
Abstract
Recent advances in human embryonic and induced pluripotent stem cell-based therapies in animal models of hepatic failure have led to an increased appreciation of the need to translate the proof-of-principle concepts into more practical and feasible protocols for scale up and manufacturing of functional hepatocytes. In this study, we describe a scalable stirred-suspension bioreactor culture of functional hepatocyte-like cells (HLCs) from the human pluripotent stem cells (hPSCs). To promote the initial differentiation of hPSCs in a carrier-free suspension stirred bioreactor into definitive endoderm, we used rapamycin for “priming” phase and activin A for induction. The cells were further differentiated into HLCs in the same system. HLCs were characterized and then purified based on their physiological function, the uptake of DiI-acetylated low-density lipoprotein (LDL) by flow cytometry without genetic manipulation or antibody labeling. The sorted cells were transplanted into the spleens of mice with acute liver injury from carbon tetrachloride. The differentiated HLCs had multiple features of primary hepatocytes, for example, the expression patterns of liver-specific marker genes, albumin secretion, urea production, collagen synthesis, indocyanin green and LDL uptake, glycogen storage, and inducible cytochrome P450 activity. They increased the survival rate, engrafted successfully into the liver, and continued to present hepatic function (i.e., albumin secretion after implantation). This amenable scaling up and outlined enrichment strategy provides a new platform for generating functional HLCs. This integrated approach may facilitate biomedical applications of the hPSC-derived hepatocytes.
Introduction
H
The successful generation of functional hepatocytes from human pluripotent stem cells (hPSCs) [embryonic (hESCs) and induced pluripotent stem cells (hiPSCs)] has raised the hopes for treating poor prognostic liver disorders due to their vast potential for proliferation and differentiation [5 –17]. To develop a favorable cell-based therapy for clinical application, it is necessary to have consistent and mass generation of functional and safe hepatocytes. Although published differentiation protocols for producing hPSC-derived hepatic cells have shown the efficiency of these cells in the recovery of injured livers in experimental models, they have only reported adherent cell culture conditions for small-scale production of the desired cells.
To integrate expansion and differentiation into a continuous procedure, we explored the maintenance and hepatic differentiation of hPSCs in a single scalable stirred bioreactor. To determine whether hiPSC-derived hepatic cells retained their functionality after engraftment, the cells were sorted based on DiI-acetylated low-density lipoprotein (LDL) uptake and were transplanted into the spleens of mice suffering acute liver damage induced by carbon tetrachloride (CCl4).
Materials and Methods
Suspension expansion of hPSCs in stirred flask
Adherent colonies of hPSC lines (hiPSC, hiPSC1[18] and hESC, Royan H6[19]) were cultured on Matrigel (1:30; Sigma-Aldrich, E1270) under feeder-free culture conditions in Dulbecco's modified Eagle's medium (DMEM/F12; Gibco, 21331-020) that was supplemented with 20% Knockout Serum Replacement (KOSR; Gibco, 10828-028), 0.1 mM β-mercaptoethanol (Sigma-Aldrich; M7522), 1% penicillin and streptomycin (Gibco; 15070-063), 1 mM nonessential amino acids (Gibco; 11140-035), 2 mM L-glutamine (Gibco; 25030-024), 1% insulin–transferrin–selenium (ITS; Gibco, 41400-045), and 100 ng/mL bFGF (Royan Institute). We treated adherent colonies with 10 mM ROCK inhibitor Y-27632 (Sigma-Aldrich; Y0503) for 1 h to change the culture condition from two-dimensional (2D) to three-dimensional (3D). Next, colonies were washed with Ca2+ and Mg2+-free phosphate-buffered saline (PBS; Gibco, 21600-051) for 1 min, then incubated with 0.05% trypsin plus 0.53 mM ethylenediamine tetraacetic acid (EDTA; Gibco, 25300-054) at 37°C for 4–5 min. Stem cell colonies were harvested by a cell scraper and adequately pipetted for dissociation into single cells. We used a hemocytometer to perform cell counts and viability, the Trypan blue exclusion method was used. A total of 1.5-2×105 live cells/mL were then transferred to low adherent bacterial plates (60 mm; Griner, 628102) with 5 mL of conditioned medium (CM) from human foreskin fibroblasts (HFFs) supplemented with 100 ng/mL bFGF. The CM was obtained of hESC medium after 24 h incubation with inactivated HFFs by mitomycin C (Sigma-Aldrich; M0503). The plates were incubated under standard conditions (37°C, 5% CO2, and saturated humidity); after 48 h, single cells were aggregated and loosely attached to the plate surface. At this time, the medium of each plate was refreshed; early aggregates were detached from the plate and suspended in medium. The spheroids were passaged regularly each 7 days. After a few subcultures, 107 cells were inoculated into 50 mL CM in a siliconized (Sigmacote; Sigma-Aldrich, SL2) stirred flask (Cellspin; Integra Biosciences).
Suspension differentiation of hPSCs into definitive endoderm
This was a two-step suspension differentiation protocol of hPSCs into definitive endoderm (DE): (i) priming with 100 nM rapamycin (Rapa; Calbiochem, 553210) for 24 h and (ii) induction with 50 ng/mL activin A (R & D Systems, Inc.; 338-AC) in bacterial dishes for 4 days under static conditions. Before priming, the medium was replaced by induction medium composed of DMEM/F12 supplemented with 0.1% KOSR, 1% vitamin A-free B27 (Gibco; 12587-010), 2 mM L-glutamine, 1 mM nonessential amino acids, and 0.1 mM β-mercaptoethanol. The ITS concentration was 0% for the first day, 0.5% for the second day, and 1.5% for the next 2 days after priming.
For this step of the study we used two control groups containing 100 ng/mL activin A (A100) for 4 days: (i) adherent condition; and (ii) suspension condition, which included suspended spheroids. The test group (Rapa-A50) was treated with Rapa for 1 day and 50 ng/mL activin A for 4 days.
To establish a practical protocol for DE formation in the dynamic phase, we considered different refreshment strategies to renew the induction medium during the static phase. In the single refreshment strategy, induction medium supplemented with 50 ng/mL activin A was entirely renewed after 24 h. For double refreshment (DR) half of the medium was refreshed every 12 h and for triple refreshment, one third of the induction medium was refreshed every 8 h.
Harvested spheroids in the DE phase were evaluated by immunofluorescent assay and flow cytometry analysis for DE-specific protein markers. Samples were collected from three independent biological repeats.
Differentiation of hPSCs into hepatocyte-like cells in stirred flask
In the dynamic phase, at 4 to 5 days after cell seeding in the stirred flask, we began KOSR tapering; after 24 h, spheroids were transferred into induction medium and primed with Rapa for 1 day. Then, for 4 days, 50% of the medium in the stirred flask was refreshed twice a day, while 50 ng/mL of activin A was added to the induction medium. The obtained spheroids were cultured for 7 days in the same induction medium that contained 10% KOSR supplemented with 10 ng/mL hepatocyte growth factor (HGF; R&D Systems, 294-HG) and 10 ng/mL fibroblast growth factor 4 (FGF4; R&D Systems, 235-F4).
Then, spheroids were transferred into Hepatocyte Basal Medium (HBM; Lonza, CC-3199) and DMEM/F12 that was mixed at a 1:1 ratio and contained 10 ng/mL oncostatin M (OSM; R&D Systems, 295OM), 0.1 μM dexamethasone (Dex; Sigma-Aldrich, D-2915), 10% KOSR, 1 mM nonessential amino acids, and L-glutamine. Spheroids were cultured for 10 days. In this stage, the medium was supplemented with HCM™ SingleQuots® (Lonza; CC-4189).
In the hepatic commitment period, we observed two different appearances in the induced spheroids, cystic that had a slow sedimentation rate and dense, with a fast sedimentation rate.
Quantitative reverse transcriptase–polymerase chain reaction
We performed quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR) for lineage-specific genes to evaluate hepatic differentiation of the cells at the transcriptional level. First, total RNA was extracted by the RNeasy Mini Kit (Qiagen; 74104) from dense spheroids. Then cDNA was produced by the RevertAid First Strand cDNA Synthesis Kit (Fermentas; K1632) based on the manufacturer's instructions. Duplicate qRT-PCR reactions were performed with the SYBR Green Master Mix (Takara Bio, Inc., SYBR® Premix Ex Taq™ II RR081Q) by the real-time PCR system (Corbett Life Science; Rotor-Gene 6000 instrument) and analyzed with Rotor-Gene 6000 analysis software (Corbett Life Science; version 1.7). The samples were collected from three independent biological replicates. The expression levels of desired genes were normalized to GAPDH as a reference gene and then calculated relative to corresponding stem cells.
Analysis was performed by the comparative CT Method (2
Transmission electron microscopy
We used transmission electron microscopy (TEM) to evaluate the hepatocyte-like cells (HLCs) ultrastructure in the dense spheroids. First, day 21 harvested spheroids were washed twice with PBS, then prefixed by 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4) for 2 h, after which they were postfixed with 1% osmium tetroxide for 1.5 h. Following dehydration with an acetone series, the spheroids were embedded in epoxy resin (TAAB). Then, 60 to 70 nm sections (silver-grade) were prepared by ultra-microtome (Leica Ultra Cut UC6), double-stained with 5% uranyl acetate and 0.5% lead citrate, and visualized with a Zeiss EM 900 TEM.
Uptake of LDL
We incubated the dense spheroids for 4 h with acetylated LDL labeled with 1, 1′-dioctadecyl-10-3, 3, 30, 30-tetramethyl-indo-carbocyanine perchlorate (DiI-Ac-LDL; Biomedical Technologies Inc., 9202K08) to evaluate cellular functionality for LDL uptake. The test was performed according to the manufacturer's instructions after which cells were visualized using a fluorescent microscope (IX71; Olympus).
Flow cytometry analysis
Spheroids were enzymatically dissociated into single cells as previously described and then fixed in 4% paraformaldehyde (Sigma-Aldrich; P6148) overnight at 4°C. These cells were permeablized in 0.1% Triton X-100 (10 min, 25°C) and blocked in 10% secondary antibody host serum in 0.5% BSA (1 h, 37°C), then incubated overnight with primary antibodies at 4°C. Supplementary Table S2 shows the antibody list. Primary and secondary antibodies were the same as those used for immunofluorescence staining. We used a BD-FACS Calibur Flow Cytometer (FACS Calibur) for analyses. For each marker there were three independent biological experiments performed. Acquired data were analyzed with CellQuest™ software.
Immunofluorescence staining
To evaluate the presence of late hepatic protein markers we washed the dense spheroids twice with PBS, and then fixed them in 4% paraformaldehyde (overnight, 4°C). Spheroids were then embedded in agar gel, followed by embedding in paraffin blocks. We used a microtome (Microm™, HM325) to cut the paraffin blocks into 6 μm sections. Sections were transferred to glass slides and slides were deparaffinized by xylene prior to hydration. Next, sections were treated by antigen retrieval (Dako). Acquired sections were permeabilized with 0.1% Triton X-100 for 10 min and blocked in 10% goat serum (SAFC Bioscience, 12306 C) in PBS for 1 h at 37°C. Slides were incubated overnight with primary antibodies at 4°C. Following incubation, they were washed and incubated with FITC-conjugated secondary antibodies for 1 h at 37°C. The nuclei were counterstained with DAPI (Sigma-Alrich; D8417), after which slides were analyzed with a fluorescent microscope (BX51; Olympus).
Indocyanin green uptake and release
Dense spheroids at day 21 were incubated with indocyanin green (ICG, CardioGreen; Sigma-Aldrich, 12633) in HCM for 1 h in an incubator under standard conditions. ICG uptake was visualized with light microscopy (IX71; Olympus).
Periodic acid-Schiff staining for glycogen storage
Glycogen storage of HLCs in dense spheroids was evaluated by Periodic acid-Schiff (PAS) staining at day 21. The hydrated 6 μm spheroid sections were oxidized in 1% periodic acid for 5 min, and then rinsed in dH2O. Then sections were treated with Schiff's reagent for 15 min following color development in dH2O for 5–10 min. Finally, slides were assessed by light microscope (BX51; Olympus).
Masson trichrome staining
The acquired sections were stained by Masson trichrome (MT) staining according to standard protocols. The relevant photos were taken by light microscope (BX51; Olympus).
Cytochrome P450 activity
The pentoxyresorufin o-dealkylase (PROD) test was performed to evaluate P450 cytochrome activity in HLCs. Pentoxyresorufin is a nonfluorescent substrate, which is o-dealkylated by the cytochrome P450 system and converted into a red fluorescent substance, resorufin. To evaluate the inducibility of CYP3A4, day-21 dense spheroids were incubated with 25 μM rifampicin for 3 days in HCM in bacterial dishes [20]. Then, spheroids were washed and transferred into medium that contained 5 μM 7-pentoxyresorufin (Sigma-Aldrich; P9049) and 80 μM dicumarol (Sigma-Aldrich; M1390) in Hank's balanced salt solution, after which plates were incubated under standard conditions in an incubator for 30 min. The supernatant of the spheroids was collected for the fluorescent intensity test before and after induction (BioTek Synergy™ 4). We used fluorescent microscopy (IX71; Olympus) to detect red areas that indicated the presence of resorufin inside the spheroids. The negative control in this assay was hPSCs (data not shown).
Albumin and urea production
We collected the dense spheroid conditioned media after 48 h on days 15, 17, 19, and 21 of the maturation phase. Media was stored at −20°C for future assay.
Samples were assayed for albumin (ALB) secretion using the hALB ELISA Kit (Bethyl, E80-129); for urea production, we used the Colorimetric Assay Kit (Pars Azmun) according to the manufacturer's instructions. The ELISA test results were analyzed by a Stat Fax® microplate reader (Awareness Technology, Inc.) and the resultant values normalized to 106 cells. Samples were collected from three independent biological experiments.
Cell sorting and preparation
Dense spheroids were sorted at day 21 from cystic ones based on differences in the sedimentation rates. Collected dense spheroids were incubated with DiI-AC-LDL, then dissociated into single cells as previously described and sorted according to LDL uptake by a cell sorter instrument (BD Bioscience, BD FACS Aria™ II) and labeled by a PKH67 Fluorescent Cell Linker Kit (Sigma-Aldrich; PKH67GL) according to the manufacturer's instructions, then suspended in PBS. Harvested cells were counted and injected into the spleens of CCl4-induced mice. The transplanted animals were immunosuppressed by daily cyclosporine 20 mg/kg/d (Novartis Pharmaceuticals) 4 days before cell transplantation and continued for 2 weeks following transplantation.
Experimental animal model
In this study there were four animal groups: control (Cntl), fibroblast (Fib), vehicle (Vhcl), and HLC transplanted (Tx). Acute liver failure (ALF) was induced in 10-week-old male NMRI mice by intraperitoneal injections of CCl4 (0.6 mL/kg). The HLC transplanted group received intrasplenic injections of 2×106 cells 24 h after induction of ALF. The fibroblast transplanted (2×106 cells) and vehicle groups received intrasplenic injections of fibroblasts and medium accordingly after the CCl4 injury, whereas the control group only received CCl4. After cell transplantation, we monitored the survival rate in all groups and collected corresponding blood sera samples. The levels of alanine transaminase (ALT) and aspartate transaminase (AST) were evaluated in mice sera on different days by commercially available kits (Biorex, BX 0203A & 0213A). At day 14 after cell transplantation, while under deep anesthesia (ketamine/xylazine) the animals were sacrificed after which their liver and blood sera were collected. Liver samples were taken for histopathologic analyses to evaluate the engrafted cell's homing abilities. Liver samples were fixed in 10% formalin for 48 h and after processing they were embedded in paraffin blocks. For histological analyses, 6 μm sections were cut. The resultant sections were mounted on glass slides followed by deparaffinization and hydration steps. The nuclei were counterstained with DAPI (Sigma D8417). The slides were visualized by a fluorescent microscope (BX51; Olympus) and stochastically selected fields of sections were taken from each animal, by using a magnification 10× objective lens. Quantification of cell homing was estimated by the percentage of PKH+ cells compared to the total numbers of cells in the fields. We performed immunofluorescent assays to evaluate the status of hALB+ cells. The percentages of positive cells (ALB+ and PKH+) compared to the total cells were calculated. We used the hALB ELISA Kit (Bethyl; E80-129) to determine the presence of hALB in mice sera.
Statistical analysis
The data are presented as mean±standard deviation (SD). One-way repeated measures analysis of variance (One-way ANOVA) followed by the Tukey post hoc test multiple group comparison was used to analyze group differences of the subsequent data. Differences between groups were considered statistically significant at P<0.05. Data analysis was performed by SPSS 16.0 software for Windows.
Results
Carrier-free expansion of hPSCs in stirred bioreactor
In the current study, we used human foreskin fibroblast-derived conditioned medium (HFF-CM) for long-term maintenance of hPSCs in a microcarrier-free suspension culture as previously described [21] (Fig. 1A). Briefly, we passaged hiPSC1 cell line from adherent feeder-free Matrigel cultures by dissociation into single cells, after which the cells were inoculated into a subsequent static suspension cultures in low adherent bacterial plates at a cell density of 1.5-1.8×105 cells/mL in the presence of 10 mM ROCK inhibitor. The culture medium was DMEM/F12 that had been conditioned on HFF for 1 day and supplemented with bFGF (100 ng/mL) and ROCK inhibitor. After 1 week, suspended spheroids of hiPSCs were subcultured 1:3 and transferred into new plates at the same cell concentration and medium volume. Cell aggregates were treated with ROCK inhibitor 1 h prior to enzymatic dissociation by Accumax™ or Accutase™. Then, 1×107 of the single cells from static suspension cultures were conducted in the siliconized vessel to 50 mL medium of a stirred bioreactor. The cell expansion rate was 4-5-folds for hiPSCs [21].
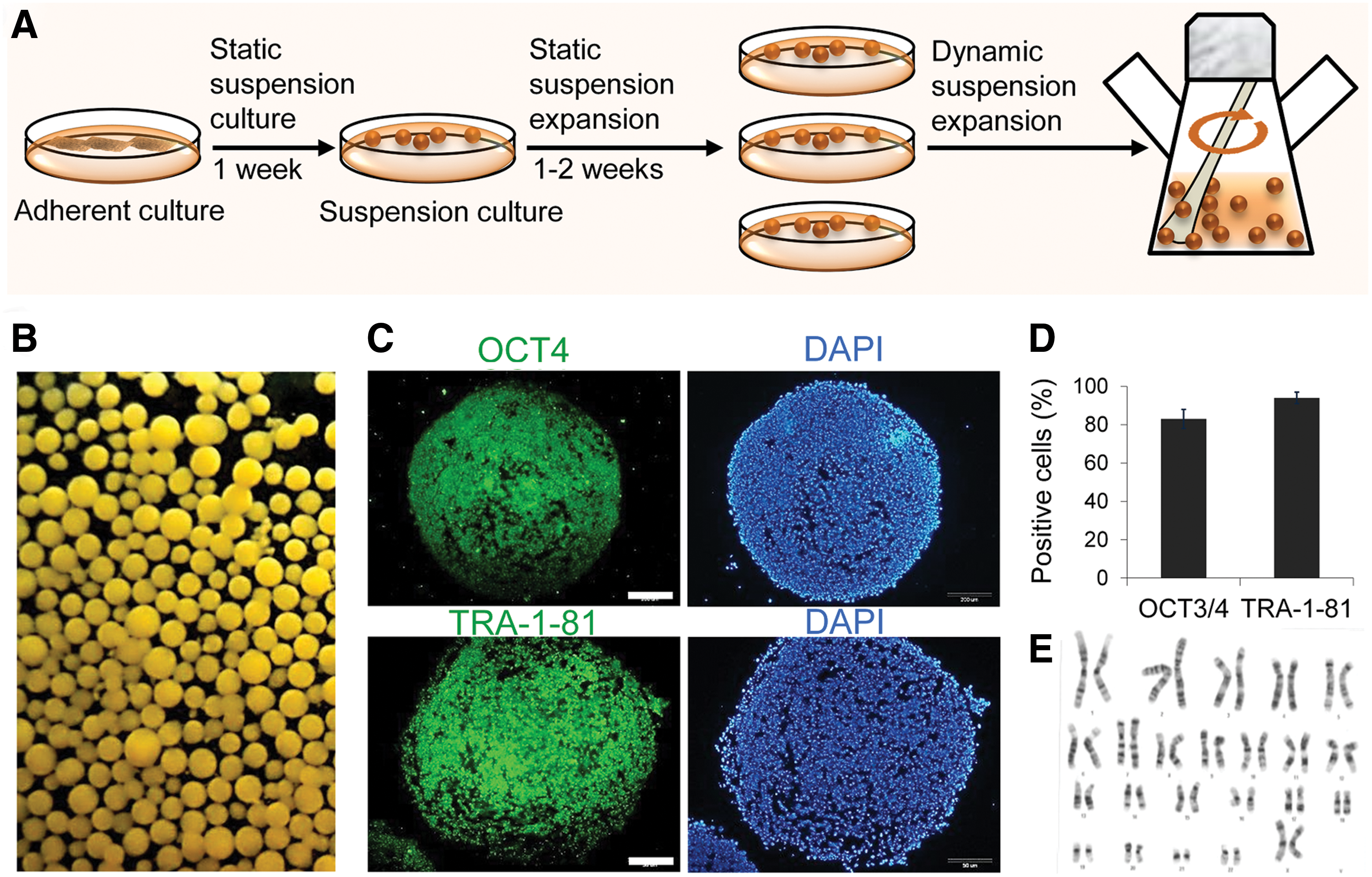
Expansion and characterization of human induced pluripotent stem cells (hiPSCs) in a dynamic suspension culture.
We characterized these cells in suspension condition for self-renewal and pluripotency by immunofluorescent labeling and flow cytometry as previously described [21,22]. The suspension expanded cells expressed the pluripotency markers OCT4 and TRA-1-81 (Fig. 1C). As seen in Fig. 1D, flow cytometry analysis showed that most cells were positive for the nuclear marker OCT4 (85%±12%) and surface marker TRA-1-81 (92%±5%). Moreover, the expression of OCT4 and NANOG was similar in both cell culture condition (Supplementary Fig. S1A). Karyotype analysis following suspension culture demonstrated a normal karyotype (Fig. 1E). The suspension expanded cells had potential of embryoid body formation and differentiated spontaneously (Supplementary Fig. S1B). Moreover, we showed previously the cultivation of pluripotent hESCs and hiPSCs in suspension resulted in successful maintenance of the aggregates in an undifferentiated state and pluripotency. Moreover, transcriptome analysis of the aggregates showed very similar expression profiles when compared with the adherent culture [21,22].
These data indicated that the pluripotent cell lines could be successfully expanded in the HFF-CM dynamic suspension culture protocol while maintaining their self-renewal and pluripotency.
Suspension differentiation of hPSCs into DE
To assess the efficiency of our DE induction protocol in suspension culture, we used qRT-PCR to analyze the expression of two endoderm-specific markers [SOX17 and FOXA2 (also known as HNF3b)], [23] a visceral endoderm marker (SOX7), and a mesoderm marker (BRA). To induce direct differentiation of hPSCs into DE, we replaced CM with non-CM DMEM/F12 supplemented with vitamin A-free B27 (1%), 0.1% KOSR, and activin A (100 ng/mL) in static suspension bacterial dish cultures (Fig. 2A). We used 4-day spheres after cell inoculation for endoderm induction. The average aggregate diameter was 130±40 μm with 2000±500 cells. This size of spheroids is ideal for induction toward endoderm fate [24].
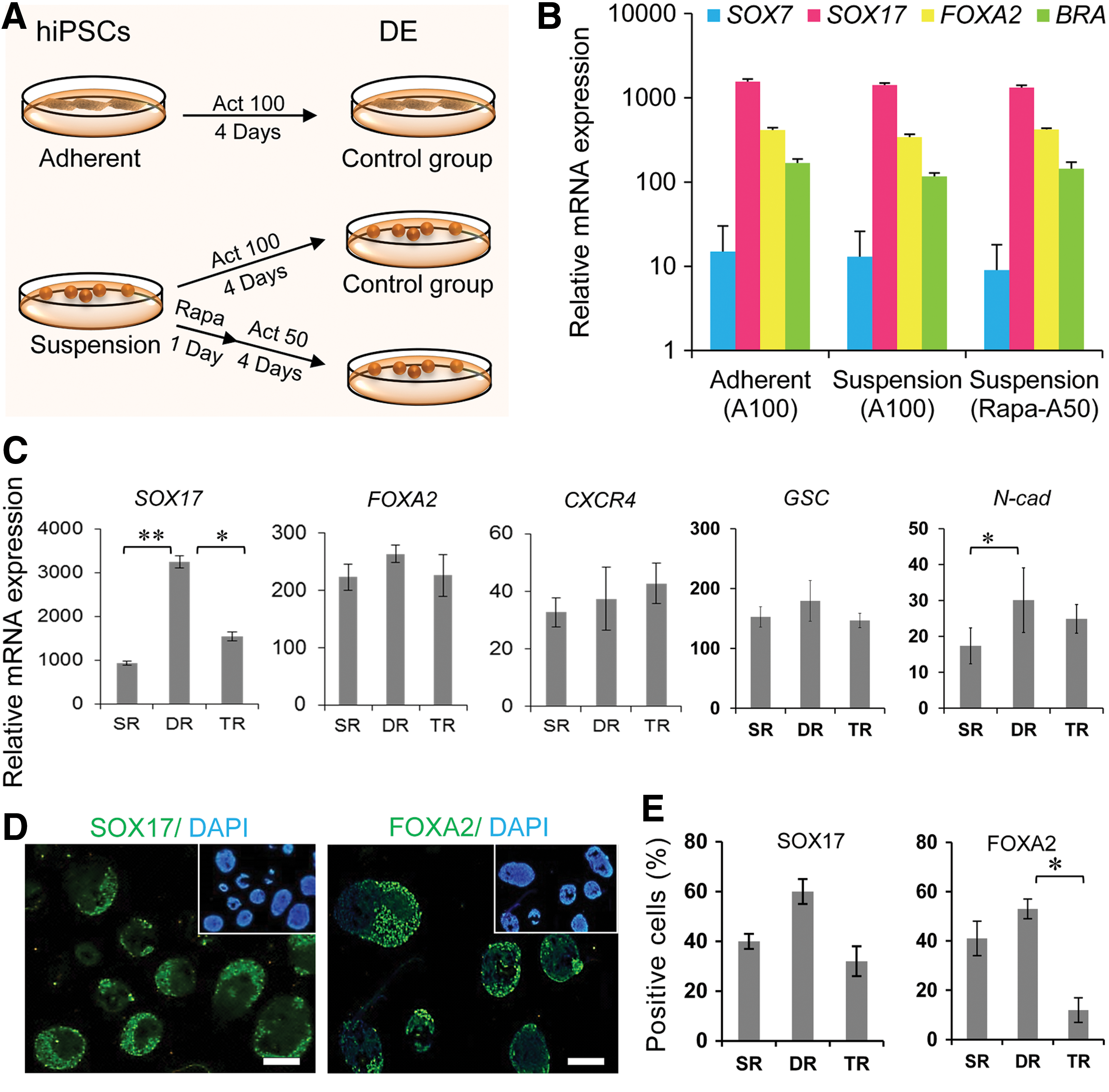
Induction of hiPSCs into definitive endoderm.
To develop a cost effective, efficient process for DE differentiation from hPSCs, we recently described a protocol that uses Rapa (100 nM) for 1 day to prime human pluripotent cells followed by activin A (50 ng/mL, 4 days) for DE induction (Fig. 2A). This treatment leads to an efficient DE differentiation of hPSCs with hepatic lineage competency [25]. With this protocol the activin A concentration is decreased by half. Therefore, we used this approach to differentiate the suspension hiPSC cultures. The comparison of both suspension groups and the adherent control showed similar expressions of the aforementioned genes (Fig. 2B).
We monitored the effects of refreshment condition of the induction medium on DE formation in suspension cultures. We compared single, double, and triple refreshing of the induction medium for 4 days after Rapa administration.
The resultant cells were evaluated for expressions of DE markers at the mRNA and protein levels. DR, or renewal of half of the DE induction medium every 12 h caused increased expression of DE-related genes, SOX17, N-Cad, GSC, and FOXA2, when compared to single or triple refreshing conditions as shown by qRT-PCR, immunostaining, and flow cytometry (Fig. 2C–E).
Similar results were obtained from same experiment with hESC (Supplementary Fig. S2A, B).
Therefore, we continued the stirred bioreactor culturing strategy as a starting platform for large-scale hepatic lineage differentiation and began DE formation by Rapa priming and DR of induction medium.
Generation of HLCs from hPSCs in a stirred bioreactor
Figure 3A illustrates the procedure for hPSC differentiation in a stirred bioreactor. The spheroids of hPSCs were induced to a hepatic fate in a stepwise protocol composed of: cell priming with Rapa for 1 day, followed by DR of induction medium supplemented with activin A (50 ng/mL) for 4 days to DE specification, hepatic induction, and hepatic maturation. To protect cells against activin-induced cell death [26], prior to priming with Rapa, the medium was replaced with non-CM supplemented with tapering concentrations of KOSR (20% to 0.1%) for 1 day. Cells were then treated with activin A (50 ng/mL) for 4 days in non-CM supplemented with 0.1% KOSR. For the next 7 days cells were treated with FGF4 and HGF in the same medium with 10% KOSR. Then spheroids were transferred into HCM with OSM and Dex up to day 21.
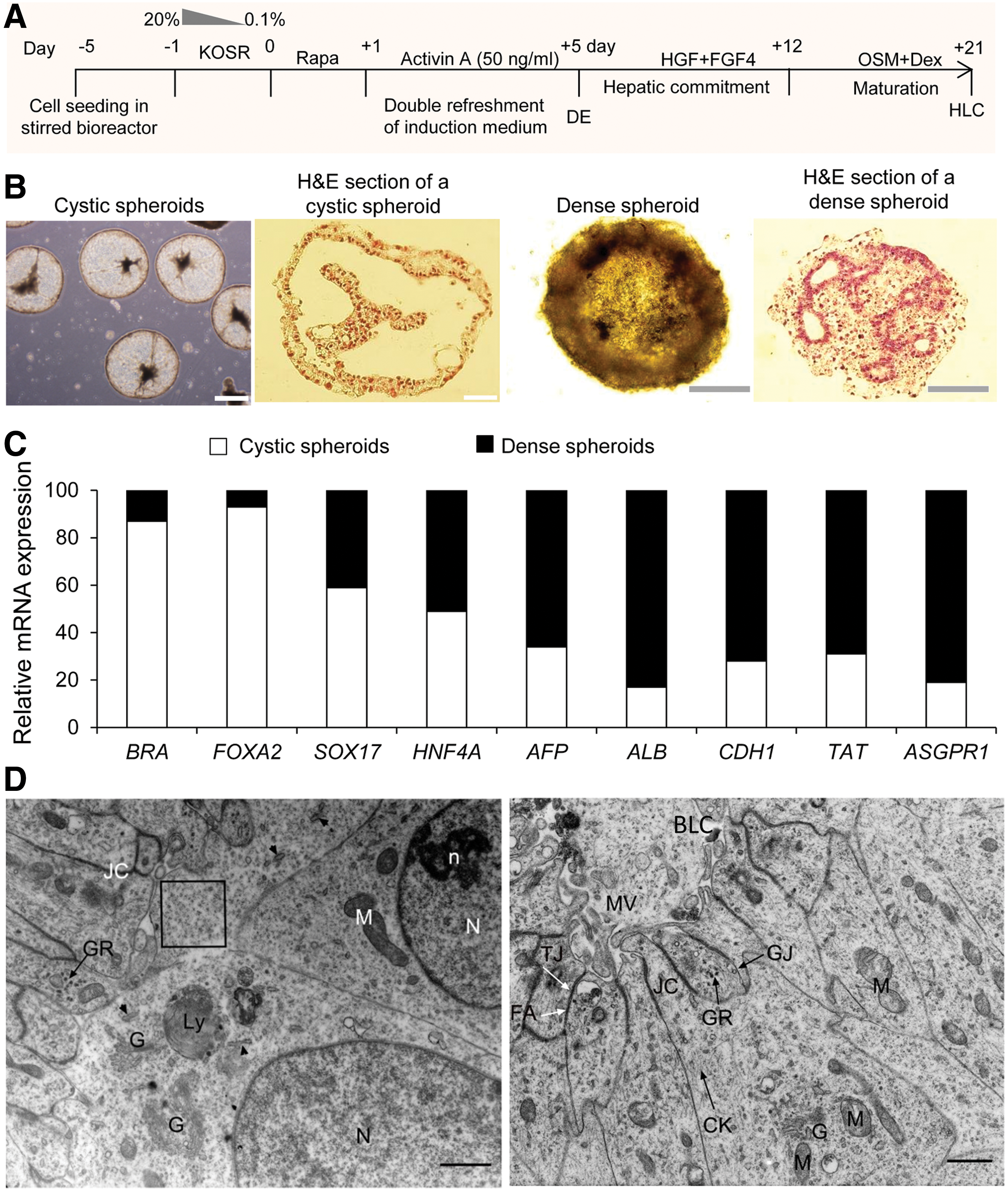
Differentiation of hepatocyte-like cells (HLCs) from hiPSCs in the stirred bioreactor.
In the hepatic commitment period, we observed two different appearances in the induced spheroids, cystic and dense with slow and fast sedimentation rate, respectively. The cross section of the spheroids showed that large and small cavities were surrounded by epithelioid cells in cystic and dense spheroids, respectively (Fig. 3B).
Expression analyses of early and late hepatic lineage-specific genes on day 21 indicated that cystic spheroids expressed genes known to promote mesoendodermal fates (BRA, FAXA2, and SOX17) more than dense spheroids (Fig. 3C). In contrast, the expressions of early and late hepatic fate genes (HNF4α, AFP, ALB, TAT, CDH1, known as E-cadherin, and ASGPR1) showed more upregulation in dense spheroids (Fig. 3C).
Electron microscopy studies of differentiated cells in dense spheroids at day 21 revealed that the cell ultrastructure was similar to immature HLCs (Fig. 3D). The nuclei were euchromatin with almost prominent nucleoli. Mitochondria, developing Golgi apparati, lysosomes, and small rough endoplasmic reticuli were observed in the cytoplasm of differentiated cells. Glycogen granules and intermediate filaments were detected throughout the cytoplasms. Tight junctions, gap junctions, and zonula adherens or adherens junction were observed between adjacent cells. Bile-like canaliculi were also occasionally observed in the center of adjacent epithelioid cells covered with microvilli.
Consequently, the obtained data indicated that dense spheroids consisted of immature HLCs. Therefore, these spheroids were evaluated for additional characterizations.
Characterization of differentiated dense spheroids in the stirred bioreactor
Temporal expression pattern of mesoendodermal markers showed upregulation of these genes by DE formation and their downregulation in subsequent phases of differentiation (Fig. 4A).
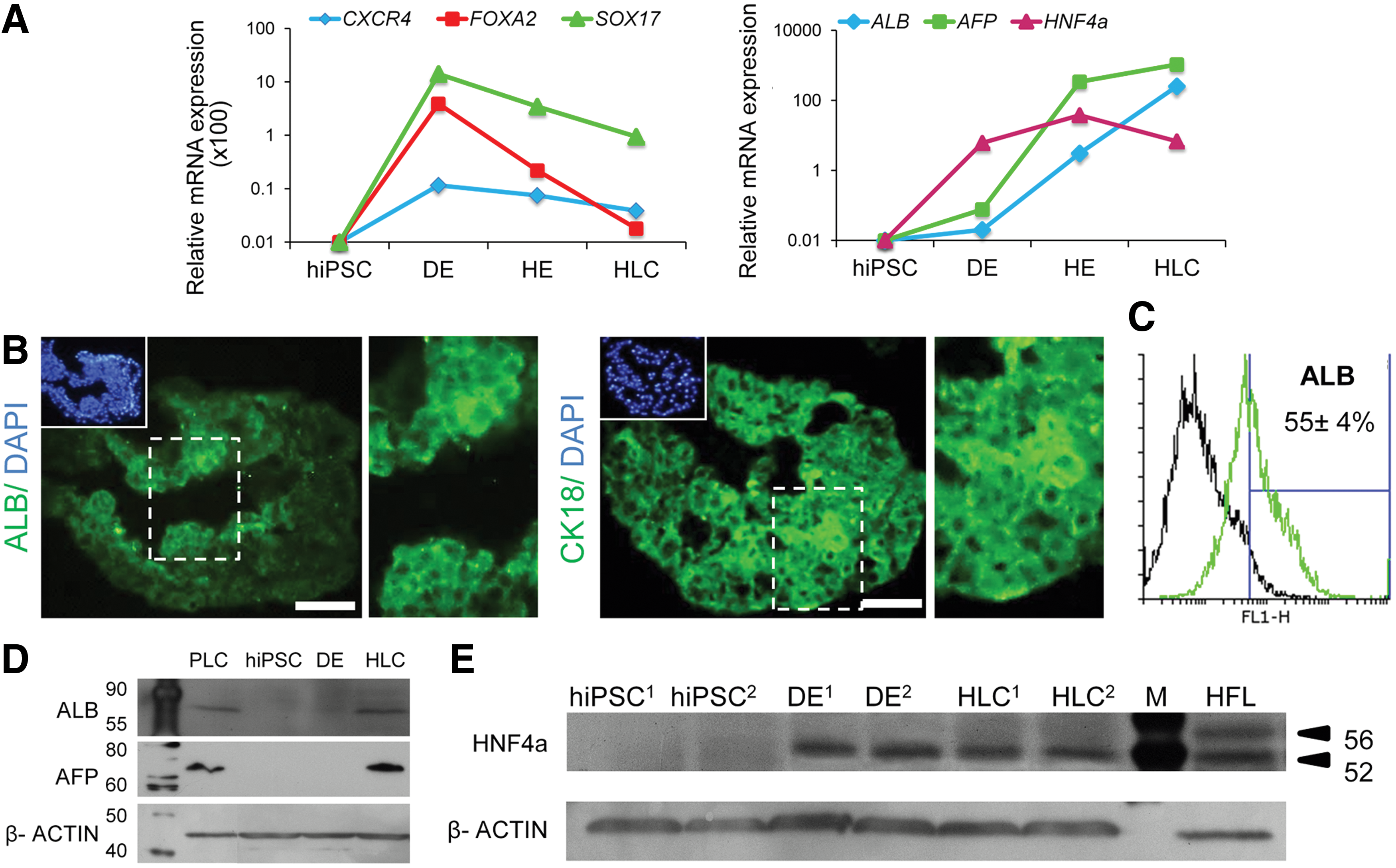
Gene and protein expression analyses of hiPSC-HLCs in dense spheroids in the stirred bioreactor.
To confirm that these differentiated dense spheroids developed into endoderm and then HLCs, we performed several experiments (Figs. 4 and 5). qRT-PCR analysis of the cells at various differentiation steps showed that expression levels of endodermal markers (SOX17, CXCR4, and FOXA2) for both hiPSC and hESC lines were at their highest level on the fifth day after priming, which suggested the induction of an endodermal fate at this stage (Fig. 4A and Supplementary Fig. S2C). Although the endodermal genes gradually downregulated during the differentiation period, the early hepatic markers, HNF4a, AFP, and ALB upregulated from days 5 to 21 (Fig. 4A and Supplementary Fig. S2C). Therefore, these expressions indicated the transition from an endodermal to a hepatic fate.
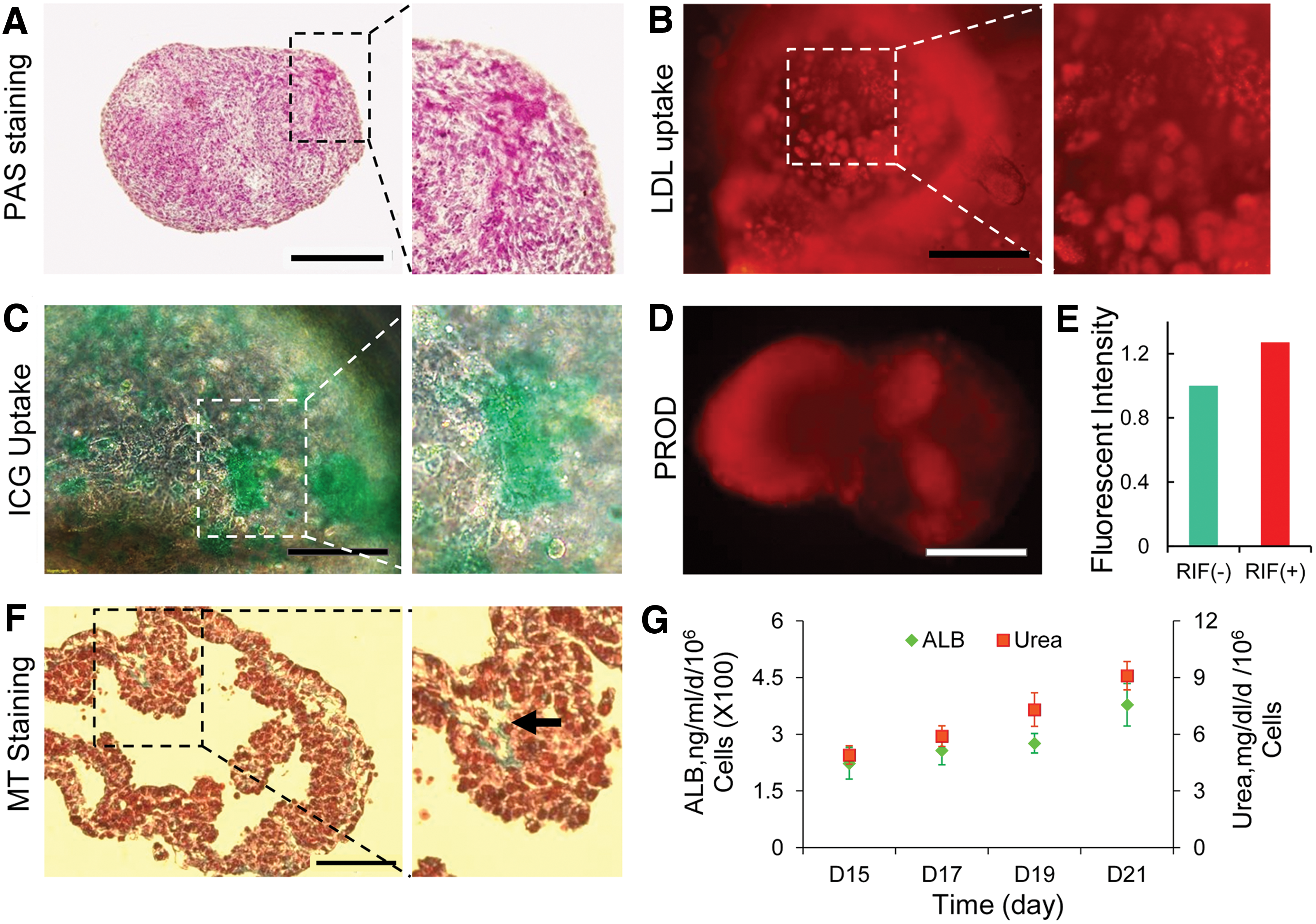
Functional analysis of hiPSC-differentiated dense spheroids in the stirred bioreactor.
Immunofluorescence staining showed that the cells at day 21 expressed ALB and Cytokeratin 18 (Fig. 4B). According to flow cytometry analysis up to 55%±4% (n=3) of the cells expressed ALB (Fig. 4C). Western blot analysis demonstrated that the levels of AFP and ALB increased to match those achieved by the primary liver cell carcinoma line (PLC) (Fig. 4D). Since HNF4a is a crucial transcription factor in hepatogenesis [27], we showed the expression of HNF4a by western blot (Fig. 4E).
Similar results were observed in hESC-derived HLCs (Supplementary Fig. S2D, E). Further, 18%±7% of hiPSC-derived cells expressed ASGPR1, a surface marker for mature hepatocytes. Numerous cells had evidence of pervasively glycogen accumulation (Fig. 5A). These HLCs at day 21 also exhibited other characteristics of hepatocytes for example, Dil-Ac-LDL (Fig. 5B) and ICG uptake (Fig. 5C). In addition, hiPSC-HLCs exhibited CYP450 metabolic activity and dealkylation of pentoxyresorufin to resorufin (a red fluorescent compound), which is a specific measure of CYP3A4-mediated metabolism. Also, the inducible hepatic CYP3A4-mediated 7-pentoxyresorufin-Odealkylase (PROD) activity was demonstrated before and after rifampicin (RIF) induction. The fluorescence intensity of cell supernatant increased after RIF induction in dense spheroids compared with the control group (i.e., before induction of spheroids; Fig. 5E). The differentiated cells secreted collagen to extracellular matrix, which was similar to collagen fibers in space of Disse, as detected by MT staining (Fig. 5F). Cells were also assessed for their ability to secret human ALB and urea production in culture. The results showed the levels of ALB and urea increased in a time-dependent manner throughout the differentiation period (Fig. 5G).
According to the results, hiPSC-HLCs from dense spheroids in the stirred bioreactor were functional in vitro.
Enrichment and transplantation of hiPSC-HLCs based on DiI-Ac-LDL uptake
The enrichment and transplantation procedures of the cells are illustrated in Fig. 6A. To isolate the population of cells with a differentiated hepatocyte phenotype and decrease or eliminate undifferentiated cells or those that might have differentiated toward other cell lineages, dense spheroid derived HLCs were enriched using fluorescent activated cell sorting based on their uptake of DiI-Ac-LDL. Labeling cells with this marker did not affect cell viability and before sorting the percent of DiI-Ac-LDL-positive cells (49%±7%) matched with ALB-positive cells (55%±4%; Fig. 4D) in the same population. After sorting, there were 75% of cells positive for DiI-Ac-LDL and approximately the same percent expressed ALB.
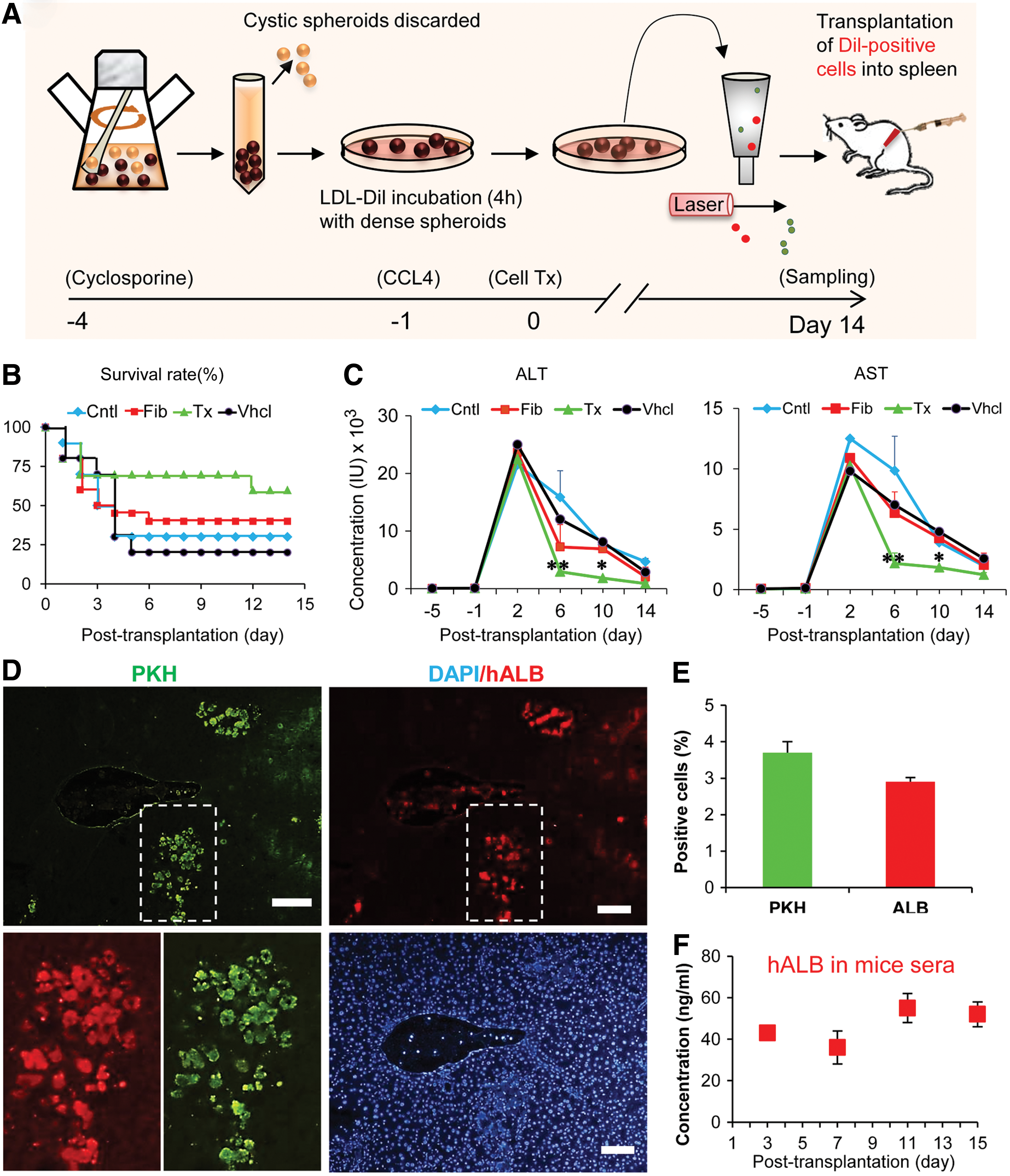
Engraftment and functionality of sorted cells for low density lipoprotein uptake in injured liver parenchyma.
To investigate whether the differentiated cells were engraftable, we transplanted 2×106 DiI-Ac-LDL-sorted cells into the spleens of cyclosporine-treated mice suffering acute liver damage after CCl4 treatment. Animal survival was evaluated up to 15 days after CCl4 injection (Fig. 6B). In the HLC transplanted group, 70% survived by day 10 compared to 20% of vehicle-treated, 40% of fibroblast transplanted, and 30% of the control mice. On day 14, 60% of mice survived in the hiPSC-HLC transplanted group compared to 20% of vehicle, 40% of fibroblast transplanted, and 30% of control groups (Fig. 6B).
In addition, we measured changes in liver enzymes (AST and ALT) 14 days after CCl4 administration (Fig. 6C). Significant reductions were found in the plasma levels of AST and ALT on the day sixth (P<0.01) and tenth (P<0.05) after transplantation in animals treated with HLCs compared with the other control groups (Fig. 6C).
To explore the engraftment of hiPSC-HLCs, we labeled cells with PKH prior to infusion to assess homing of the transplanted cells. Two weeks later, in cell transplanted animals, we detected PKH-labeled cells in recipient livers by fluorescent microscope (Fig. 6D). Engrafted cells were found around the pericentral and periportal veins of the hepatic lobules; only a few were spread throughout the lobules. Within the recipient livers, 3%–4% of cells were PKH+ per section (Fig. 6E).
To further explore their fate, transplanted cells were assessed in corresponding serial sections. Double PKH/hALB-positive cells were considered as indicators of functional activity of the transplanted HLCs. hiPSC-derived hepatic cells engrafted in the mouse liver with an efficiency that ranged from 3% to 4%, with a frequency similar to PKH-counted cells in 30 sections from different areas in the livers of all recipients (Fig. 6E). At 2 weeks post-transplantation, 40–60 ng/mL human ALB was detected in the sera of cell-transplanted mice (Fig. 6F).
No teratoma or tumors were identified after cell transplantation in the livers or other visceral organs of recipient mice during our study at 9 weeks post-transplantation by gross or histologic examinations. To confirm nontumorigenicity of the transplanted cells, we transplanted 2×106 sorted cells into nude mice. At 15 weeks following cell transplantation, there was no evidence of any teratoma or tumor formation in the liver or other major organs.
These results suggested that hiPSC-derived hepatic cells could engraft into the mouse liver and retain the ability to produce hALB with a very low chance of teratoma formation.
Discussion
In this study, we have shown the first demonstration of large-scale integrated manufacturing of HLCs from hPSC-based differentiation and treatment process for hepatic failure, which can be used for other possible biomedical applications (Fig. 7). Human PSCs were expanded in enormous population in suspension and result in aggregates of undifferentiated cells, which could be differentiated into functional hepatocytes in suspension en masse in the stirred bioreactor. This approach differs from traditional differentiation of embryoid bodies, which is stochastic at best and incapable of directing uniform specification. The suspended spheroids contained HLCs that expressed various liver markers, including HNF4α, AFP, ALB, CDH1, TAT, CK18, and ASGPR1. These cells possessed liver-specific functions for example, ALB secretion, urea production, collagen synthesis, ICG and LDL uptake, glycogen storage, and inducible cytochrome P450 activity. The ALB secretion in our study (0.15–0.45 μg/day/106 cell) was comparable with other experiments in 2D and 3D differentiation of hPSCs into HLCs [15,28]; however, primary hepatocytes produce 30–40 μg/day/106 cell in 3D scaffolds [29]. Additionally, the concentration of urea production in the current study (4–10 mg/dL/day/106 cell) was in range of results of other experiments while primary hepatocytes produced urea 10-folds more [15]. Therefore, in terms of gene expression, ultrastructure, and functional analysis, these cells have the properties of immature hepatocytes, and therefore need additional maturation processes to acquire more functional phenotypes.
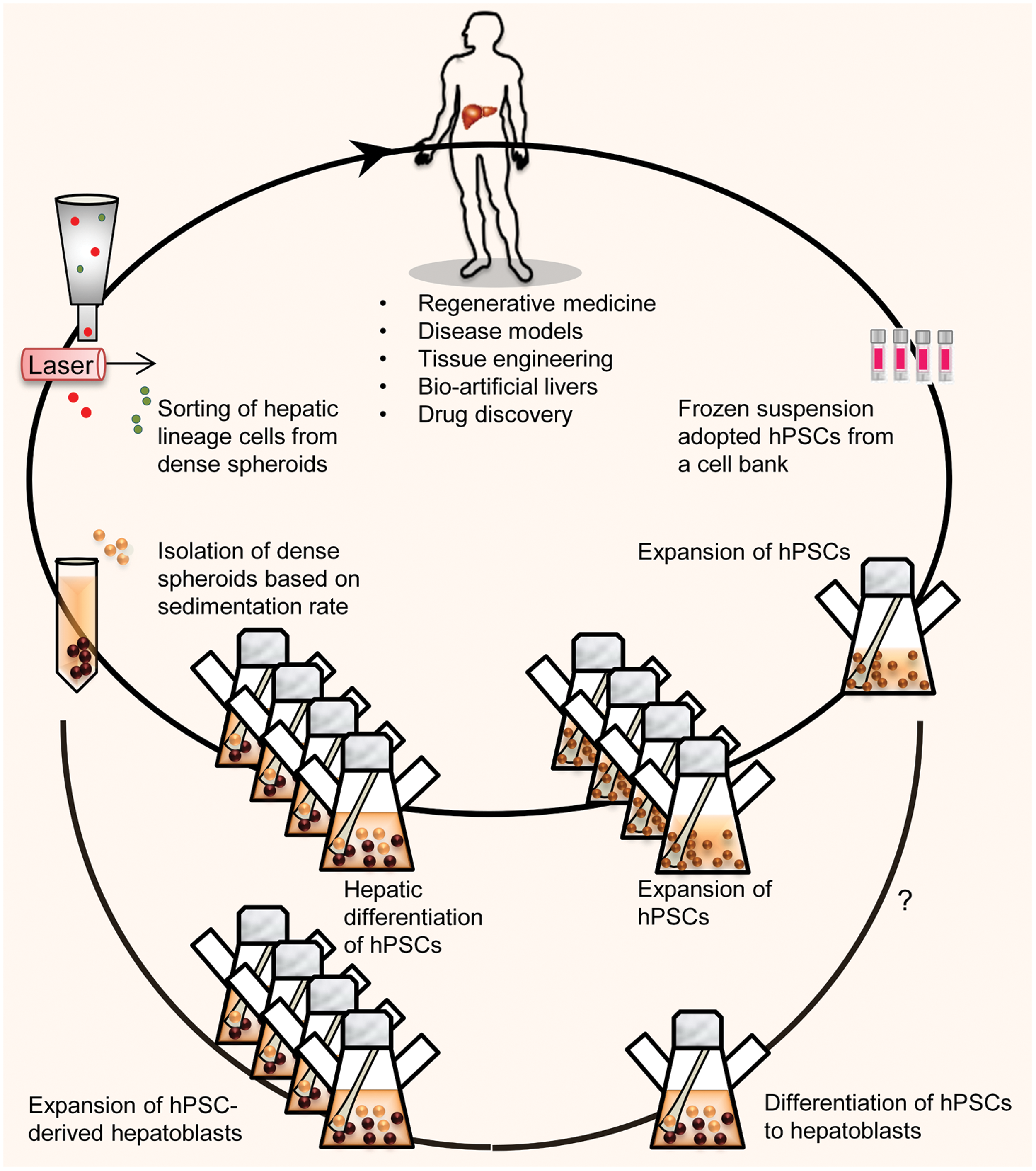
Schematic presentation shows the manufacturing process for mass production of hPSCs or differentiated hepatic progenitors (hepatoblasts) in a stirred suspension bioreactor. By using the proposed scale-up platform, a number of frozen vials of hPSCs were thawed to provide adequate starting material for regenerative medicine. Qualified lots of undifferentiated hPSCs are amenable for further scaling via additional passages and later used for possible biomedical applications. By this method, frozen vials that included a total of 10×106 hPSCs could theoretically be thawed and 4 weeks later, up to 2×109 hPSCs could be produced [21]. A clinically relevant cell number of hPSC-HLCs (∼5–8×108 cells) could be produced in four spinner flasks that contain a total of 400 mL of medium over a 3 week period. Another option might be the in vitro expansion of differentiated hepatic progenitors (hepatoblasts) derived from hPSCs. Color images available online at
This suspension hepatic differentiation is comparable with the efficiency of hepatic differentiation in adherent cultures or by embryoid body formation and replating, as has been previously described for the differentiation of hESCs and hiPSCs (for review see [30 –32]). The advantages of suspension differentiation of hPSC aggregates include high scalability, feasible handling, reasonable costs, and elimination of exogenous extracellular matrix.
By microwell-based technology hPSC aggregates have been induced to differentiate into DE [33,34] or functional pancreatic progenitors in a dynamic rotational suspension culture in plates [35]. Further, spheroid culture of human hepatocytes resulted in their long-term maintenance in vitro [36]. Three-dimensional suspension cultures of hESC-derived DE in a static 3D culture of Algimatrix scaffolds [37] or collagen [38] also significantly improved hepatocyte differentiation and function when compared with the monolayer culture [28]. Hyaluronan-embedded human fetal liver cells in a perfusion environment or cultures of rat hepatocytes as 3D structures on a stirred bioreactor have resulted in better conditions for liver cell survival, organization, and function compared with traditional monolayer cultures [39,40]. Recently, hepatocyte lineage-committed cells were generated from mouse ESCs in spinner flask culture system [41].
In our study, the suspended differentiated aggregates showed two distinct morphologies, cystic and dense. Dense spheroids according to qRT-PCR analysis expressed higher hepatic lineage markers than cystic spheroids, whereas later aggregates showed predominantly mesoendodermal. The size of the spheroid might be important in this regard. Recently, Takayama et al., have established a hepatocyte differentiation method that used 3D hPSCs spheroid cultures and a Nanopillar plate to control spheroid size and configuration to generate more matured HLCs than seen in 2D cultures [42].
In large spheroids, directed controlled differentiation with exogenous factors and oxygen supply is restricted due to diffusion limitations [43,44]. Unwanted intercellular signaling inside aggregates [45] results in low purity [35] of desired cells. This may lead to formation of cystic spheroids that remain in the early stages of differentiation and express endodermal markers. The microparticulate-mediated growth factor delivery systems may enhance efficiency of the permeation and localization of exogenous factors inside spheroids by facilitating diffusion through intercellular spaces and overcome this problem [46,47].
Next, we explored whether the differentiated cells were engraftable and functional in vivo by transplanting hiPSCs-HLCs into a CCl4-injured mouse model. Our HLCs in the dense spheroids enriched up to 75% by vital staining of DiI-Ac-LDL uptake. This simple method is reproducible, free of genetic manipulation and antibody labeling, and based on natural function of cells (i.e., DiI-Ac-LDL uptake). Additionally, undifferentiated hPSCs do not uptake DiI-Ac-LDL and selected negatively [48,49]. The lipoprotein that is labeled with DiI-Ac-LDL is degraded by lysosomal enzymes and the DiI fluorescent probe accumulates in the intracellular membranes. The equal abundance of DiI-Ac-LDL and hALB-positive cells has shown that the majority of these cells may be human HLCs. HLCs enrichment has been previously performed based on ASGPR1 [8] or ICG uptake [50].
The CCl4-injured mouse model is well defined and widely used to investigate the early phase of liver regeneration. We have observed that sorted cells engrafted into the recipient's liver and increased survival rate. Also, in HLCs transplanted group, serum ALT and AST levels reduced markedly. The presence of human ALB in the serum of cell-transplanted mice is reflective of an acceptable correlation between engraftment efficiency and the physiological function of transplanted cells with regard to ALB secretion. This recovery may be due to not only functional activity of cell replacement but also the delivery of trophic paracrine factors that support and facilitate endogenous liver regeneration [50].
Tumor formation has been a concern associated with hESC- or hiPSC-based cellular therapy. A previous grafting study demonstrated the risk of tumor formation following intrasplenic transplantation of hESC-HLCs into NOD-SCID mice [8]. We did not observe any tumor formation in the recipient livers or other major organs for the whole duration of this study (up to 9 weeks in immunosuppressed NMRI mice or 15 weeks after transplantation in nude mice). The lack of tumor formation may relate to the purity of the transplanted cells or recipient mouse strain used in this study. Recently, it has been reported that hESC-HLCs purified by ICG staining contribute to the recovery of CCl4-injured liver tissues in mice and did not produce any tumor after intrasplenic transplantation of 2×106 cells into BALB/c nude mice [50]. However, more studies should be performed for long-term follow-up to assess the safety and functionality of engrafted cells.
In conclusion, the findings presented here, for the first time, provide a new platform for efficient development of functional HLCs from hPSCs by a simple and reproducible method in a carrier-free suspension stirred bioreactor and outlined enrichment strategy. This integrated approach in scaling up functional hPSC-derived HLCs may open new windows in clinical application or pharmaceutical industries.
Footnotes
Acknowledgments
We express our appreciation to Abbas Piryaei, Keyhan Azadmnaesh, Mehran Rezai, Zahra Farzaneh, Mostafa Najarasl, Azam Samadian, Fazel Sahraneshin Samani, Shahab Mirshahvaladi, Ehsan Janzamin, and Mohammad Chehrazi at Royan Institute for technical assistance and data analyses. We also thank Katherina Psathaki for taking TEM micrographs from Max Planck Institute for Molecular Biomedicine, Münster, Germany. This study was funded as a PhD program grant provided by Royan Institute and Pasteur Institute of Iran.
Author Disclosure Statement
The authors declare they have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.