
Abstract
Mesenchymal stem cells (MSCs) are attractive candidates for novel cell-therapy applications. However, the in vitro expansion of MSCs typically depends on the presence of fetal bovine serum (FBS) and coating materials derived from animal sources, which may cause contamination in clinical applications. In this study, we investigated whether human umbilical cord extract (UCE) could serve as a serum replacement and whether collagen purified from umbilical cord (UC-collagen) could act as an extracellular matrix (ECM) for the in vitro culture of MSCs derived from human UC (UC-MSCs). A total of 5.61±0.54 mg UCE and 18.41±2.42 mg collagen were extracted, and 1.3±0.2×105 cells were isolated from 1 g of UC, as determined by the expression of typical MSC surface markers. Importantly, the proliferation and stemness of the UC-MSCs cultured with the UCE media were similar to those cultured under FBS conditions on UC-collagen-treated plates for eight passages. Based on these results, we suggest that UCs, which are discarded as medical waste, represent a viable alternative source of xeno-free biomaterials to replace animal-derived serum and ECM materials for the cultivation of various cell types, including UC-MSCs, adipose tissue-derived MSCs, bone marrow-derived MSCs, and fibroblasts. This innovative xeno-free MSCs culture system can overcome many of the problems associated with immunogenicity, and it will further contribute to the enhancement of treatment efficiency.
Introduction
H
Although hMSCs exhibit immunosuppressive capabilities [7 –9] in terms of increasing the efficacy of cell treatment, patient-derived autologous MSCs have been recognized as optimal therapeutic cells. In addition, to ensure the safety of hMSCs without xeno-contamination in vitro, clinical-grade cells should be cultured under xeno-free conditions that exclude animal-derived serum and extracellular matrix (ECM) materials. One source of patient-derived hMSCs is bone marrow (BM) [10]. However, the harvest of BM-derived MSCs (BM-MSCs) involves invasive surgery, which can cause infection, bleeding, and chronic pain. Moreover, the age of the patient can significantly accelerate cell senescence [11], thereby restricting the use of BM-MSCs as source cells for clinical applications.
To overcome the limitations of BM-MSCs, substitute hMSCs have been derived from perinatal and other adult tissues, including peripheral blood, umbilical cord (UC) matrix, placenta, and UC blood [12 –14]. These cells show traditional BM-MSC characteristics such as CD73, CD90, CD105, and HLA-ABC expression and differentiation into various cell types, including osteocytes, chondrocytes, and adipocytes [14 –16]. Thus, these cells could replace BM-MSCs and be useful for therapeutic purposes [17,18]. In general, hMSCs cultured in vitro are maintained and expanded on ECM materials such as gelatin- and collagen-coated plates supplemented with medium containing fetal bovine serum (FBS) derived from animal sources. One critical disadvantage is that the properties of hMSCs vary significantly depending on batch-to-batch variations in FBS production conditions. The use of hMSCs cultured in FBS before clinical application is a serious concern, because there is a risk of transmitting known and unknown xeno-pathogens and xeno-immunogenic substances along with the components of FBS [19,20]. Therefore, xeno-free culture conditions represent a viable alternative to the use of animal-derived products in regenerative medicine settings.
The UC contains two arteries and one vein surrounded by mucoid connective tissue, known as Wharton's jelly [21,22]. During gestation, the human UC grows till it is 30–60 cm in length, weighing approximately 50 g at birth. The UC is physiologically and genetically a part of the fetus, and it supplies the fetus with oxygenated, nutrient-rich blood from the placenta for prenatal development [23]. The UC contains fibroblast-like cells with properties that are similar to hMSCs, and it may represent a rich source of primitive cells [24 –28]. Specifically, Wharton's jelly contains several growth factors, including bFGF, EGF, PDGF-AB, FGF, and IGF-1, and it is rich in ECM components, mainly collagen and glycosaminoglycan [29 –32]. Based on previous reports, we hypothesized that discarded UCs which are considered medical waste represent a good alternative xeno-free source of biomaterials for cell cultures.
In this study, we developed a safe and efficient culture condition for hMSCs that can be applied for clinical applications. First, we isolated MSCs, collagen, and umbilical cord extract (UCE) from human UC matrix. UC-MSCs were then cultured on UC-collagen-coated plates in UCE-supplemented medium to compare the expansion and stemness of these cells with MSCs cultured using standard culture conditions (commercial collagen with FBS supplementation) for approximately eight passages. The results showed that our novel culture system supports the primary characteristics of MSCs in a manner that is comparable to animal source-derived products. In addition, various cell types, including adipose tissue-derived MSCs (ADSCs), BM-MSCs, and fibroblasts, could be cultured using these xeno-free conditions. Furthermore, developing this system into a generalized method could lead to innovations in the cell-therapy industry.
Materials and Methods
Isolation and extraction of UC-MSC, UCE, and UC-collagen from human UCs
This study was performed with the approval of an Institutional Review Board from the CHA General Hospital. All information pertaining to subjects and all human samples were used in compliance with Korean legislation, and all human participants provided informed written consent [33]. Healthy volunteers at the CHA General Hospital donated human UC samples, and the samples were used within 24 h, as described in Fig. 1. To isolate UC-MSCs, Wharton's jelly was sliced into 5-mm explants after removing the umbilical vessels, and the slices were subsequently attached and cultured in minimum essential medium Eagle-alpha modification (α-MEM) (Hyclone) supplemented with 10% FBS (Hyclone, Cat# SH30919.03), 100 μg/mL streptomycin, and 100 IU/mL penicillin (Hyclone) on culture plates. The medium was changed every 3 days, and UC-MSCs cell populations appeared as outgrowths from the UC fragments at day 6. After 15 days, the UC fragments were discarded, and the cells were passaged with TrypLE (Invitrogen, Cat# 12604-054) and expanded until they reached sub-confluence (80%–90%) [34]. We performed a fluorescence activated cell sorting (FACS) analysis on isolated UC-MSCs from six different donor UCs. For UCE extraction, human UCs were dissociated into 1-cm pieces using sterile surgical scissors and stirred in phosphate-buffered saline (PBS) (Hyclone) on a magnetic stirrer for 24 h at 4°C. The stirred suspensions were centrifuged at 16,000g for 15 min at 4°C, and the collected supernatants, termed “UCEs,” were filtered through 0.2-μm filters to remove bacteria and particulate matter before use in cell culture [35]. A Bradford reagent (Bio-Rad Laboratories) was used for the protein assays. Extracts (fresh, frozen, or lyophilized) were then stored at −20°C for future use. UC-collagen was extracted by dissociating human UCs into 1-cm pieces using sterile surgical scissors and washed a minimum of thrice in distilled water. For virus inactivation, the dissociated tissues were transferred to 70% ethanol for 24 h at 4°C [8]. The tissues were washed in distilled water and stirred in 3% H2O2 on a magnetic stirrer for 24 h at 4°C. The tissues were then washed twice in distilled water, homogenized in 0.5 M acetic acid, and transferred into 10% pepsin (Sigma-Aldrich, Cat# 77161) for 24 h at 4°C [36]. For pepsin inactivation and desalting, the tissue suspensions were centrifuged at 15,000g for 30 min at 4°C with NaOH (pH 7.1). Supernatant proteins were precipitated in 2.4 M NaCl for 12 h. The mixture was clarified at 15,000g for 30 min at 4°C, and the supernatant was subsequently desalted and concentrated using an ultrafiltration system [31]. Finally, the collagen was lyophilized, and the manufactured collagen was analyzed using a hydroxyproline method [37].
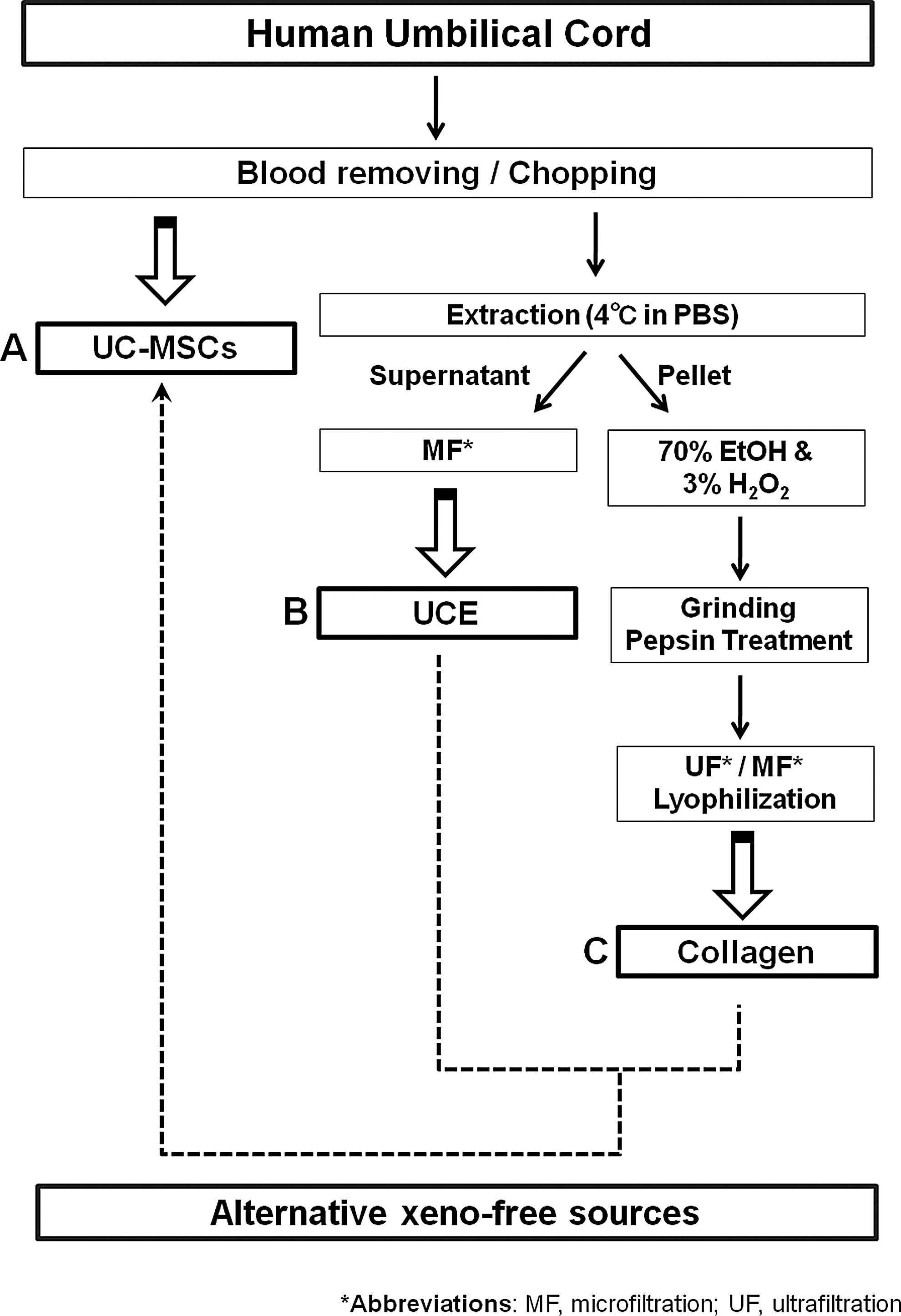
Overall strategy for the development of a xeno-free culture system using human umbilical cord (UC).
Flow cytometry
For flow cytometric analyses, cells were dissociated with trypsin/EDTA (Hyclone) and washed with PBS (Hyclone). Subsequently, 3×105 harvested cells were incubated with 10 μL of the indicated antibody for 20 min on ice, resuspended in 200 μL of PBS, and incubated with 5 ng/mL propidium iodide (Sigma-Aldrich) to detect nonviable cells. The fluorescence intensities of the cells were evaluated using a flow cytometer (FACScalibur; BD Biosciences), and the data were analyzed using WinMDI 2.9 software (J. Trotter. Scripps Institute) [38]. MSC characteristics were identified using fluorescein isothiocyanate-conjugated mouse anti-human CD31, CD45, CD34, CD9, CD105, and HLA-DR antibodies (BD Biosciences) and PE-conjugated mouse anti-human CD29, CD166, CD73, and HLA-ABC antibodies (BD Biosciences).
Analysis of UCE components
Protein profiles of the UCEs were analyzed for 507 growth factors and cytokines using the RayBio® Biotin-label-based human antibody array I (RayBiotech, Inc.), according to the manufacturer's protocols. Briefly, the array membranes were blocked for 1 h at room temperature and then incubated with 1 mL of biotin-labeled UCE for 2 h at room temperature. Samples were washed thrice with the provided buffer, and membranes were incubated with horseradish peroxidase-conjugated streptavidin for 2 h at room temperature [39]. The chemiluminescence signals on the array membrane were detected using an LAS-3000 device (Fujifilm), and the signal intensity of each spot was quantified using Multi Gauge V3.0 software (Fujifilm).
Analysis of UC-collagen purity by SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to the method described by Laemmli [40], using 6% native-polyacrylamide gels and 5% stacking gels at room temperature. After heating at 95°C for 5 min, the samples (20 μg) were loaded into the wells along with molecular-weight markers and run in a gel electrophoresis system (Bio-Rad). The gel was stained for protein with 0.1% Coomassie brilliant blue R-250 and destained in 10% methanol and 10% acetic acid.
Analysis of the cell proliferation in different UC-collagen concentrations
To test the proliferative ability of various cell lines on UC-collagen-coated dishes, UC-MSCs were cultured in α-MEM, and human ADSCs (ATCC; PCS-500-011, passage #6), human BM-MSCs (Stem Cell Technologies; MSC-001F, passage #7), and human fibroblasts (ATCC; PCS-201-012, passage #6) were cultured in Dulbecco's modified Eagle medium containing 10% FBS for optimal proliferation conditions in different concentrations of UC-collagen-coated (1, 5, and 50 μg/mL) plates [9,41,42]. Cell numbers and viability were determined using a hemocytometer and trypan blue (Sigma-Aldrich) staining, respectively.
Optimization of the UCE concentration for cell culture comparable to 10% FBS
Various cell types (UC-MSCs, ADSCs, BM-MSCs, and human fibroblasts) were seeded at 2.5×103 cells/cm2 in 24-well plates and allowed to attach for 24 h. The medium was removed and replaced with 10% FBS medium or medium containing different concentrations of UCE (0.1 mg/mL, 0.3 mg/mL, 0.5 mg/mL, and 1 mg/mL) from three donors; these media were changed every 3 days. Cells were incubated for 2 h in medium with 10% (v/v) WST-1 (Daeillab Service; EZ-3000) to assess cell proliferation. Absorbance at 450 nm was measured using a spectrophotometric plate reader (Epoch; BioTek), and each experiment was performed in triplicate.
Cell proliferation analysis
To measure the doubling time (dT), UC-MSCs were seeded at 2×103 cells/cm2 in six-well plates and were subcultured every 3 days. The cells from passages 1 to 8 were harvested and counted using a hemocytometer and trypan blue (Sigma-Aldrich) staining. The dT was calculated using the following formula: dT=log2×T/(log NH–log NI), where T represents culture time and NH and NI represent the cell harvest number and initial cell number, respectively [43].
Isolation of total RNA
Total RNA was isolated using the easy-spin Tri-reagent (iNtRON Biotechnology). Harvested cells were homogenized by passing the lysate through a 20-gauge needle using a syringe. The samples were then processed according to the manufacturer's protocols (iNtRON Biotechnology). RNAs were eluted with 50 μL of RNase-free water by centrifugation for 1 min at 7,000g. The obtained RNAs had A260/A280 ratios of >1.8, as measured using an ND-1000 spectrophotometer (NanoDrop).
Reverse transcription-polymerase chain reaction analysis
cDNA synthesis was performed with 1 μg of pure total RNA using a reverse transcription master premix (ELPIS), followed by PCR amplification. The amplification was performed under the following conditions: predenaturation at 94°C for 10 min; 30 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, extension at 72°C for 30 s, and a final extension at 72°C for 10 min. PCR products were visualized by electrophoresis on a 1.5% (w/v) agarose gel stained with ethidium bromide. Primer sequences are listed in Table 1. cDNA from human embryonic stem cells was used as a positive control for pluripotent gene expression [44].
LPL, lipoprotein lipase; ALP, alkaline phosphatase; FABP4, fatty acid binding protein 4; KLF-4, Kruppel-like factor 4; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Microarray assays
For each cDNA sample, 750 ng of cDNA labeled with biotin-NTP was hybridized to a WG-6 expression v.2 bead array for 16–18 h at 58°C, according to the manufacturer's instructions (Illumina, Inc.). Array signals were detected using Amersham fluorolink streptavidin-Cy3 (GE Healthcare Bio-Sciences), according to the bead array manual instructions. The arrays were scanned using an Illumina Bead Array Reader confocal scanner, according to the manufacturer's instructions. The quality of the hybridization and overall bead array performance were monitored by visual inspection of both the internal quality controls and the raw scanned data. The raw data were extracted using the software provided by the manufacturer (Illumina Genome Studio v2011.1 [Gene Expression Module v1.9.0]). Array probes that had detection P-values>0.05 in more than 50% of the samples were filtered out. Selected gene signal values were log transformed and normalized using a quantile method. The significance of the expression data was determined according to the fold change. Gene enrichment and functional annotation analysis of the significant probe list was performed using DAVID (
Characterization of UC-MSC differentiation potential
For adipogenic differentiation, UC-MSCs were seeded at 1×104 cells/cm2 in six-well plates and cultured for 14 days using the STEMPRO® Adipogenesis Differentiation Kit (Invitrogen). Adipocytes derived from UC-MSCs were evaluated using RT-PCR analysis of adipocyte-specific genes (Table 1) and identified using oil red O (Sigma) staining to detect the inclusion of lipids in differentiated cells [46]. Osteogenic differentiation was conducted by seeding UC-MSCs at 1×104 cells/cm2 in a six-well plate and culturing with the STEMPRO Osteogenesis Differentiation Kit (Invitrogen) for 14 days. Differentiated cells were characterized by RT-PCR analysis of osteocyte-specific genes (Table 1) and Alizarin Red S Solution (Sigma-Aldrich) staining [47]. The stained samples were observed using an Olympus CKX 41 microscope (Olympus).
Statistical analyses
Quantitative results are shown as the mean±standard deviation for each experiment. Statistical analyses were performed using the t-test for pair-wise comparisons within Sigma Plot 10.0 software (Systat Software). P<0.05 was considered significant.
Results
Characterization of UC-MSC, UCE, and UC-collagen
After isolating MSC, UCE, and collagen from their corresponding biological sources (Fig. 1, as described in the Materials and Methods), their characteristics were analyzed. Using our system, we obtained average yields of 5.6±0.5 mg UCE, 18.4±2.4 mg collagen (Supplementary Fig. S1; Supplementary Data are available online at
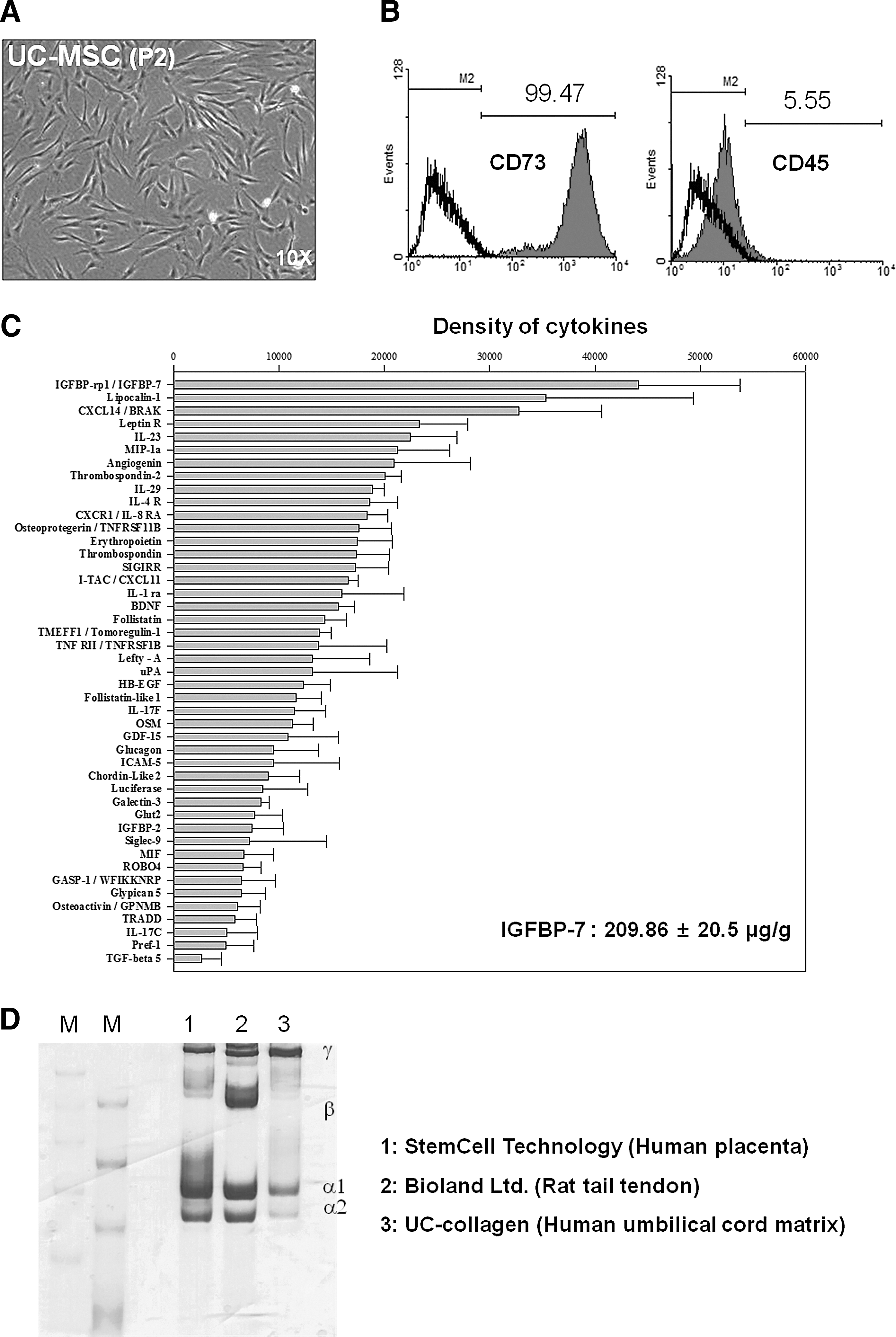
Characterization of UC-Mesenchymal stem cells (MSCs), UCE, and UC-collagen isolated from human UC
Evaluation of UC-collagen and UCE as coating/adhesive agents for MSC culture
To test the viability of cells using UC-collagen as a coating material, we tested the attachment ratios of various cell types, including ADSCs, BM-MSCs, fibroblasts, and UC-MSCs, on various concentrations of UC-collagen (1, 5, and 50 μg/mL). After 2 days, all cell types adhered to the surface without any observable differences. Furthermore, the attachment ratios of all groups were significantly higher on the 50 μg/mL UC-collagen-treated plates compared with the nontreated group (Fig. 3A). In addition, there were no significant differences in the cell attachment rate or cell proliferation on the 50 μg/mL UC-collagen-coated plates compared with commercialized animal source-derived collagen-coated plates (Supplementary Fig. S3). To evaluate the use of UCE as a replacement product for animal-sourced serum, we examined cell proliferation ratios after 8 days on 50 μg/mL UC-collagen-coated plates. Cell numbers increased with increasing UCE concentrations, although the proliferation patterns for the cell types differed (Fig. 3B). Among the tested concentrations, we selected 0.3 mg/mL UCE as the optimal concentration, because this condition showed cell growth curves and morphological changes that were not detectably different from the 10% FBS treatment group for all cell types tested (Fig. 3B, C).

Effects of UC-collagen and UCE on cell culture.
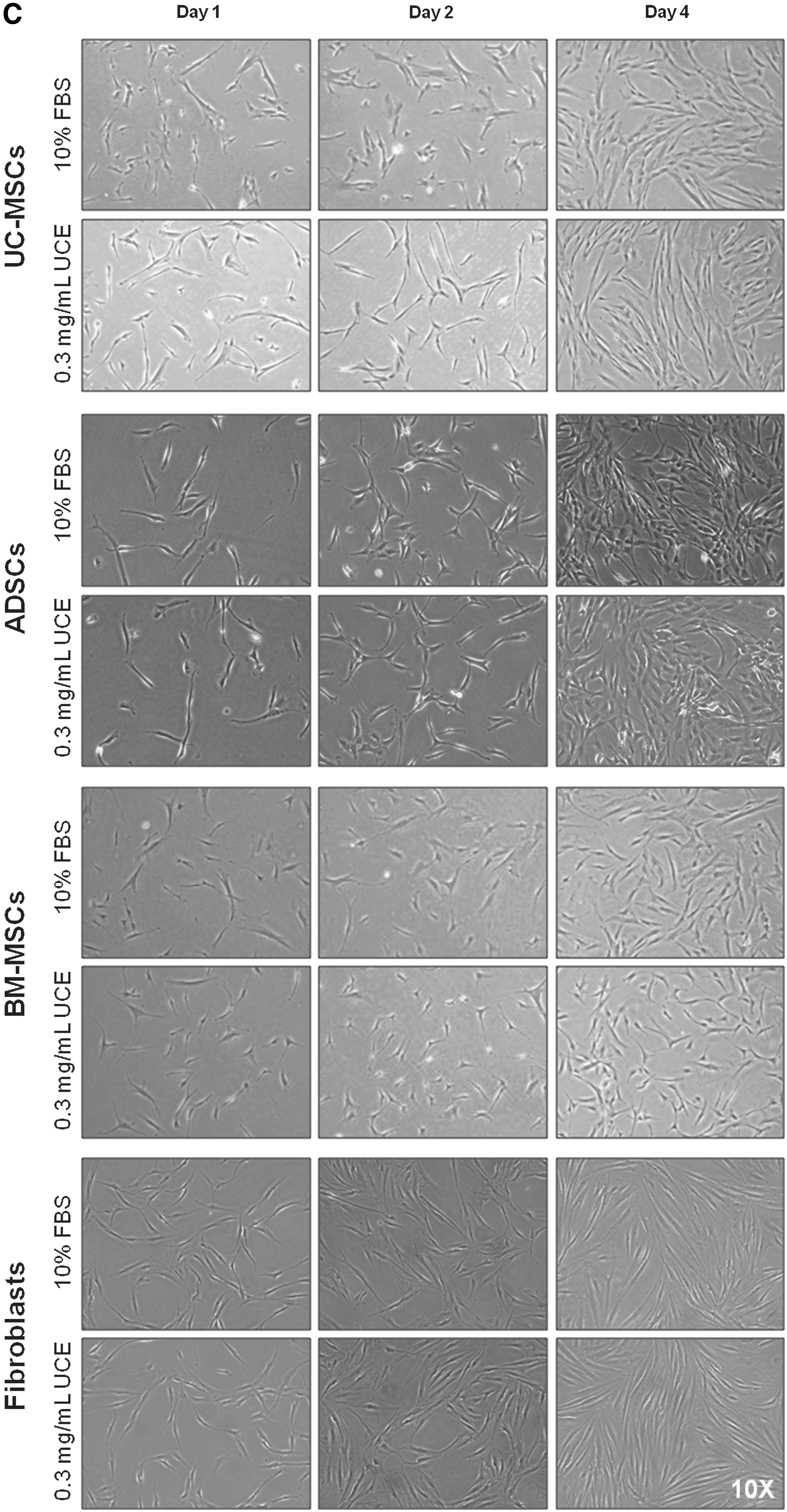
Maintenance of UC-MSCs with UCE on UC-collagen-coated plates
To investigate the stability of UCE for the maintenance of UC-MSCs, we compared the growth properties of UC-MSCs between FBS (10%)- and UCE (0.3 mg/mL)-supplemented medium on 50 μg/mL UC-collagen-treated culture plates for eight passages. We did not detect any morphological changes in the UC-MSCs cultured with UCE medium compared with the FBS-supplemented medium over eight passages (Fig. 4A). UC-MSCs cultured in both FBS- and UCE-supplemented media displayed similar dTs for each passage. On average, UC-MSCs underwent 30.2±0.1 doublings in UCE-supplemented medium and 29.7±1.0 doublings in FBS-supplemented medium, indicating no significant difference between the media over eight passages (Fig. 4B, right panel). To determine the global gene expression pattern for UC-MSCs cultured under the two different media conditions, we performed a microarray analysis of cultured UC-MSCs at passage 6. In general, the gene expression patterns in UC-MSCs cultured in both media were similar (Fig. 4C). These results verified that it is possible to maintain UC-MSCs on UC-collagen-coated plates using UCE-supplemented media, as no changes in morphology, growth rate, or global gene expression were observed compared with UC-MSCs grown in FBS-supplemented medium.
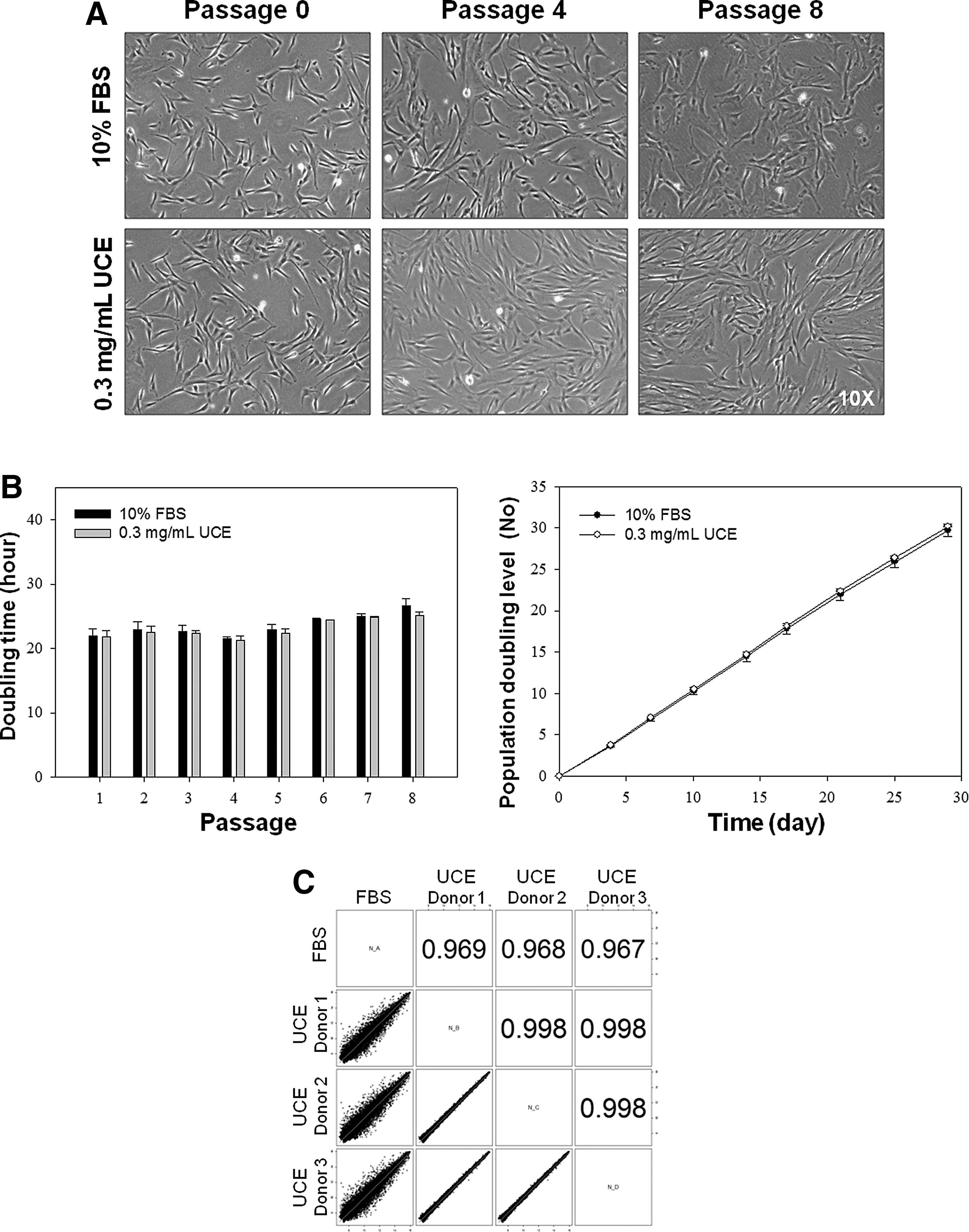
Evaluation of UCE as an alternative serum source for UC-MSC maintenance.
Characterization of UC-MSCs cultured in UCE-supplemented medium on UC-collagen-coated plates
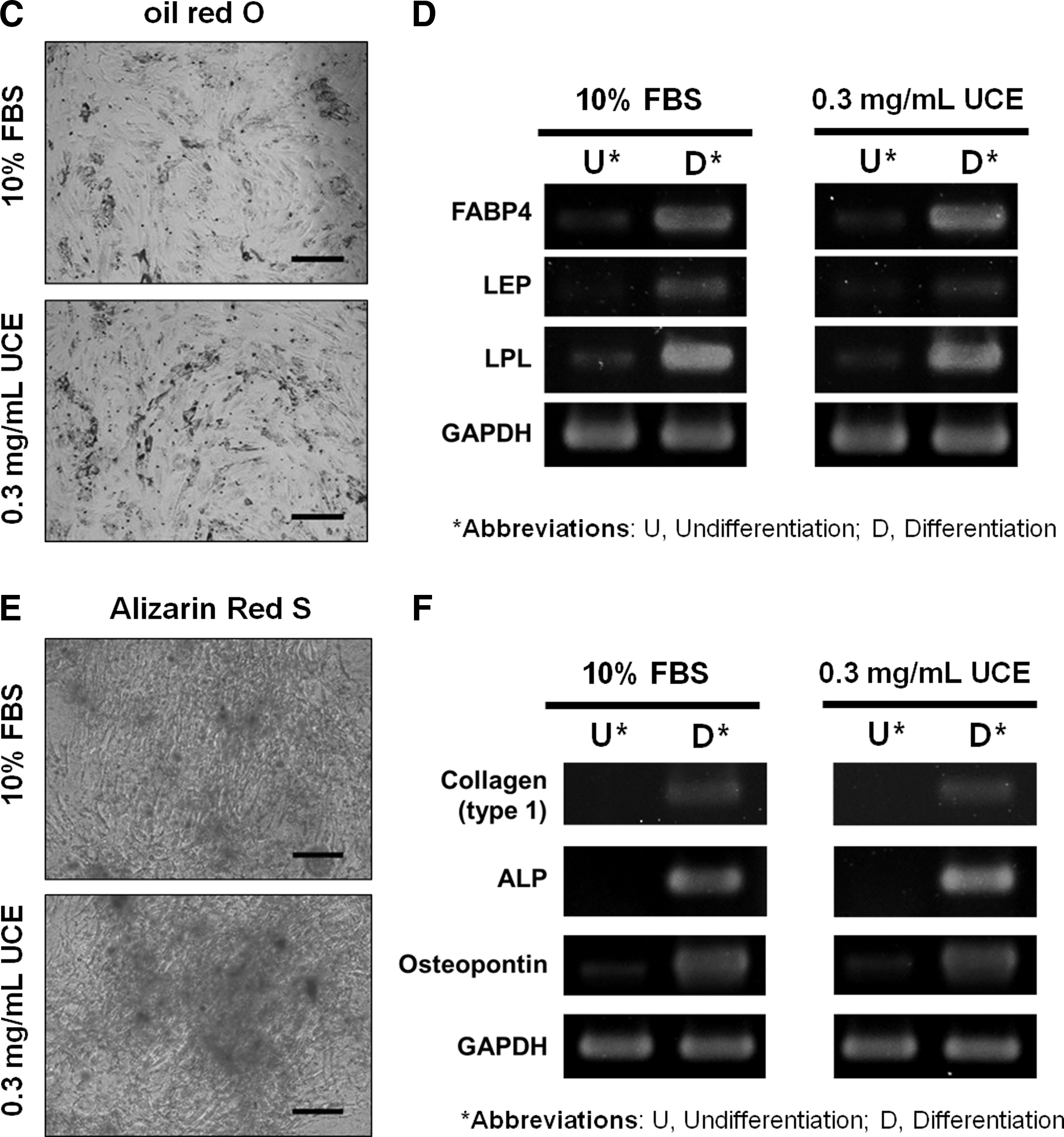
To further evaluate the quality of UCE and UC-collagen, we characterized UC-MSCs maintained using UCE medium on UC-collagen-coated plates. For eight passages, stemness markers, such as Oct-4, Sox 2, and KLF-4, were continuously expressed in UC-MSCs cultured using UCE medium at similar levels to UC-MSCs cultured in FBS medium (Fig. 5A) [48 –50]. Furthermore, a phenotypic analysis of UC-MSC at passage 6 by FACS revealed that UC-MSCs cultured using UCE medium not only showed a higher expression of representative MSC-positive markers, such as CD9, CD29, CD73, CD105, CD166, and HLA-ABC, but also had lower expression of MSC-negative markers, such as CD31, CD34, and CD45, compared with UC-MSCs cultured in FBS (Fig. 5B). These results indicate that UCE could replace animal-derived FBS for UC-MSC culture and maintenance without changing their characteristics in vitro. We also attempted to induce UC-MSC differentiation into adipocytes and osteoblasts to characterize the multi-lineage differentiation potential of MSCs in vitro. Using UC-MSCs grown under two different culture conditions at passage 6, we detected similar differentiation capacities for the formation of adipocytes, as assessed by oil red O staining (Fig. 5C). In both groups, we observed enhanced expression of adipogenesis-related genes, including FABP4, LEP, and LPL, relative to the undifferentiated state (Fig. 5D). In addition, osteoblasts derived from UC-MSCs contained bone-like nodules, as indicated by Alizarin Red S staining (Fig. 5E). Further, RT-PCR demonstrated the presence of the collagen type I, ALP, and osteopontin genes, which were not observed in undifferentiated UC-MSCs (Fig. 5F]. From these multi-lineage differentiation assessments of UC-MSCs, we conclude that the UCE-mediated culture condition is fully capable of preserving the differentiation characteristics of UC-MSCs.
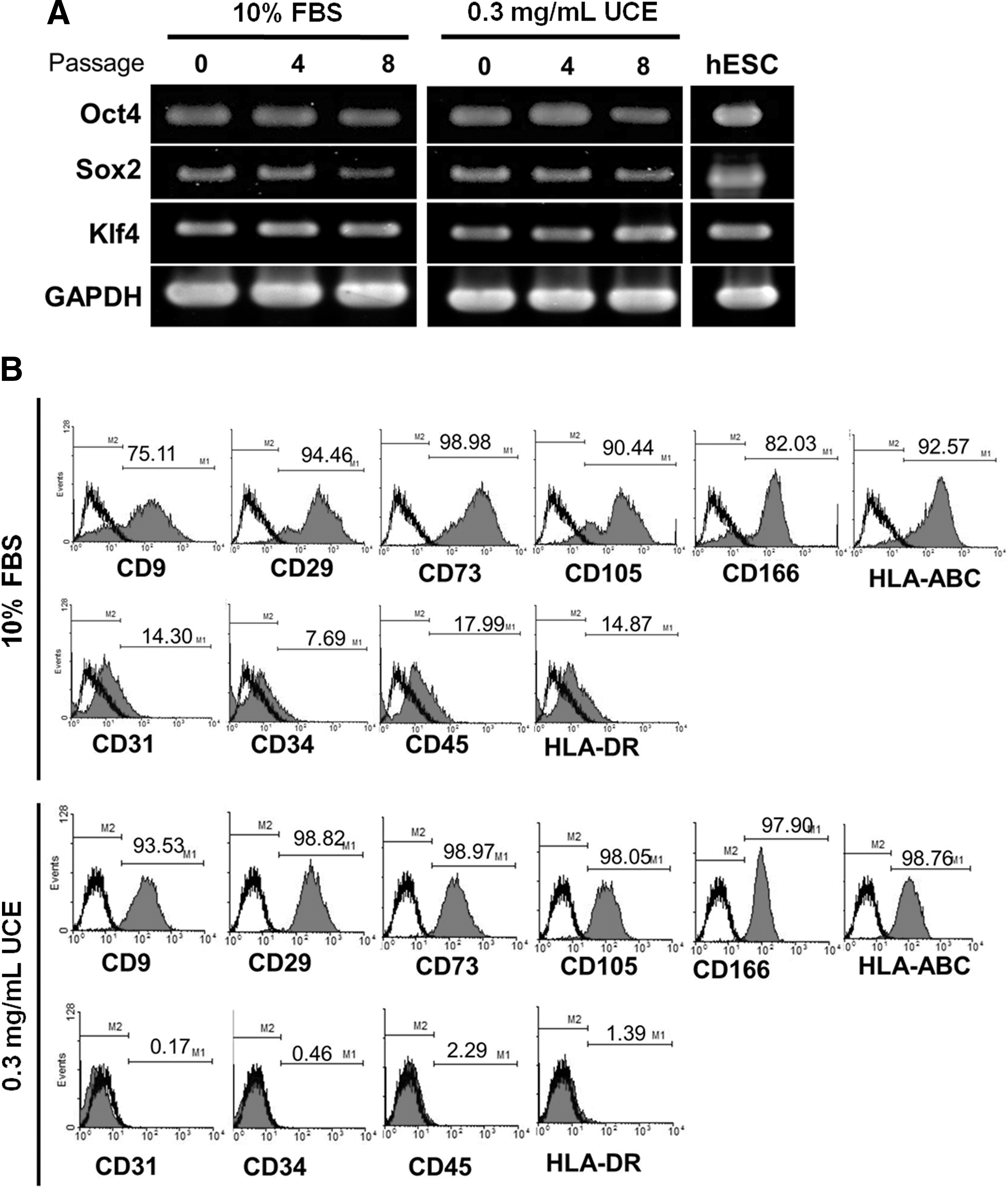
Characterization of UC-MSCs cultured with 0.3 mg/mL UCE on UC-collagen-coated plates.
Effect of sterilized UCE on UC-MSC culture
To further develop UC-collagen and UCE as industrial products for clinical cell culture, sterilization is a prerequisite for eliminating pathogens. In this study, we analyzed the effect of UCE sterilization using typical methods, such as pasteurization and autoclave treatment, on UC-MSC culture compared with filtered and nonsterilized UCE (Fig. 6). SDS-PAGE analysis revealed that unlike the autoclaved UCE, the pasteurized UCE showed similar expression patterns to control UCE (Fig. 6A). While pasteurized UCE reduced UC-MSC proliferation compared with FBS or native-type UCE, the pasteurized product could be used in cell culture as an alternative to sterilized serum, unlike autoclaved UCE (Fig. 6B).
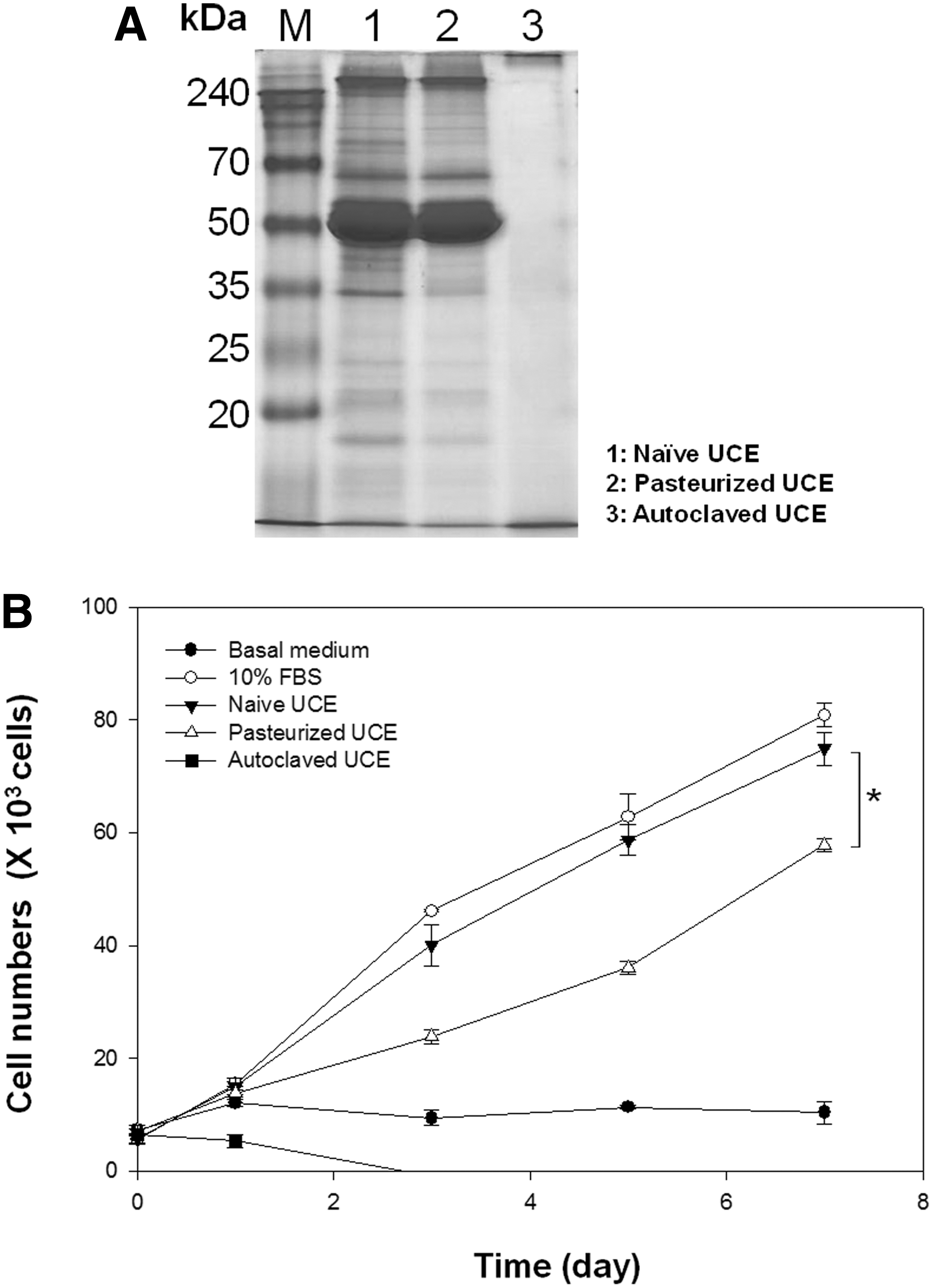
Effect of sterilized UCE on UC-MSC culture.
Discussion
Currently, maximizing the therapeutic effects of MSCs by suppressing immune rejection and safe culture technique are the most important issues for the development of cell therapies in regenerative medicine [51,52]. Interest in hMSCs as a cell therapy resource is increasing due to their capabilities for self-renewal, multi-lineage differentiation, and low major histocompatibility complex class I expression [38,53]. In addition, autologous hMSCs, which can be used to overcome the problems associated with immune rejection, can be obtained by isolation from BM, adipose tissue, UC blood, and the UC matrix [38].
Despite the advantages of hMSCs, optimal conditions for their culture require animal source-derived serum and coating materials, such as collagen and gelatin, which raises concern over immunological reactions caused by these animal-derived materials [54,55]. Spees et al. reported that proteins in FBS, the most common source of serum, are easily absorbed by cells, making them difficult to remove by washing [56]. Conventional culture methods that use animal serum for cell therapy have several risks, including immune inflammatory rejection and pathogen transmission due to contamination with animal-derived proteins. These proteins cannot be effectively eliminated through processing after culture for clinical use [57]. Therefore, this issue should be addressed before providing therapeutic treatments.
To overcome this problem, Bocelli-Tyndall et al. reported that FGF2 and PDGF, but not platelet lysate, induce proliferation-dependent, functional class II major histocompatibility complex antigens in hMSCs [58]. However, the analysis of individual growth factors for hMSC culture is complicated and poorly understood. Shahdadfar et al. reported that hMSCs isolated and expanded in 10% human autologous serum were equally effective in stimulating growth compared with cells grown in 10% FBS, indicating that human autologous serum is a safe and effective FBS alternative for therapy [59]. However, human serum is in short supply; thus, it is not widely used for cell culture in vitro.
In general, to support various aspects of cell biology, plate surfaces are routinely coated with animal-derived ECM materials such as collagen and gelatin [60,61]. Human-derived ECM materials can effectively replace the use of animal-derived serum, but a high cost and lack of availability limit their use for clinical applications. In this study, we used discarded human UCs to isolate hMSCs that exhibited the general characteristics MSCs (Fig. 2). To provide a xeno-free ECM for in vitro maintenance of UC-MSCs, we purified collagen derived from the Wharton's jelly of the UC, and we demonstrated that 50 μg/mL collagen coating was sufficient to support the proliferation of various cell types in a manner comparable to conventional coating materials that consist of animal derived-collagen (Fig. 3). In addition, we provide evidence that various cytokines and growth factors in UCE supported UC-MSCs, suggesting that UCE is an alternative serum to animal-derived FBS. As shown in Fig. 3, various cell types displaced similar proliferation rates in 0.3 mg/mL UCE compared with a conventional culture condition with 10% FBS. In addition, several important factors were expressed at high levels in UCE, including IGFBP-7 and thrombospondin-2. The IGFBP family (eg, Igfbp2, 3, 5, 6, and 7), which binds and sequesters insulin growth factor, is a potent stimulant of epidermal proliferation [62,63]. Thrombospondin is necessary for cell adhesion and cell growth [64], and human hematopoietic progenitor cells of all three lineages (erythrocytes, megakaryocytes, and granulocytes) use thrombospondin as an attachment protein [65]. In addition, UCE could be stored for 8 weeks after lyophilization without losing effectiveness (Supplementary Fig. S4).
In recent years, several studies have attempted to replace FBS with human-derived medium additives, such as human serum albumin, human platelet lysate, and UC blood serum [66,67]. Although human growth supplements have a major advantage because they avoid any risk of secondary effects, the possibility of contamination by these blood-derived products due to harmful substances, undiscovered pathogens, or donor effects pose other major risk factors. Therefore, various strategies have been developed to address these shortcomings. In general, filtration through 0.2-μm filters may eliminate certain bacteria and particulate matter, but they are usually unable to remove pathogens smaller than 0.1 μm.
In summary, our newly developed culture system is strikingly similar to animal source-derived products for serum and ECM coating. When UC-MSCs were cultured using UCE on UC-collagen-coated plates for eight passages, the proliferation and stemness capacities of UC-MSCs were maintained. Interestingly, the expression of representative MSC surface markers, such as CD9, CD29, CD105, CD166, and HLA-ABC, was higher in UCE culture conditions compared with FBS culture conditions (Fig. 5B). Furthermore, to prove that UCE and UC-collagen are xeno free and could be used for clinical applications, we derived UC-MSCs using 0.3 mg/mL UCE media without FBS on UC-collagen-coated plates. Under these conditions, we observed UC-MSCs of a typical spindle-shaped fibroblastic morphology (Supplementary Fig. S5A) that weakly expressed CD45 and strongly expressed CD73 (Supplementary Fig. S5B). In addition, we compared the isolated cell populations from 1 g UC in 10% FBS and 0.3 mg/mL UCE media conditions. Interestingly, the UCE medium condition produced more UC-MSC populations than the FBS medium condition (Supplementary Fig. S5C). These results indicate that UCE may be an alternative to FBS for MSC culture. In the past, UCs were considered medical waste but a few companies in the world (
Our development of UCE and UC-collagen as resources for MSC culture overcomes many of the problems associated with the clinical application of cell therapy. In this study, we extracted 5.61±0.54 mg UCE and 18.41±2.42 mg collagen from 1 g of UC (Supplementary Fig. S1). The average weight of a human UC is ∼40 g; thus, 800 mg UC-collagen and 240 mg UCE could potentially be obtained per UC, enabling the production of 16 L of UC-collagen coating solution and 800 mL of pasteurized UCE medium, respectively. Therefore, to develop a scalable mass culture system for the xeno-free MSCs culture, a large quantity of UCE and collagen can be extracted from a single UC, which could be used to expand MSCs for human clinical applications (up to 109 cells).
In conclusion, we developed a xeno-free culture system that uses alternative resources from discarded human UCs. We established that UCE and UC collagen can serve as replacements for serum and ECM, respectively, for the in vitro culture of UC-MSCs. In addition, we investigated the synergistic effects of UC-MSCs with UCE medium on UC-collagen-coated plates for the development of a xeno-free culture system. Based on this study, we suggest that discarded UCs represent a novel alternative xeno-free source of biomaterials to replace animal-derived serum and ECM materials for cell culture. If this new system is generalized through further development, it could lead to innovation in the cell-therapy industry.
Footnotes
Acknowledgments
This research was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (project no. A121965). In addition, this research was supported by the Space Core Technology Development Program (no. 2011-0030754), funded by the Ministry of Education Science and Technology (MEST).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.