
Abstract
Human induced pluripotent stem (iPS) and embryonic stem (ES) cells can differentiate into a variety of cell types. We reported on adipogenic potential of human iPS and ES cells in vitro. In the present study, we investigate the survival and maintenance of adipocytes differentiated in vitro from human iPS and ES cells after transplantation. Following adipogenic induction in vitro, the differentiated cells exhibited functional properties of adipocytes such as lipid storage, lipolysis, and insulin responsiveness. Subsequently, Matrigel containing the differentiated human iPS and ES cells was transplanted into the subcutaneous tissue of nude mice. After 1–4 weeks, the cells with adipocyte-like features were observed in transplanted Matrigel by histological analysis. The human origin of the cells, their lipid accumulation, and gene expression of adipocyte markers in transplanted cells were then confirmed, suggesting the presence of adipocytes in transplanted Matrigel. When the relative areas of these cells were calculated by dividing the adipocyte areas by the total Matrigel areas, we found that they peaked at 2 weeks after transplantation, and that the adipocytes persisted at 4 weeks. The present study demonstrates that human iPS and ES cells can differentiate into adipocytes with functional properties and that adipocytes derived from human iPS and ES cells can survive and maintain the differentiated properties of adipocytes for at least 4 weeks after transplantation. Adipocytes derived from human iPS and ES cells thus have the potential to open new avenues for stem cell-based research into metabolic diseases and future therapeutic applications.
Introduction
Lipodystrophy is a rare syndrome characterized by loss of adipose tissue, which causes insulin resistance, diabetes, dyslipidemia, and ectopic fat accumulation [18,19]. Transplantation of adipose tissue ameliorates the phenotype of lipodystrophy [20]. Further, adipose tissue, adipocytes, and adipose-derived stem cells are therapeutically useful for soft tissue reconstruction after tumor resection [21 –24]. Adipocytes derived from human PS cells can be a possible source of cell therapy for lipodystrophy and soft tissue reconstruction. To date, there have been several reports of using scaffolds to construct adipose-like tissue derived from human adult stem cells and embryonic germ cells [25 –28]; however, construction of adipose-like tissue derived from human iPS and ES cells has not been fully demonstrated.
In the present study, we initially assessed functional properties of adipocytes such as lipid storage, lipolysis, and insulin responsiveness in vitro in differentiated human iPS and ES cells. We then studied survival and maintenance of adipocytes derived from human iPS and ES cells following their transplantation.
Materials and Methods
Cell culture
Human iPS cells: 253G4 (G4), 201B7 (B7), and W12, were maintained in a Primate ES medium (ReproCELL) supplemented with 4 ng/mL recombinant human basic fibroblast growth factor (Invitrogen) as we previously described [13]. G4 and B7 were kindly provided by Shinya Yamanaka [29]. Human ES cells (H9 and KhES-1) were cultured as described previously [29]. H9 was purchased from Wicell Research Institute, Inc. KhES-1 was kindly provided by Norio Nakatsuji [30]. W12 as well as B7 were generated from human dermal fibroblasts from a 36-year-old Caucasian woman (Cell Applications, Inc.) by introducing four factors, such as Oct3/4, Sox2, Klf4, and c-Myc. Expression of human ES cell markers and pluripotency were examined by immunocytochemistry, in vitro differentiation, and teratoma formation (Supplementary Fig. S1A–K; Supplementary Data are available online at
Adipogenic differentiation of human iPS and ES cells
Human iPS and ES cells were differentiated into the adipocyte lineage via embryoid body (EB) formation using a modified version of the protocol, which we previously described [13]. Briefly, adipogenic differentiation was initiated by aggregation of iPS and ES cells to form EBs. From day 2–5, retinoic acid (SIGMA-Aldrich) with the concentration of 100 nM was supplemented. From day 8–11, 1 μg/mL insulin (Roche Diagnostics) and 1 μM pioglitazone (SIGMA-Aldrich) were supplemented. After 11 days of EB culture, EBs were transferred to plates coated with type IV collagen (BD Biosciences). Adipogenic differentiation was induced for additional 3–5 days of culture using 10% fetal bovine serum/Dulbecco's Modified Eagle Medium (DMEM) containing 0.5 mM 3-isobutyl-1-methylxanthine (Nacalai Tesque), 0.25 μM dexamethasone (Nacalai Tesque), 1 μg/mL insulin, and 1 μM pioglitazone.
Oil Red O staining
Differentiated human iPS and ES cells were washed with phosphate-buffered saline (PBS) twice, fixed in 3.7% formaldehyde for 1 h, and then stained with a 0.6% (w/v) Oil Red O solution (60% isopropanol and 40% water) for 2 h at room temperature. Cells were then washed with water to remove unbound dye. Optical sections were obtained with BZ-9000 (KEYENCE). Oil Red O-stained areas were calculated as the percentage of the area divided by the total area.
Immunocytochemistry
Differentiated human iPS and ES cells were washed with PBS and fixed for 10 min with 4% paraformaldehyde. Then, the cells were permeabilized by 0.1% Triton X in PBS for 5 min and incubated by Protein Block (DAKO) for 30 min at room temperature. The cells were immunostained with a 1:200 dilution of a primary antibody against peroxisome proliferator-activated receptor γ (PPARγ; Cell Signaling Technology). After washed with PBS, Alexa 546-conjugated anti-rabbit IgG (Molecular Probes) was used as a secondary antibody with the concentration of 10 μg/mL. Then, after being washed with PBS, BODIPY 493/503 (Molecular Probes) was added for 15 min for lipid staining. The cells were mounted in the medium with 4′,6-diamidino-2-phenylindole after washed with PBS twice (Vector Labs). Then, human iPS cells were stained with primary antibodies against Nanog (1:20; R&D Systems), TRA-1-60 (1:100; Millipore), SSEA-4 (1:100; Santa Cruz Biotechnology, Inc.), β3-tubulin (1:100; Millipore), α-smooth muscle actin (pre-diluted; DAKO), and α-fetoprotein (1:100; Millipore) according to the protocol described previously [20]. Alexa Fluor 488-conjugated anti-mouse IgG, Alexa Fluor 546-conjugated anti-goat IgG, and Alexa Fluor 546-conjugated anti-mouse IgG antibodies (Molecular Probes) were used as secondary antibodies with a concentration of 10 μg/mL. An alkaline phosphatase activity was detected using a BCIP/NBT substrate system (DAKO). 3T3-L1 cells and human mesenchymal stem cells were stained with a primary antibody against vimentin (1:100; DAKO). Alexa Fluor 546-conjugated anti-mouse IgG was used as a secondary antibody. Optical sections were obtained with BZ-9000.
Lipolysis assays
Before lipolysis assays, differentiated human iPS and ES cells were incubated in the serum-free DMEM with 0.5% fatty acid-free bovine serum albumin for 4 h. For lipolysis assays, differentiated cells were stimulated with 10 μM forskolin for 6 h. The medium was then collected and established procedures were used to quantify the glycerol as instructed by the manufacturer (SIGMA-Aldrich). The protein content was determined using Protein Assay (BIORAD).
Western blot analysis
Preparation of total cell lysates and western blot analysis were performed as we previously described [31]. Briefly, cells were harvested in the lysis buffer. For western blot analysis, proteins were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were immunoblotted with the primary antibodies against AKT and phospho AKT (Ser 473) (Cell Signaling Technology). Membranes were reacted with the secondary antibody (GE Healthcare) and developed with ECL plus (GE Healthcare) as instructed by the manufacturer. The signal on the blot was detected with ImageQuant LAS 4000 System (GE Healthcare).
Flow cytometric analysis
At 3 days after attachment of EBs and supplementation of adipogenic cocktails, cells were harvested. Single-cell suspensions were labeled for 30 min on ice with the mouse fluorescence-conjugated antibodies against PE-CD73 and PE-CD105 (eBioscience). The cells were analyzed by FACS-Aria II (BD Biosciences).
Transplantation of derivatives from human iPS cells and ES cells
This study was performed after approval of the Kyoto University Graduate School and Faculty of Medicine, Ethics Committee (No. 824 and ES6). All animal experiments were performed in strict accordance with the guidelines for animal experiments of Kyoto University. Matrigel (BD Biosciences) was mixed with a suspension of differentiated or undifferentiated human iPS and ES cells harvested from a confluent 100-mm dish. Matrigel incorporating 2×107 differentiated or 1×107 undifferentiated PS cells was then carefully implanted into the subcutaneous tissue on the backs of 8-week-old male BALB/cA nude mice (CLEA Japan) using a syringe with a 21-gauge needle. Samples of skin tissue, including the implanted Matrigel were harvested for further studies at 1 day, 1 week, 2 weeks, and 4 weeks after transplantation. Matrigel incorporating adipocytes derived from 2×106 human bone marrow-derived mesenchymal stem cells was also implanted into the subcutaneous tissue on the backs of the BALB/cA nude mice as a positive control study.
Immunohistochemistry
Human adipose tissue biopsy was performed after approval of the Kyoto University Graduate School and Faculty of Medicine, Ethics Committee (No. 553). Each tissue specimen was fixed in 10% neutralized formalin solution, embedded in paraffin, sectioned at the central portion of implanted Matrigel, and followed by staining with hematoxylin and eosin (HE) or immunohistochemical studies. The paraffin sections were immunostained using the Polymer Immunocomplex System (DAKO) with a 1:50 dilution of a primary antibody against human vimentin (DAKO). Phosphohistone H3 was stained with a 1:100 dilution of the primary antibody (Cell Signaling Technology). Cryosections from the samples were fixed with a 10% neutralized formalin solution before embedding in optimal cutting temperature compound. Lipid accumulation in transplanted cells was assessed by Oil Red O staining. Morphometric analyses were performed using BZ-9000. The relative adipocyte area was expressed as the ratio of the adipocyte area to the total Matrigel area in each section.
Reverse transcription polymerase chain reaction
Total RNA was extracted using the TRIzol reagent (Invitrogen). For reverse transcription polymerase chain reaction (RT-PCR) assay, cDNA was synthesized by iScript (BIORAD). Semiquantitative PCR was carried out using GeneAmp PCR System 9700 as instructed by the manufacturer (Applied Biosystems). TaqMan PCR was performed using Step One Plus Real-Time PCR System as instructed by the manufacturer (Applied Biosystems). Relative levels of mRNA were normalized to the mRNA level of GAPDH. The primers and probes used were listed in Supplementary Fig. S2.
Statistical analysis
The data are presented as mean±standard error. The Student's t-test was used as the statistical analysis. The level of significant difference was the p-value<0.05.
Results
Adipogenic differentiation of human iPS and ES cells in vitro
Human iPS (G4, B7, and W12) and ES (H9) cells were differentiated into the adipocyte lineage in vitro. Differentiated iPS and ES cells exhibited prominent lipid accumulation by Oil Red O staining, while lipid accumulation was rarely observed in undifferentiated cells (Fig. 1A–E). After differentiation in vitro, gene expression of a set of adipocyte markers, such as PPARγ2, CCAAT/enhancer-binding protein (C/EBP) α, fatty acid-binding protein-4 (FABP4), and leptin, was detected by RT-PCR analysis. Gene expression of Nanog in differentiated cells was markedly lower than in undifferentiated cells (Fig. 1F). Immunohistochemical analysis showed that the PPARγ protein was present within the nuclei of a portion of the differentiated iPS and ES cells containing lipid droplets, while the PPARγ protein was not detected in undifferentiated cells (Fig. 1G–O). Further, these differentiated cells were examined whether they have lipolytic responses and insulin responsiveness. In differentiated cells from both iPS and ES cells, 10 μM forskolin significantly enhanced glycerol release into the culture medium (∼1.6-fold and ∼1.8-fold increase, respectively), as compared to vehicle-treated groups. In undifferentiated iPS and ES cells, 10 μM forskolin did not significantly increase glycerol release (Fig. 1P). In addition, 100 nM or 1 μM insulin remarkably increased AKT phosphorylation in differentiated iPS and ES cells as compared to vehicle-treated groups. AKT phosphorylation was also slightly enhanced in undifferentiated iPS and ES cells (Fig. 1Q). These findings suggest the presence of adipocytes with functional properties in differentiated iPS and ES cells.
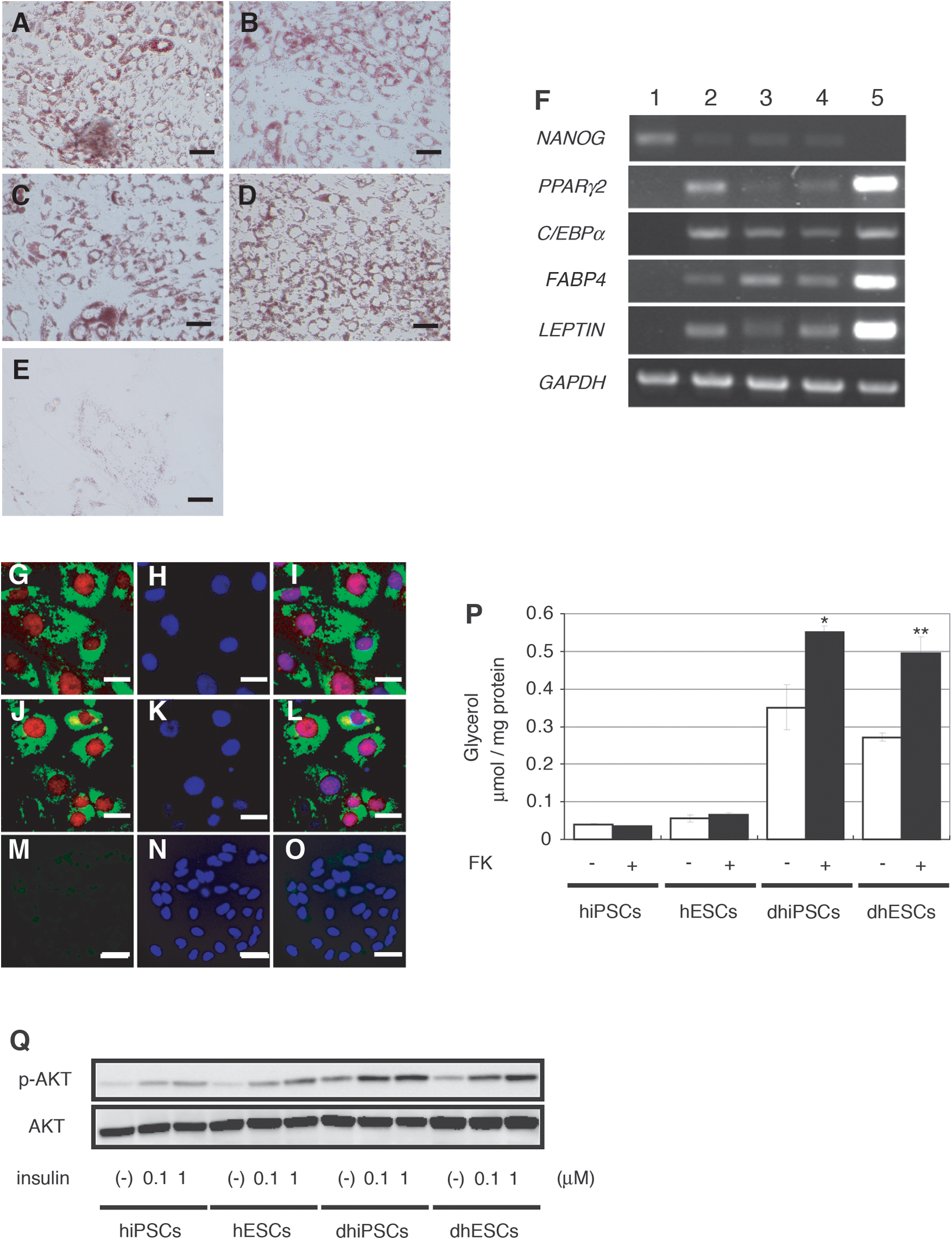
Transplantation of adipocytes derived from human iPS and ES cells
After 14 days of in vitro differentiation, Matrigel containing differentiated human iPS (G4) or ES (H9) cells was transplanted into the subcutaneous tissue on the backs of 8-week-old BALBc/A nude mice (Fig. 2A). Matrigel containing undifferentiated iPS cells or cell-free Matrigel was transplanted as a negative control, while Matrigel containing adipocytes derived from human bone marrow-derived mesenchymal stem cells was transplanted as a positive control. At 1 day, 1 week, 2 weeks, and 4 weeks after transplantation, samples of skin tissue containing the Matrigel were harvested. Grossly, small blood vessels could be seen distributed in Matrigel (Fig. 2B). Timeline of the transplantation study was demonstrated (Fig. 2C). Preparation of histological sections and HE staining of specimens collected at 1 week after transplantation revealed that the differentiated iPS or ES cells possess adipocyte-like features, including thin rims of cytoplasm surrounding the vacuole and flattened nucleus (Fig. 2D, E). Similar histological findings were observed at 2 and 4 weeks after transplantation of differentiated iPS and ES cells (Fig. 2F–I). These cells were also noted at 4 weeks after transplantation of adipocytes derived from human mesenchymal stem cells (Fig. 2J), but no adipocytes described above were observed in cell-free Matrigel at 4 weeks after transplantation (Fig. 2K). By contrast, adipocytes derived from human iPS and ES cells were rarely seen at 1 day after transplantation (data not shown), which may indicate the loss of lipid droplets caused by the transplantation procedure and insufficient vascularization in the Matrigel at that time. Histological findings also showed that Matrigel with undifferentiated human iPS cells mainly contained immature neuroectodermal cells such as neural tube cells at 1, 2, and 4 weeks after transplantation (Fig. 2L–N). No adipocytes described above were detected throughout the transplantation period.
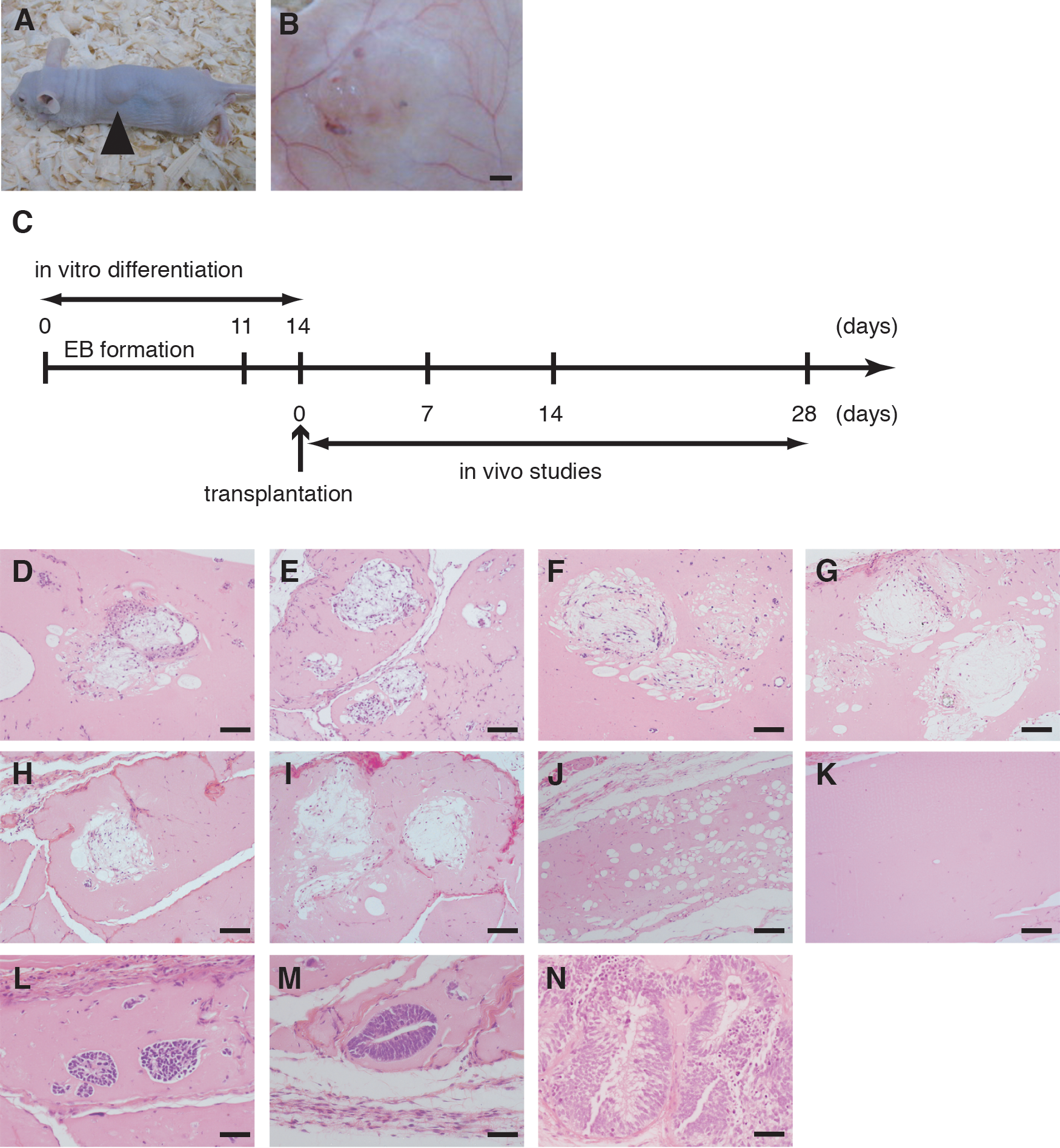
Transplantation of adipocytes derived from human iPS cells and ES cells.
Characterization of transplanted cells derived from human iPS and ES cells
We next calculated the relative areas of the adipocytes by dividing the adipocyte area by the total Matrigel area in sections of skin tissue collected at 1 day, 1 week, 2 weeks, and 4 weeks after transplantation of differentiated human iPS and ES cells. The areas were 0.63%±0.10% (G4) and 0.42%±0.17% (H9) on day 1 after transplantation. At 1 week after transplantation, the relative areas were 2.27%±1.19% (G4) and 9.39%±1.36% (H9), and by 2 weeks after transplantation, they had increased to 5.26%±0.46% (G4) and 12.9%±5.32% (H9). However, the areas had declined to 2.17%±1.28% (G4) and 6.97%±1.66% (H9) at 4 weeks after transplantation (Fig. 3A). Thus, the adipocytes were clearly present at 1–4 weeks after transplantation of differentiated iPS or ES cells, and relative adipocyte areas were maximal at 2 weeks after transplantation.
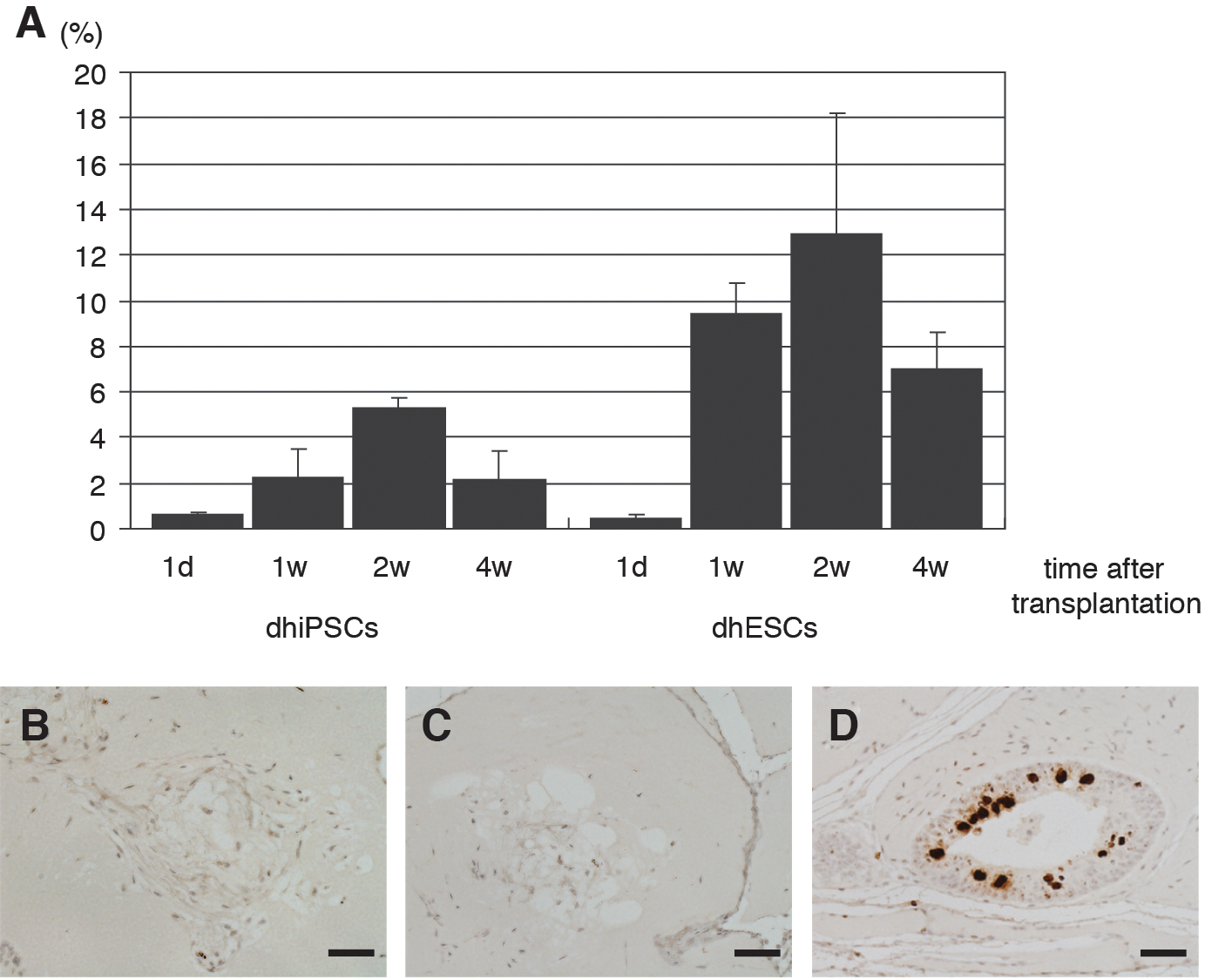
Quantification of adipocyte areas and proliferative capacity of transplanted cells.
To assess the proliferative capacity of the transplanted cells, the cells were immunostained with an antibody against phosphohistone H3 at 2 weeks after transplantation. Adipocytes derived from human iPS and ES cells exhibited little or no proliferative capacity (Fig. 3B, C), whereas the immature neuroectodermal cells exhibited a high-proliferative capacity in transplantation of undifferentiated human iPS cells (Fig. 3D).
The origin of the adipocytes in the Matrigel was studied by immunostaining with an antibody against human vimentin. According to the manufacturer's guide and an earlier report [32], the antibody used does not cross react with mouse vimentin. We also evaluated the cross reactivity of the antibody with mouse vimentin. Vimentin was stained with the antibody in human mesenchymal stem cells, while it was not stained in mouse 3T3-L1 cells (Supplementary Fig. S3A–D). Moreover, human subcutaneous adipose tissue was stained with the antibody (Supplementary Fig. S3E), but mouse subcutaneous adipose tissue was not (Supplementary Fig. S3F).
When we then assessed the human vimentin immunoreactivity of the cells within the Matrigel, we found that adipocytes derived from both iPS and ES cells were labeled by the anti-vimentin antibody at 2 and 4 weeks after transplantation (Fig. 4A–H). Further, lipid accumulation in the cells was demonstrated by staining frozen sections with Oil Red O at 2 and 4 weeks after transplantation (Fig. 4I–L). Similar histological findings were observed at 2 weeks after transplantation of adipocytes derived from human bone marrow-derived mesenchymal stem cells (Fig. 4M–O).
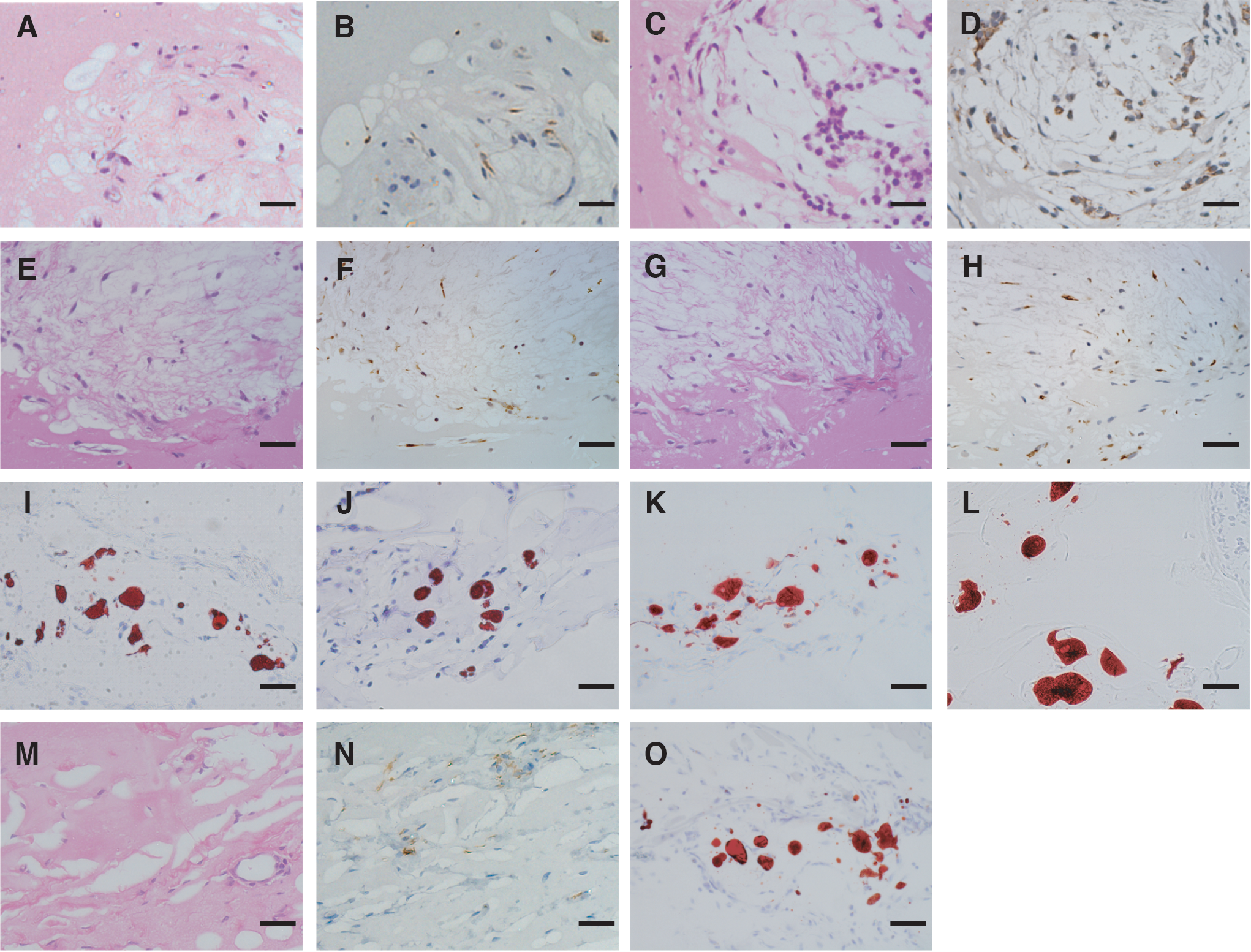
Human origin and lipid accumulation in transplanted cells.
Gene expression of a set of adipocyte markers in transplanted differentiated iPS and ES cells was then investigated by PCR analyses. Gene expression of PPARγ2, C/EBPα, aP2, and leptin was detected at 2 and 4 weeks after transplantation (Fig. 5A, B). Quantitative real-time PCR analyses carried out with the samples after transplantation revealed that leptin and PPARγ2 were expressed at 1, 2, and 4 weeks after transplantation. In both iPS and ES cells, mRNA levels of leptin at 1 and 2 weeks after transplantation were higher than those at 4 weeks after transplantation (Fig. 5C). Meanwhile, mRNA levels of PPARγ2 at 4 weeks after transplantation were maintained in both cell lines as compared with those at 2 weeks (Fig. 5D).
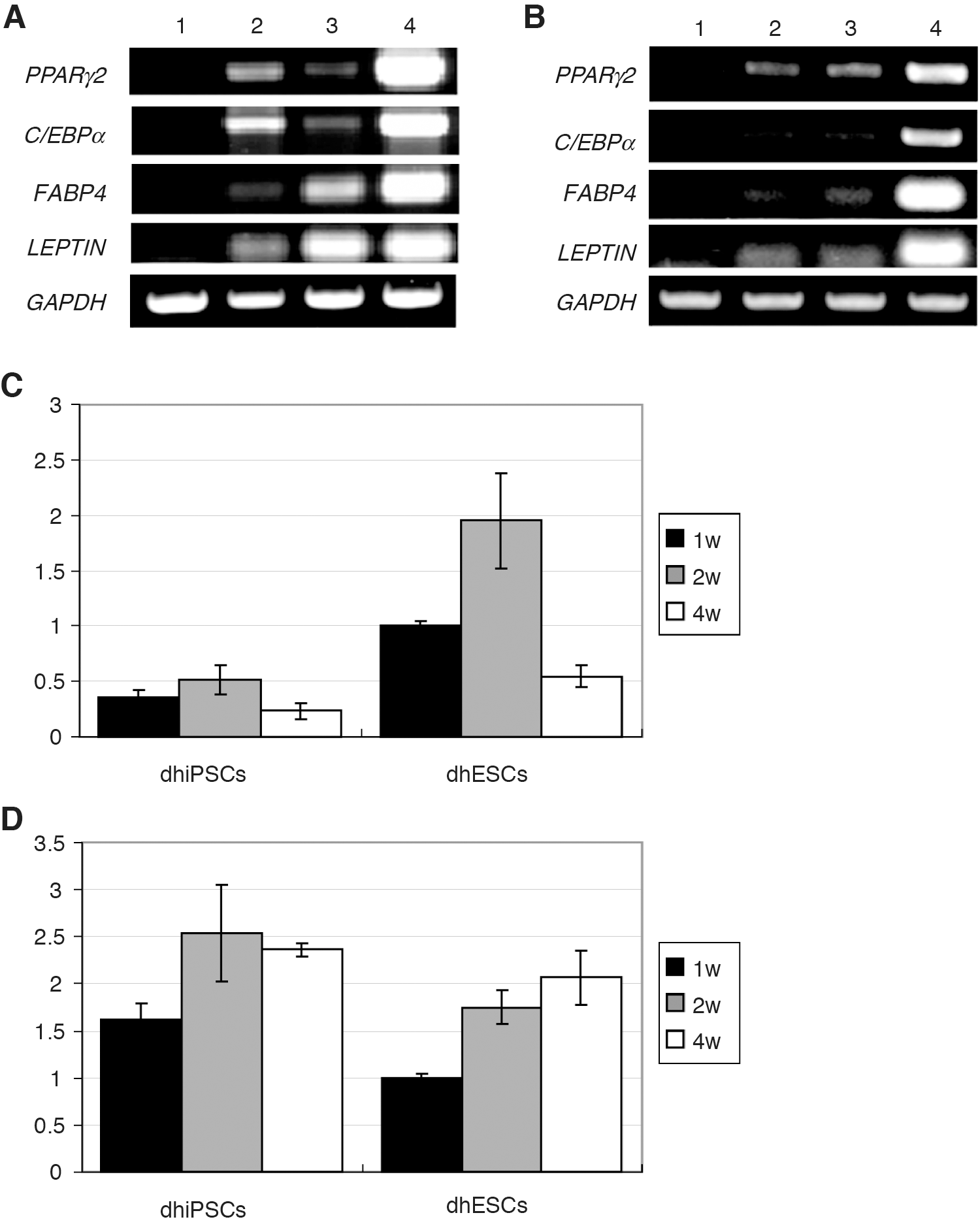
Gene expression of adipocyte markers in transplanted cells.
Transplantation of other human pluripotent stem cell lines
When we similarly transplanted Matrigel containing other differentiated human iPS cell line (B7 and W12) and another human ES cell line (KhES-1), adipocytes described above were observed at 2 weeks after transplantation, suggesting that adipocytes derived from other PS cells can survive after transplantation (Fig. 6A–C). At that time, the relative areas of adipocytes derived from B7, W12, and KhES-1 were 1.11%±0.57%, 1.45%±0.48%, and 2.41%±1.42%, respectively (Fig. 6D), much less than were seen with G4 and H9. To examine the adipogenic differentiation rate of in vitro differentiated iPS and ES cells before transplantation, we quantified Oil Red O-stained areas of the differentiated iPS and ES cells. The areas of differentiated G4 and H9 were 23.89%±1.28% and 26.51%±1.29%, respectively. Moreover, those of differentiated B7, W12, and KhES-1 were 20.92%±2.77%, 22.92%±0.56%, and 21.25%±2.17%, respectively (Supplementary Fig. S4), smaller than those of differentiated G4 and H9.
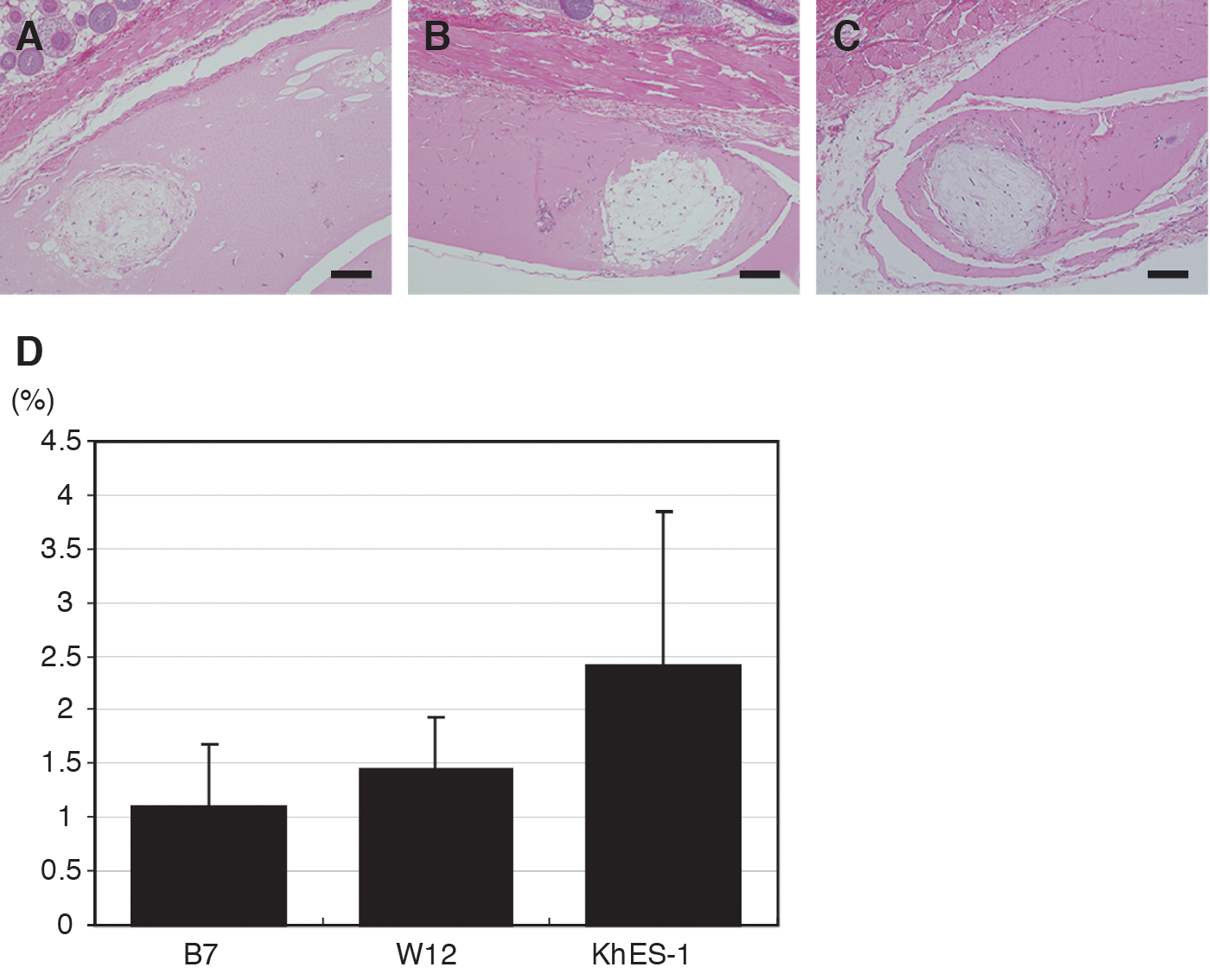
Transplantation of adipocytes derived from other human pluripotent stem cells.
Discussion
The present study demonstrates that human iPS and ES cells can differentiate into adipocytes with functional properties in vitro and that adipocytes derived from human iPS and ES cells can survive and maintain the differentiated properties in vivo for at least 4 weeks after transplantation.
We and others have reported that human iPS [13,33] and ES [34 –40] cells have adipogenic potential in vitro. However, the functional properties of adipocytes derived from human iPS cells have not yet been fully characterized. Lipid storage and lipolysis are considered as major functions of mature adipocytes. Adipocytes store triacylglycerol when energy is in excess, and hydrolyze triacylglycerol to release fatty acids for utilization in other cells in response to energy demand. Triacylglycerol synthesis occurs in various tissues, while lipolysis predominantly occurs in adipose tissue [41]. The present study shows that PPARγ is localized in the nuclei of differentiated human iPS cells containing lipid droplets, and that these cells exhibit forskolin-stimulated lipolytic responses and insulin-induced AKT phosphorylation, suggesting that adipocytes with functional properties, such as lipid storage, lipolysis, and insulin responsiveness, can be differentiated from human iPS cells.
We also confirmed the presence of adipocytes after transplantation of differentiated human iPS and ES cells. We studied whether in vitro-differentiated adipocyte-derived human PS cells survived or not. When human iPS and ES cells are subjected to in vitro adipogenic induction via EB formation, the derivatives make up a heterogeneous population that includes adipocytes, preadipocytes, residual undifferentiated cells, and other cell types. Thus, adipocytes in transplanted Matrigel could be derived from any of these cell types after transplantation.
Time course analysis showed that the presence of adipocytes was confirmed at 1 week after transplantation of differentiated iPS and ES cells. Then, relative adipocyte areas peaked at 2 weeks. The adipocytes at 2 weeks after transplantation exhibited little or no proliferative capacity compared to immature neuroectodermal cells derived from undifferentiated iPS cells. Adipocytes were still present at 4 weeks. These findings indicate that adipocytes survive and maintain the differentiated state for 4 weeks after transplantation. Further, adipocytes described above were not seen in transplantation of undifferentiated iPS cells.
In that context, we hypothesize that the presence of adipocytes in transplanted Matrigel reflects the survival and maintenance of iPS and ES cells differentiated in vitro, although in vivo differentiation of preadipocytes may also contribute to the adipocytes seen in Matrigel after transplantation. Adipocytes as well as skeletal muscle cells, osteocytes, and chondrocytes are thought to be derived from mesenchymal progenitor cells [42,43]. Human mesenchymal stem cells have a characteristic surface antigen profile. Mesenchymal progenitor cells derived from human PS cells are enriched by sorting CD73- or CD105-positive cells [14,33,34,38]. We investigated the presence of mesenchymal progenitor cells in differentiated iPS and ES cells with adipogenic cocktail just before the transplantation. The expression of these surface antigens was analyzed in differentiated iPS and ES cells after the treatment with adipogenic cocktail. However, these representative surface antigens of human mesenchymal stem cells were not detected in differentiated iPS and ES cells (Supplementary Fig. S5), suggesting that mesenchymal progenitor cells are rare populations in differentiated iPS and ES cells after adipogenic induction at that point although mesenchymal progenitor cells may reside in derivatives from human PS cells at an earlier time point of this differentiation protocol.
Transplantation of mature adipocytes often results in graft loss caused by direct reduction of the number of viable adipocytes [22]. Transplantation of mature adipocytes together with adipose-derived stem cells significantly improves survival times and graft volumes, as compared with transplantation of mature adipocytes alone [21,44]. Thus, cotransplantation of mature adipocytes and preadipocytes derived from human iPS and ES cells may be advantageous for graft survival. Establishing a lineage-specific adipocyte differentiation protocol and the methods for the purification of adipocyte progenitors derived from human PS cells will be essential for the success of future cell therapies using adipocytes derived from human iPS cells.
Adipose tissue is now known to be a bona fide endocrine organ, which secretes a variety of adipocytokines, including leptin. Generalized lipodystrophy is caused by a profound deficiency in adipose tissue, which leads to diabetes with marked insulin resistance, hypertriglyceridemia, and ectopic lipid accumulation. We and others have established the efficacy and safety of long-term leptin replacement therapy for generalized lipodystrophy [45 –50], but this therapy does not rescue these patients from their generalized lack of adipose tissue. The only complete cure for these patients would be replenishment of adipose tissue or adipocytes. Indeed, transplantation of adipose tissue or adipocyte progenitors has been demonstrated to ameliorate metabolic disorders in animal models of lipodystrophy [20,51]. Generalized lipodystrophy is classified into two types, congenital and acquired lipodystrophy. In the case of congenital lipodystrophy, we need to repair gene mutation of the patient-specific iPS cells for cell therapy. On the other hand, in the case of acquired lipodystrophy, iPS cells are expected to differentiate into adipocytes. However, successful engraftment of adipocytes derived from human iPS cells may be affected by host factors. Then, in consideration of allogeneic transplantation, iPS cell banking is now discussed, and some groups proposed clinical application of iPS cells from HLA homologous donors [52,53]. Transplantation using those allogeneic iPS cells can decrease or minimize the risk of immune rejection. Human iPS cell-derived adipocytes from patients or HLA homologous donors are a new strategy for the treatment of lipodystrophy.
In our transplantation studies, adipocytes derived from differentiated iPS (G4) and ES (H9) cells were clearly observed, whereas adipocytes derived from other human iPS cell lines (B7 and W12) or another human ES cell line (KhES-1) were observed less frequently. This suggests there is diversity among these cell lines with respect to the survival and maintenance of adipocytes. It was previously reported that there are marked differences in differentiation propensity among human ES cell lines [54]. One possible explanation is that these differences are attributable to the difference of in vitro adipogenic differentiation potential among these cell lines caused by their genetic backgrounds, sites of transgene integration, and epigenetic states. Further studies will be needed to clarify the mechanism underlying the observed differences.
In summary, the present study demonstrates that human iPS and ES cells can differentiate into adipocytes with functional properties and that adipocytes derived from human iPS and ES cells can survive and maintain the differentiated properties in vivo for at least 4 weeks after transplantation. Establishment of refined adipocyte differentiation protocol of human iPS and ES cells and the transplantation method of adipocytes derived from human iPS and ES cells will contribute to understanding the pathophysiology of metabolic diseases such as obesity and lipodystrophy as well as to future therapeutic applications.
Footnotes
Acknowledgments
We thank S. Yamanaka and K. Takahashi (Center for iPS Cell Research and Application) for providing human iPS cells and technical assistance of generating human iPS cells. We thank N. Nakatsuji (Institute for Frontier Medical Science, Kyoto University) for providing human ES cells. We thank A. Ryu and M. Nagamoto for technical assistance. This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, including Grant-in-Aid for Scientific Research on Innovative Areas (research in a proposed research area). “Molecular Basis and Disorders of Control of Appetite and Fat Accumulation” research project no. 22126001; the Ministry of Health, Labour and Welfare of Japan; the Takeda Medical Research Foundation; the Smoking Research Foundation; Suzuken Memorial Foundation; Japan Foundation of Applied Enzymology; Novo Nordisk Insulin Research Award; and Lilly Education and Research Grant office.
Author Disclosure Statement
The authors declare no potential conflict of interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.