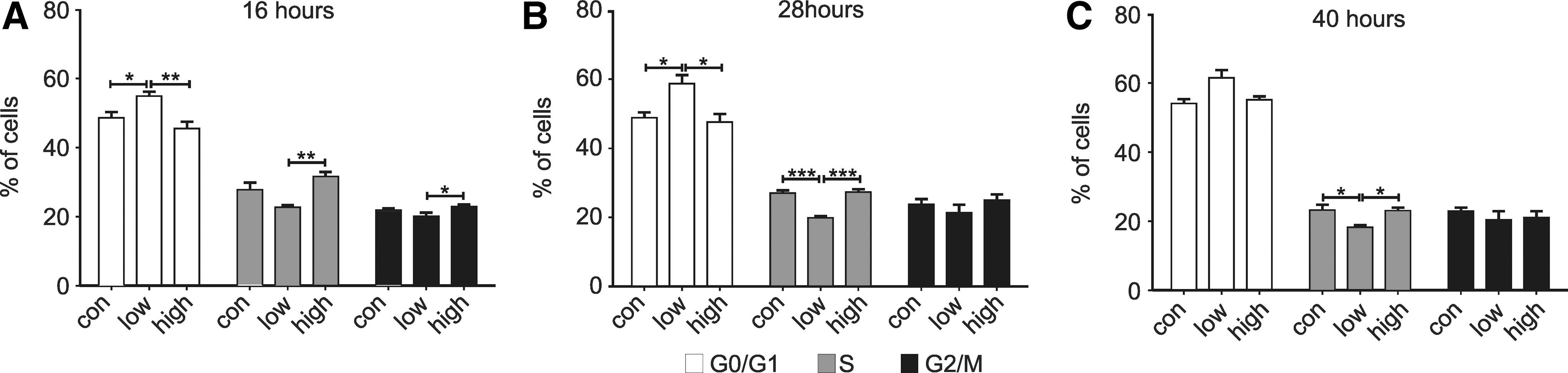
Distribution of different current types of hNPCs. Cell cycle distribution after
Both lengthening of G1 phase and shortening of S phase have been associated with neurogenesis [31,32]. Therefore, we asked whether low [K+]E has an influence on neuronal differentiaton of hNPCs. Cells were proliferated under control, low, and high [K+]E conditions for 16 h before the induction of differentiation in medium with control [K+]E. After 3 days of differentiation, the number of βIII-tub positive cells was analyzed using flow cytometry. No statistically significant differences were found between the conditions with altered [K+]E (control: 3.8%±0.04%; low K+: 3.3%±0.2%; high K+: 3.7%±0.2%; Fig. 7D, left panel). To examine whether [K+]E had any effect on neural development after differentiation, the cells were allowed 24 h to exit the cell cycle before switching to control, low, or high [K+]E condition. The analysis of βIII-tub-positive cells at 3DD revealed no statistically significant difference (control: 3.5%±0.1%; low K+: 3.3%±0.1%; high K+: 3.4%±0.2%; Fig. 7D, right panel).
Discussion
The range of developmental processes involving Ca2+ signaling suggests that understanding how these signals are regulated in different stem cell lines will be beneficial in the development of stem cell therapies. However, the wide range of potential mechanisms and our limited knowledge about such signaling mechanisms in stem cells presents a significant hurdle. In this study, we have shown that TRP channel activity appears to be the main source of Ca2+ influx during proliferation and differentiation of hNPCs, and it could specifically identify a change in TRPV2 and V3 expression on switching from proliferation to differentiation. The level of Ca2+ signaling in culture could be controlled by adjusting the extracellular K+ concentration, and an increase in Ca2+ signaling correlated with a slowing of the cell cycle, with features that have been associated with increased neurogenesis, though no such effect was apparent in subsequent differentiation in the hNPCs used in this study.
TRP channels underlie spontaneous Ca2+ signals in hNPCs
The pharmacological properties of the Ca2+ transients described here strongly suggest that TRP channels are the underlying mechanism for almost all the spontaneous activity seen in both proliferating and differentiating hNPCs.
In proliferating cells, the inhibition of activity by RuR combined with the Ca2+ influx caused by 2-APB indicate that TRPV1, V2, and or V3 channels were present, and that they contributed to the spontaneous activity. This finding differs from results obtained in another human proliferative NPC population, one derived from ESCs in vitro, in which RuR had no effect on spontaneous activity while Ni2+ inhibited activity [6]. Although 2-APB also reduced activity in the hNPCs, its targets are not clear, as apart from being a TRPC channel inhibitor, it also acts on internal stores. Furthermore, the effect size was similar to that of RuR, and may be due to inhibition of TRPV channels by continuous stimulation. A second TRPC channel inhibitor, SKF-96365, had no effect.
It is also apparent that other mechanisms contribute to the activity in proliferating cells, as high-frequency oscillations were seen in some proliferating hNPCs (Fig. 1A, D); however, these were too infrequent for further characterization in this study.
In differentiating cells, the inhibition by Gd3+ and La3+, plus the ineffectiveness of P2×receptor and CaV channel antagonists, also implicated TRP channels in the spontaneous activity. However, no other antagonists had any effect, indicating that the channel properties are distinct from those in proliferating cells. It appears that TRPV channels no longer contributed to the spontaneous activity, as RuR had no effect, and the lack of response to 2-APB application, along with the observed reduction of TRPV2 and TRPV3 protein expression, suggests that channel expression itself is reduced, rather than a reduction in activation. Unlike TRPV2 and TRPV3, TRPV1 was increased by a tendency in differentiating cells, though given the lack of 2-APB response it may be that these channels are either present at much lower levels, in a nonfunctional isoform, or not located in the plasma membrane.
Curiously, in differentiating cells, application of the P2 receptor antagonists PPADS and Suramin, or the ATP-degrading enzyme apyrase slightly increased the level of spontaneous activity. That both treatments had similar effects despite acting via unrelated mechanisms suggests that there is a biological effect, but via what potential mechanism is unclear. The apparent increase in activity by 2-APB in differentiating cells could be explained by the increase in baseline Ca2+ seen after 2-APB is applied (Fig. 3B), which is probably due to Ca2+ leak from internal stores and inhibition of mitochondrial Ca2+ uptake [33], and that could hyperpolarize cells through activation of KCa channels. Alternatively, Ca2+-sensitive Ca2+ channels could be activated, such as InsP3-Rs, or potentially the channel(s) underlying the spontaneous activity.
How specific signaling mechanisms are to species, cell type, and developmental stage versus functional purpose remains unclear, and will be important to clarify whether we are able to exploit these in the many cell lines that will be required for stem cell-based therapies.
Regulation of proliferation by [K+]E
We regulated Ca2+ signaling in culture by altering the extracellular K+ concentration. Although highly nonspecific, this method has the advantage of amplifying or suppressing existing signals, rather than imposing them on the system. This was supported by the observation that the activity in low [K+]E could still be abolished by La3+ and Gd3+. Lowering extracellular K+ could increase both the number of active cells and the frequency of activity for individual cells, while increasing K+ had the opposite effect. The fact that the frequency of transients in individual cells increased suggests that the effect was not simply due to a change in the electrochemical gradient, but rather that other factors, such as a voltage-dependent change in channel conductance, a gain modulation by an additional secondary activation of internal stores, or an increase in the signals activating the channels were involved.
When cultured in low [K+]E medium, the proliferation rate of the cells was dramatically slowed; whereas when proliferated in high [K+]E medium, there was no, or possibly marginal, effect on proliferation rate. The effect of low [K+]E on proliferation corresponded to a general slowing of the cell cycle without inducing arrest, while also lengthening G1 and shortening S phase relative to control medium. This could be interpreted as being consistent with the effect of K+ on Ca2+ signaling, where under control conditions Ca2+ signaling is low, and lowering [K+]E produces a dramatic increase. However, this interpretation is complicated by the slowing of proliferation by La3+, which inhibits the Ca2+ transients and would, therefore, be expected to counteract the effect of [K+]E on Ca2+ signaling. Both K+ channel activity and Ca2+ signaling are involved in proliferation [1,30,34,35], and lowering [K+]E will affect multiple cellular processes. In most, but not all, cell types, K+ channel block inhibits proliferation [30,35,36] and a transient hyperpolarization is required for progression through early G1 [36]. Somewhat contradictorily, most highly proliferative cells have depolarized membrane potentials, and it has long been known that depolarization increases proliferation rate [37 –39]. Therefore, it appears that many of the actions of K+ channels on proliferation are independent of membrane potential.
Other cellular processes that could account for the slowing of the cell cycle include volume regulation, pH, and slowing of metabolic and protein synthesis rates. In proliferating cells, volume increases during G1 phase, and inhibition of this process slows proliferation [40,41]. It is also possible that low [K+]E will cause acidification of the cytosol through reduced Na+/K+-ATPase activity. This would occur through increased intracellular Na+, which, in turn, would oppose the export of protons via the Na+/H+ exchanger, lowering intracellular pH [42]. Many cellular processes have an alkaline optimal pH, including protein and DNA synthesis [43 –45]; slowing which would slow the accumulation of cell cycle proteins; and, therefore, proliferation. Consistent with this hypothesis, Na+/H+ exchanger activity is positively linked to proliferation [46]. Ion homeostasis is not independent of membrane potential or Ca2+, and can be altered via ion channels in the cell membrane. Almost all TRP channels are non-selective or weakly Ca2+ selective cation channels; therefore, their activation will enable K+ and Na+ movement in addition to Ca2+. The interlinking of K+, cell volume, pH, membrane potential, and Ca2+ makes isolating the direct consequences of particular mechanisms difficult. This is further complicated by the methods that are available to probe these channels, as all the pharmacological antagonists available have other, nonspecific actions, and channel knock-down via RNAi can cause compensatory changes in channel expression and may lead to the absence of effects related to channel regulation. For these reasons, although TRP channel-mediated Ca2+ signals have been associated with increased proliferation in many cell types, including human and rat NSCs [6,15] and cancer cells [47 –49], their precise roles have yet to be identified.
Adjusting the ionic composition of cell culture media can have wide-ranging effects on cellular processes, and may provide a simple method for optimizing conditions for NPC development [29,50]. The effects on the cell cycle may be of particular interest, as a lengthening of G1 phase and a shortening of S phase accompany neurogenesis in the developing rodent brain [31,32,51]. Taken together, these results suggest a link between external K+ concentration, spontaneous Ca2+ transients, and cell cycle distribution, which are able to influence the fate of stem and progenitor cells.
Footnotes
Acknowledgment
The authors thank Ellen Ewald and Norman Krüger for their excellent technical support.
Author Disclosure Statement
No competing financial interests exist.